


 , Hui Wu 2,3,4, ShengYu Gong 5,*
, Hui Wu 2,3,4, ShengYu Gong 5,* , Gang Zhou 2,3,4, Qing Zhang 2,3,4, Song Li 2,3,4, Wei Yang 2,3,4
, Gang Zhou 2,3,4, Qing Zhang 2,3,4, Song Li 2,3,4, Wei Yang 2,3,41 Interventional Diagnosis and Treatment Center, Yichang Central People's Hospital, 443000 Yichang, Hubei, China
2 Institute of Cardiovascular Disease, China Three Gorges University, 443005 Yichang, Hubei, China
3 Department of Cardiology, Yichang Central People's Hospital, 443000 Yichang, Hubei, China
4 Central Laboratory, The First College of Clinical Medical Science, China Three Gorges University, 443005 Yichang, Hubei, China
5 Pharmaceutical Department, Yichang Central People's Hospital, 443000 Yichang, Hubei, China
Abstract
Myocardial ischemia-reperfusion injury is a significant complication of reperfusion therapy and a primary cause of mortality in patients with acute myocardial infarction. The pathogenic mechanism involved in myocardial ischemia-reperfusion injury is intricate, and effective preventive and therapeutic strategies remain limited in clinical practice. Recently, pyroptosis has emerged as a novel regulatory form of cell death and has attracted widespread attention as a key focus in the study of disease mechanisms and therapeutic targets. Studies indicate a close association between pyroptosis and the pathophysiological processes underlying myocardial ischemia-reperfusion injury. This article provides a comprehensive review of recent advances in research on pyroptosis in the context of myocardial ischemia-reperfusion injury. Therefore, this review aims to offer new insights into the prevention and treatment of myocardial ischemia-reperfusion injury while minimizing redundancy in the existing literature.
Keywords
- pyroptosis
- acute myocardial infarction
- myocardial ischemia reperfusion injury
- NOD-like receptor protein 3
- reactive oxygen species
Acute myocardial infarction (AMI) is a severe cardiovascular event typically caused by the sudden occlusion of a coronary artery, usually due to thrombus formation, rupture of an atherosclerotic plaque, or other factors [1]. Abrupt arterial blockage restricts blood flow to the myocardium, leading to cellular damage from oxygen and nutrient deprivation. Even with prompt treatment, myocardial infarction can result in cardiomyocyte necrosis. Following myocardial necrosis, cardiac function may be impaired, potentially leading to serious complications such as heart failure [2]. Common treatments for AMI include thrombolytic medications, percutaneous coronary intervention, and coronary artery bypass grafting. Restoration of coronary blood flow during reperfusion reintroduces oxygen and nutrients to the ischemic myocardium [3]. However, this process may also cause additional damage, known as myocardial ischemia-reperfusion injury (MIRI) [4].
Pyroptosis, identified in recent years, is a distinct form of cell death that differs from both apoptosis and necrosis [5]. As an inflammatory form of cell death, pyroptosis is closely associated with immune activation and the release of inflammatory mediators, playing a crucial role in combating infectious pathogens and cellular damage [6]. Pyroptosis typically involves the formation and activation of inflammasomes, which are intracellular multiprotein complexes. Upon sensing infection- or damage-associated signals, inflammasomes are activated, triggering pyroptosis [7]. This process promotes the release of inflammatory mediators into the surrounding environment, thereby activating immune cells and inflammatory responses, aiding in defense against infections. However, under certain conditions, pyroptosis can also cause excessive inflammation and tissue damage [8]. While pyroptosis plays a vital role in immune responses and inflammation, the dysregulation or overactivation of this process can have adverse effects on health and is associated with various diseases, including cardiovascular diseases [9], infectious diseases [10], and autoimmune disorders [11].
Recently, pyroptosis has been shown to be closely associated with MIRI [12]. However, the precise mechanisms underlying MIRI remain incompletely understood. Thus, this review primarily focuses on the relationship between MIRI and pyroptosis-associated signaling pathways, aiming to provide a theoretical basis for future research and the development of novel clinical therapeutic targets.
During myocardial ischemia-reperfusion, insufficient oxygen and nutrient supply disrupt cardiomyocyte homeostasis, leading to an imbalance in the intracellular redox state [13]. Upon reperfusion, there is an increased generation of intracellular reactive oxygen species (ROS), including superoxide radicals, hydrogen peroxide, and hydroxyl radicals [14, 15]. Under normal circumstances, moderate ROS levels help activate inflammatory signaling pathways and promote the synthesis and secretion of inflammatory mediators, thereby supporting a normal inflammatory response [16]. However, during myocardial ischemia-reperfusion, excessive ROS (e.g., superoxide) not only cause direct cellular damage but also activate the NLR family pyrin domain-containing 3 (NLRP3) inflammasome via several molecular intermediates [17]. For example, ROS promote the dissociation of thioredoxin-interacting protein (TXNIP) from thioredoxin, enabling TXNIP to bind and activate NLRP3 [18]. Additionally, ROS can induce mitochondrial dysfunction, leading to the release of mitochondrial DNA and cardiolipin, which further activate NLRP3 inflammasome assembly [19]. These events facilitate the recruitment of apoptosis-associated speck-like protein containing a CARD (ASC) and pro-caspase-1, resulting in caspase-1 activation, gasdermin D (GSDMD) cleavage, and pyroptotic cell death, thereby exacerbating MIRI [13, 20]. Furthermore, oxidative stress can cause membrane damage, impair mitochondrial function, and ultimately lead to cardiomyocyte and tissue injury or even death [21]. Studies have found that, in a myocardial infarction model, inducing ROS activation with nitrogen oxide (NOx), followed by ROS overexpression, can promote the formation of inflammasomes composed of caspase-1/NLRP3/ASC, thereby inducing pyroptosis in cardiomyocytes and worsening reperfusion injury [22]. Consistently, in a mouse model of myocardial infarction, geniposide was reported to prevent cleavage of the key pyroptosis protein GSDMD, thereby inhibiting pyroptosis and improving cardiac function [23]. Collectively, these studies suggest that excessive ROS can induce pyroptosis in cardiomyocytes and exacerbate MIRI.
Early reports have suggested a close relationship between calcium overload and pyroptosis [24]. During myocardial ischemia, the function of cell membrane channels is impaired, leading to the accumulation of intracellular calcium ions (Ca2+). Upon reperfusion, a significant influx of Ca2+ into cells triggers a series of adverse biochemical reactions, including the activation of oxidative stress responses and inflammation, ultimately leading to cell death [25]. Studies have shown that Ca2+ overload can also impair mitochondrial function, induce mitochondrial permeability transition, and promote the release of pro-death signaling molecules stored within mitochondria. These signaling molecules, in turn, can activate pyroptotic pathways, ultimately resulting in MIRI [26]. Notably, some research teams have treated rat cardiomyocytes with hydrogen peroxide (H2O2) and observed increased Ca2+ levels and excessive ROS production, which eventually damage the cell membrane. Following membrane disruption, cardiomyocytes release significant amounts of cytokines, interferons, chemokines, and other factors, thereby inducing pyroptosis and exacerbating myocardial injury [27]. In summary, there is substantial evidence to infer that Ca2+ overload is closely associated with pyroptosis and can exacerbate MIRI.
In myocardial ischemia, reduced blood flow and inadequate oxygen and nutrient supply not only damage cardiomyocytes but also affect endothelial cells in the surrounding blood vessels [21]. When coronary arteries are reperfused, the rapid restoration of oxygen and nutrient supply to the myocardium can paradoxically exacerbate endothelial cell damage. During reperfusion, cells may undergo pathological processes, including the generation of oxygen-free radicals, inflammatory responses, and pyroptosis [28, 29]. It has been found that inhibiting microvascular endothelial cell damage and pyroptosis in MIRI mouse models can alleviate myocardial ischemia/reperfusion injury and protect cardiovascular microvascular function [30]. Furthermore, in a foundational study using a reperfusion injury model, endothelial injury was shown to be mediated by regulating caspase family activation through downregulation of the Beclin 1 (BECN1) gene [31]. Based on these findings, we infer that pyroptosis may disrupt endothelial cell function and thereby contribute to the regulation of MIRI.
It is well known that mitochondria are the cellular powerhouses responsible for
energy production, and the stability of mitochondrial function is crucial for the
normal physiological activities of cardiac cells [32]. During MIRI, mitochondrial
dysfunction occurs, often leading to impaired adenosine triphosphate (ATP) synthesis. ATP is a key energy molecule essential for maintaining
cell survival and function [33]. Upon restoration of blood flow to the infarcted
area, a significant and instantaneous replenishment of ATP occurs, a major cause
of ATP disruption during ischemia [33]. Normal ATP levels help maintain
intracellular potassium ion (K+) concentrations, thereby preventing GSDMD
activation and membrane rupture, ultimately reducing pyroptosis [34]. Once ATP is
released into the extracellular space, the molecule can act as a danger signal
that triggers inflammasome activation [35]. The inflammasome is a multiprotein
complex, including the NLRP3 inflammasome, which can activate the production of
the proinflammatory cytokines interleukin-1
In summary, pyroptosis in MIRI is not an independent process but is influenced by multiple interrelated factors (Fig. 1).
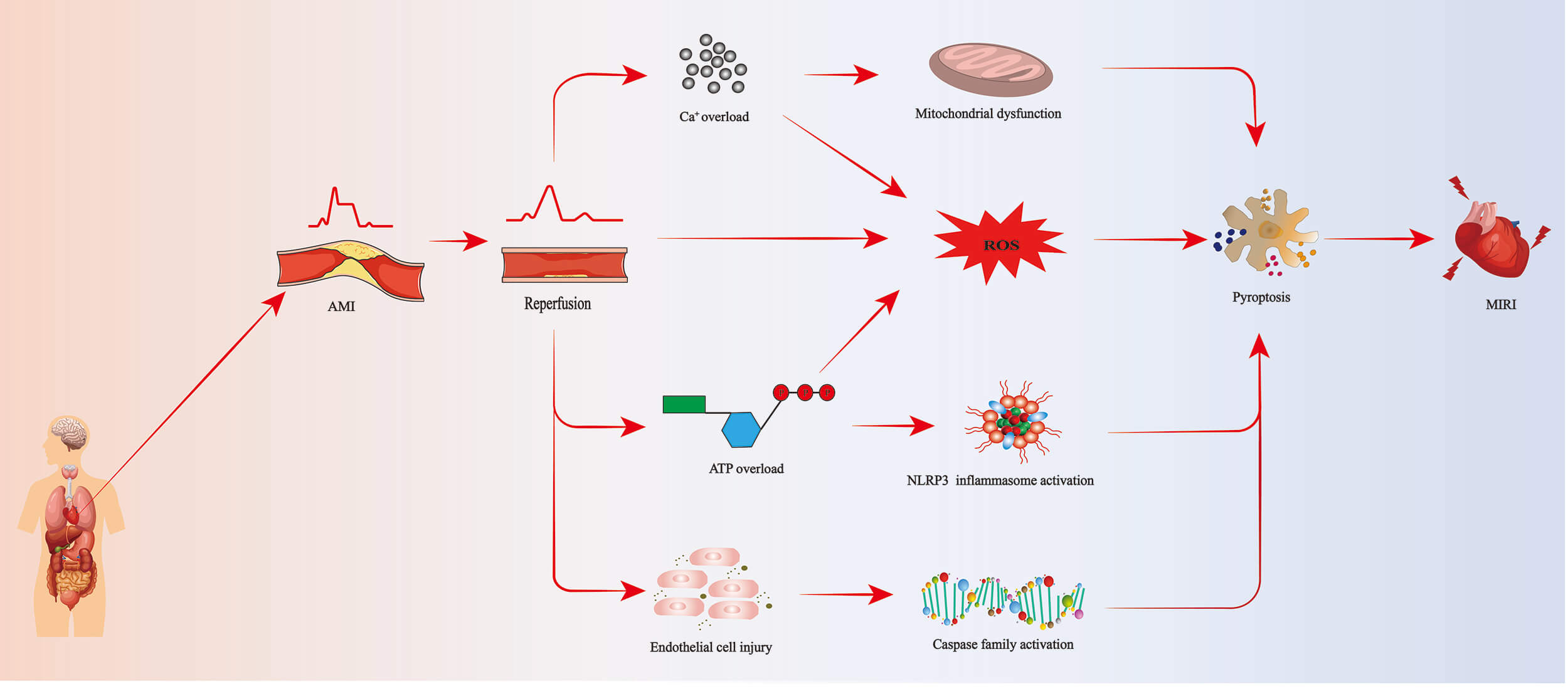 Fig. 1.
Fig. 1.
Pyroptosis pathway leading to MIRI. AMI, acute myocardial infarction; ROS, reactive oxygen species; ATP, adenosine triphosphate; NLRP3, NLR family pyrin domain-containing 3; MIRI, myocardial ischemia reperfusion injury.
Pyroptosis exhibits specific morphological characteristics distinct from other
forms of cell death, such as apoptosis, autophagy, and necrosis. During
pyroptosis, damaged cells undergo pronounced cytoplasmic swelling, leading to a
significant increase in cell volume, attributed to abnormal accumulation of
intracellular water and ions [39]. Meanwhile, pyroptosis is typically associated
with cell membrane rupture, primarily due to the formation of pores or channels.
This differs from the release of cytoplasmic bodies during apoptosis.
Additionally, pyroptosis induces DNA fragmentation, producing characteristic DNA
fragments that are distinct from those produced by the gradual degradation of DNA
in apoptosis [40]. These pores are often mediated by GSDMD, which is cleaved to
produce the membrane-pore-forming GSDMD-N. This fragment, in turn, forms pores on
the cell membrane, leading to membrane disruption and the release of
intracellular contents into the extracellular space. This controlled pore
formation is a crucial distinction between pyroptosis and necrosis, in which
plasma membrane rupture is uncontrolled [41, 42]. Pyroptosis is also distinct from
autophagy, where double-membraned autophagosomes encapsulate cytoplasmic
materials and then fuse with lysosomes for content digestion [43]. Moreover,
pyroptosis is an inflammatory form of cell death, typically accompanied by
inflammasome activation. Inflammasomes are multiprotein complexes activated upon
sensing infection or cellular damage signals. Additionally, inflammasomes trigger
an inflammatory response, leading to the release of proinflammatory cytokines
such as IL-1
| Classification | Key stimuli | Key proteins | Morphological changes |
| Pyroptosis | LPS, ATP, ROS, bacterial infection | Caspase-1/4/11, IL-18, GSDMD, IL-1 |
Cell swelling, plasma membrane pore formation, release of cellular contents, intact nucleus with DNA fragmentation |
| Apoptosis | DNA damage, growth factor withdrawal, TNF | Caspase-3/8/9, p53, Bcl-2 family | Cell shrinkage, chromatin condensation, nuclear fragmentation, formation of apoptotic bodies, intact plasma membrane |
| Autophagy | Nutrient starvation, ER stress, rapamycin | ATG5, ATG7, beclin-1, LC3, p62 | Formation of double-membraned autophagosomes, cytoplasmic vacuolization, degradation of organelles, partial chromatin condensation |
| Necroptosis | TNF, LPS, viral infection | RIPK1, RIPK3, MLKL | Organelle swelling, plasma membrane rupture, moderate chromatin condensation, release of DAMPs |
| Necrosis | Physical/chemical trauma, abrupt ATP depletion | Nonspecific (accidental) | Rapid cell swelling, loss of membrane integrity, organelle disintegration, random DNA degradation, no typical apoptotic or autophagic features |
GSDMD, gasdermin D; IL, interleukin; DAMPs, damage-associated molecular patterns; LPS, lipopolysaccharide; TNF, tumor necrosis factor; ER, endoplasmic reticulum; ATG, autophagy related; LC, microtubule-associated protein 1 light; RIPK, receptor-interacting protein kinase; MLKL, mixed lineage kinase domain-like protein.
Pyroptosis is a distinct form of cell death with unique molecular characteristics that can be used to distinguish this process from other modes of cell death. The following are the key molecular features of pyroptosis.
Caspases constitute a conserved family of cysteine proteases, with a structure
comprising an N-terminal caspase recruitment domain, a central large catalytic
domain, and a C-terminal small catalytic subunit domain. Together, these domains
collaborate to form the enzymatic active site [45]. Functionally, caspases can be
categorized based on the associated role and activity, including those associated
with inflammation, such as caspase-1/3/4/5/6/7/8/11 and 12 [46], which mediate
pyroptosis through distinct structures and activities [47]. Caspases related to
apoptosis include caspase-2, 8, 9, and 10 [48]. Meanwhile, caspase-8, which
participates in both pyroptosis and apoptosis, can activate NLRP3 and cleave
GSDMD [49]. One study found that caspase-1, while binding to the N- and
C-terminal connectors of GSDMD, also interacts with the hydrophobic pocket at the
distant end of the N-terminal domain through anti-parallel
The formation of inflammasomes is a critical feature of pyroptosis, in which, upon sensing danger signals inside and outside the cell, inflammasomes assemble and activate, triggering inflammatory responses and pyroptosis [51]. Pyroptosis is typically triggered by specific stimuli, such as those from infectious pathogens, injury, or other danger signals. These signals are recognized by intracellular sensors, primarily members of the pattern recognition receptor (PRRs) family, including nucleotide-binding oligomerization domain (NOD) and leucine-rich repeat (LRR) proteins, such as NOD-like receptor (NLR) family members, the absent in melanoma 2 (AIM2) receptor, and the tripartite motif (TRIM) family protein pyrin, among which NLRP1, NLRP3, NLR family CARD domain containing 4 (NLRC4), and the mouse proteins Nlrp1a and Nlrp1b are confirmed to form inflammasomes [52]. Once inflammatory signals are sensed, NLRP3 and ASC, along with caspase-1, assemble to form the inflammasome complex [53]. Pyroptosis can contribute to ox-LDL-induced macrophage death, promoting the formation of late necrotic cores in atherosclerotic plaques and increasing the instability of fibrous plaques by activating NLRP3 inflammasomes [54]. Studies further suggest that pyroptosis in endothelial cells, vascular smooth muscle cells, and macrophages, among others, contributes to the formation and progression of atherosclerosis by inducing NLRP3 inflammasome formation and regulating pyroptotic cell death [55]. Collectively, these findings highlight the crucial role of inflammasomes in the initiation and execution of pyroptosis.
GSDMD is a crucial molecule in pyroptosis, comprising highly conserved N- and
C-terminal functional domains. The N-terminal domain typically targets the cell
membrane, leading to cell swelling, rupture, and the release of intracellular
inflammatory factors [56]. In the resting state, the C-terminal and N-terminal
are in an autoinhibitory state, preventing pyroptosis and maintaining cellular
homeostasis [57]. During pyroptosis, damage-associated molecular patterns (DAMPs)
released by damaged cells are recognized by inflammasomes in immune cells,
thereby activating the classical pyroptotic pathway through caspase-1. Once
activated, caspase-1 selectively cleaves GSDMD, generating the N-terminal GSDMD
fragment, a pore-forming peptide. This peptide binds to phospholipids in the cell
membrane, causing swelling and rupture of the immune cell membrane, thereby
promoting the release of inflammatory factors such as IL-1
In summary, cell pyroptosis can be triggered by multiple proteins and signaling pathways that are both distinct and interrelated (Fig. 2). Thus, elucidating the molecular mechanisms of cell pyroptosis will facilitate the development of targeted strategies to modulate this form of cell death, thereby protecting the body from pyroptosis-associated damage.
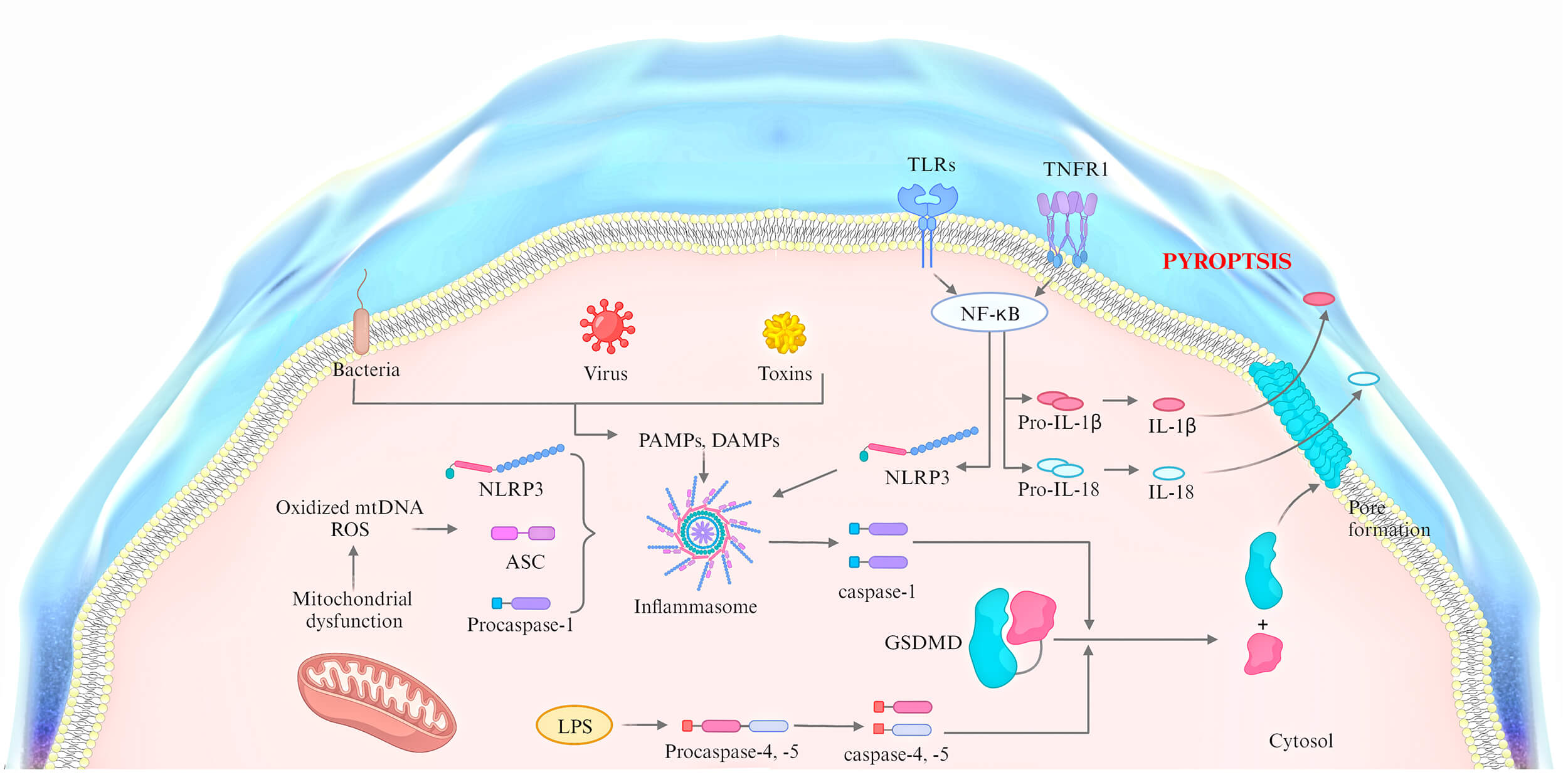 Fig. 2.
Fig. 2.
The mechanisms of pyroptosis. ASC, apoptosis-associated speck-like protein containing a CARD; TLRs, toll-like receptors; TNFR, tumor necrosis factor receptor; PAMPs, pathogen-associated molecular patterns.
Cuproptosis is a novel form of regulated cell death. The main mechanism of copper-induced death involves the intracellular accumulation of copper ions, which directly bind to lipoyl moieties within the tricarboxylic acid (TCA) cycle, leading to protein aggregation, dysregulation, TCA cycle disruption, protein toxicity stress, and induction of cell death [60]. Studies have shown that levels of cell death-related genes and proteins increase significantly with elevated copper levels in porcine jejunal epithelial cells treated with excess copper, indicating that copper overload can induce cell death [61]. In addition, copper overload has been shown to mediate macrophage pyroptosis and regulate the inflammatory response by activating the NLRP3 inflammasome. In a mouse model of NLRP3 activation-induced inflammation by intraperitoneal injection of Escherichia coli-derived lipopolysaccharide after pretreatment with the copper chelator Trillium tschonoskii Maxim (TTM), a decrease in serum caspase-1-dependent cytokines was observed, whereas caspase-1-independent cytokines were not affected by TTM pretreatment [62]. Recent research has moved beyond conceptual links to define specific cuprototic pathways active in the heart. Indeed, a pivotal 2025 study revealed that in MIRI, the upregulated protein proprotein convertase subtilisin/kexin type 9 (PCSK9) directly binds to lipoyl synthase (LIAS), a key mitochondrial enzyme, and triggers cuproptosis in cardiomyocytes. Therapeutic inhibition of PCSK9 with evolocumab disrupted this interaction, alleviated cardiac damage, and improved function in mice, identifying a novel cardioprotective target [63]. These studies suggest a close connection between cuproptosis and pyroptosis.
N6-methyladenosine (m6A) is the most prevalent RNA modification, present in the
mRNA of most eukaryotes and some viruses. Meanwhile, m6A methylation is the
process through which RNA molecules selectively add a methyl group to specific
adenine residues via the RNA methyltransferase complex [64]. Recent reports have
revealed the involvement of cell death in diabetic retinopathy and identified a
close association with m6A methylation mediated by methyltransferase-like protein
3 (METTL3) [65]. Moreover, METTL3 regulates the occurrence of pyroptosis
through m6A modification of MALAT1 [66]. Diao et al. [67] discovered the regulatory role of the
phosphatase and tensin homolog (PTEN) in cell death. The study indicated that
PTEN mRNA with m6A sites can inhibit NLRP3 inflammasome activation and
the expression of cell death-related proteins through the phosphatidylinositol
3-kinase (PI3K)/Akt/GSK-3
Ubiquitination is a process in which a class of low-molecular-weight proteins undergoes specific enzymatic reactions to categorize intracellular proteins, select target substrates, and mediate specific modifications of the target proteins [70]. These key enzymes include ubiquitin-activating enzymes, conjugating enzymes, ligases, and degrading enzymes. Ubiquitination plays a crucial role in the localization, metabolism, function, regulation, and degradation of proteins. Simultaneously, ubiquitination also participates in the regulation of nearly all vital cellular processes, including the cell cycle, proliferation, pyroptosis, DNA repair, and inflammatory immunity [71]. NLRP3, a member of the NOD-like receptor family, can broadly detect various stimuli, including pathogenic microorganisms, bacterial toxins, and inflammatory signals [72]. Activation of NLRP3 is a prerequisite for the assembly of the NLRP3 inflammasome and pyroptosis [73]. Increasing evidence suggests that the ubiquitin–proteasome system influences the assembly and activation of the NLRP3 inflammasome, and that deubiquitination of NLRP3 is crucial for activating the NLRP3 inflammasome [74]. A seminal study showed that ubiquitination of liver kinase B1 (LKB1) significantly inhibited the NLRP3 inflammasome response via the LKB1/AMPK pathway, ultimately reducing pyroptosis [75]. Additionally, ROS generation was identified as a critical link in regulating NLRP3 inflammasome activation. The accumulation of cytoplasmic ROS disrupts the interaction between NLRP3 and ubiquitin. Importantly, when cytoplasmic ROS are eliminated using N-acetylcysteine, NLRP3 reverts to a polyubiquitinated state, leading to significant downregulation of NLRP3 inflammasome-related proteins and a significant decrease in pyroptosis [76]. A novel mechanism involves the E3 ligase Listerin, which catalyzes K63-linked polyubiquitination of the cholesterol transporter ABCA1. This non-degradative ubiquitination stabilizes ABCA1, promoting cholesterol efflux from macrophages and attenuating atherosclerosis—a primary cause of ischemic events [77]. Collectively, these studies indicate that ubiquitination significantly influences the incidence of pyroptosis.
MicroRNAs (miRNAs) are a class of small single-stranded RNAs closely associated
with the regulation of gene expression [78]. Notably, miRNAs regulate pyroptosis
by binding to the 3′ untranslated regions of various pyroptosis-related
protein mRNAs, thereby inhibiting their translation or inducing their degradation
[79]. Studies have shown a significant increase in the expression of
miRNA-30d in streptozotocin-induced type 2 diabetes (T2DM) rats and
cardiomyocytes treated with high glucose. The upregulation of miRNA-30d promotes caspase-1 expression and the release of the proinflammatory cytokines
IL-1
Recent research suggests that several pyroptosis inhibitors can suppress cardiac
inflammation, improve MIRI, enhance cardiac function, and reduce myocardial
infarction. As further investigations advance, drugs targeting pathways involving
NLRP3, caspase-1, GSDMD, IL-1
In an ischemia-reperfusion model of Sprague–Dawley rats, Peng et al.
[83] discovered that the ethyl acetate extract of C. ramulus and the
associated bioactive component cinnamic acid can attenuate MIRI by inhibiting
NLRP3 inflammasome and cell pyroptosis. Cinnamaldehyde, one of the main active
compounds in cinnamon, was shown in a rat ischemia-reperfusion model to
counteract MIRI by inhibiting NLRP3 inflammasome activation and GSDMD-mediated
myocardial pyroptosis [84]. Aesculin, a hydroxycoumarin glycoside with various
biological properties, was found by Xu et al. [85] to protect
cardiomyocytes from MIRI by inhibiting NLRP3 inflammasome-mediated pyroptosis in
a rat ischemia-reperfusion model. Cathepsin B (CTSB) plays a crucial role in
regulating cell death, inflammatory responses, and angiogenesis. In a rat
ischemia-reperfusion model, Ilexsaponin I (ISI), a triterpene saponin extracted
from holly, significantly inhibited CTSB-triggered NLRP3 inflammasome activation
and reduced the maturation of IL-1
| Researcher | Year | Model | Target | Drug |
| Peng et al. [83] | 2021 | I/R-treated rat | NLRP3, IL-1 |
Ethyl acetate extract of Cinnamomi Ramulus |
| Luan et al. [84] | 2022 | I/R-treated rat | NLRP3/caspase-1/GSDMD | Cinnamaldehyde |
| Xu et al. [85] | 2021 | I/R-treated rat, OGD/R-treated NRCMs | NLRP3, Akt/GSK3 |
Aesculin |
| Wu et al. [86] | 2022 | I/R-treated rat, OGD/R-treated H9c2 | NLRP3, cathepsin B/HSP70 complex | Ilexsaponin I |
| An et al. [87] | 2023 | I/R-treated rat, H/R-treated HCMs, 293 T cell lines | Circular RNA PAN3/microRNA-29b-3p/stromal cell-derived factor 4 axis | Sevoflurane |
| Lei et al. [88] | 2022 | I/R-treated rat | NLRP3 | Piperazine ferulate |
| Wang et al. [9] | 2023 | I/R-treated rat, H/R-treated H9c2 | miR-665/MEF2D/Nrf2 axis | Dexmedetomidine |
| Mao et al. [89] | 2021 | I/R-treated C57, H/R-treated NRCMs | TXNIP | Extracellular vesicles |
| Pan et al. [90] | 2023 | I/R-treated rat | PDHA1 | Insulin |
I/R, ischemia-reperfusion; TXNIP, thioredoxin-interacting protein; OGD, oxygen-glucose deprivation; HCMs, neonatal rat cardiomyocytes; GSK, glycogen synthase kinase; MEF2D, myocyte enhancer factor 2d; ARC, apoptosis repressor with caspase recruitment domain; PDHA1, pyruvate dehydrogenase e1 subunit alpha 1.
In the context of MIRI, pyroptosis is a significant mechanism underlying myocardial damage. Therefore, targeting pyroptosis with specific drugs has the potential to reduce cardiomyocyte death and protect cardiac tissue from damage. Overall, modulation of pyroptosis with targeted agents represents a promising strategy for treating MIRI, offering additional therapeutic options in clinical practice and enhancing treatment efficacy.
Pyroptosis, a recently redefined form of regulated cell death, has been confirmed by numerous studies to play a crucial role in MIRI. Despite being a well-studied aspect within MIRI, several pressing issues remain. Firstly, during MIRI, pyroptosis coexists with other forms of cell death, such as apoptosis, necrosis, and autophagy. Therefore, investigating the relationships between cellular pyroptosis and these other cell death modalities will help elucidate the mechanisms of ferroptosis in the MIRI process and enable more effective inhibition of MIRI-triggered cardiomyocyte death. Secondly, drug development targeting cellular pyroptosis in MIRI remains largely confined to basic research. Although these substances are effective in inhibiting myocardial pyroptosis post-MIRI in animal studies, significant progress in clinical practice remains to be achieved. Moreover, research on the impact of the drugs currently approved for clinical use on pyroptosis remains limited. Thus, pharmaceutical treatment targeting pyroptosis in MIRI requires further reinforcement in clinical practice. Finally, MIRI is a dynamic process, and pyroptosis changes with the development of ischemia and reperfusion. Hence, investigating these dynamic changes will help more accurately reveal the pathophysiological mechanisms of MIRI.
In summary, pyroptosis in MIRI holds significant research value. In-depth exploration of the regulatory mechanisms underlying pyroptosis during the MIRI process will yield new perspectives and strategies for preventing and treating MIRI-related injuries.
DZ and SYG conceived the study and wrote the original draft. GZ, QZ, SL and WY participated in writing and editing the manuscript. HW reviewed the manuscript. All authors contributed to the conception and editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by grant from the National Natural Science Foundation of China (No. 81600234).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.