







 , Xin Shao 3, Zihe Zheng 1,2, Mingliang Li 1,2,4, Changbo Xiao 1,2,5, Bo Chen 1,2,6, Guowei Tu 7, Ben Huang 3,*
, Xin Shao 3, Zihe Zheng 1,2, Mingliang Li 1,2,4, Changbo Xiao 1,2,5, Bo Chen 1,2,6, Guowei Tu 7, Ben Huang 3,* , Xiaofu Dai 1,2,*
, Xiaofu Dai 1,2,*1 Department of Cardiovascular Surgery, Fujian Medical University Union Hospital, 350000 Fuzhou, Fujian, China
2 Key Laboratory of Cardio-Thoracic Surgery (Fujian Medical University), Fujian Province University, 350108 Fuzhou, Fujian, China
3 Department of Cardiac Surgery, Zhongshan Hospital, Fudan University, 200032 Shanghai, China
4 Department of Cardiovascular Surgery, The General Hospital of Ningxia Medical University, 750003 Yinchuan, Ningxia Hui Autonomous Region, China
5 Department of Cardiovascular Surgery, Chest Hospital of Zhengzhou University, 450008 Zhengzhou, Henan, China
6 Department of Cardiovascular Surgery, Gaozhou People’s Hospital, 525200 Gaozhou, Guangdong, China
7 Department of Cardiac Intensive Care Centre, Zhongshan Hospital, Fudan University, 200032 Shanghai, China
Abstract
Aortic dissection (AD) is a cardiovascular emergency with high mortality; however, the underlying molecular pathophysiology of AD remains incompletely understood. Pyroptosis, a proinflammatory form of programmed cell death, contributes to vascular injury; nonetheless, the upstream transcriptional regulation of pyroptosis in AD is similarly poorly defined.
Differentially expressed genes were identified in aortic tissues from AD patients (Gene Expression Omnibus (GEO) datasets) using bioinformatics analyses, with a focus on cell death-related candidates. In vivo AD mouse models and in vitro vascular smooth muscle cell (VSMC) systems were employed to investigate the roles of these genes in AD. Potential transcription factors for pyruvate kinase M2 (PKM2) were predicted using the Just Another Simple Array Retrieval/Simple API for Repository (JASPAR) and University of California, Santa Cruz (UCSC) databases, and validated by luciferase reporter and chromatin immunoprecipitation assays. Gain- and loss-of-function approaches were used to dissect the zinc finger protein 460 (ZNF460)–PKM2–gasdermin E (GSDME) axis and the associated impact on pyroptosis and AD progression.
PKM2 expression was markedly elevated in AD tissues. PKM2 silencing suppressed GSDME cleavage, attenuated VSMC pyroptosis, and mitigated experimental AD, whereas PKM2 overexpression aggravated these outcomes. GSDME upregulation rescued pyroptosis in PKM2-depleted cells. Mechanistically, the transcription factor ZNF460 directly bound to the PKM2 promoter, enhancing PKM2 transcription and activating downstream GSDME-mediated pyroptosis. ZNF460 knockdown reduced pyroptotic cell death and preserved aortic wall integrity in vivo.
This study identifies ZNF460 as a novel upstream regulator of PKM2 that drives GSDME-dependent pyroptosis, thereby exacerbating AD progression. Targeting the ZNF460–PKM2–GSDME axis may represent a promising therapeutic strategy for preventing pyroptosis-driven vascular damage in AD.
Keywords
- ZNF460
- PKM2
- GSDME
- pyroptosis
- aortic dissection
Aortic dissection (AD) is a catastrophic vascular disorder characterized by intimal tearing of the aorta and formation of a false lumen, often leading to acute organ ischemia, cardiac tamponade, and sudden death [1, 2]. Despite advances in imaging, surgical techniques, and endovascular therapies, its incidence and mortality remain high [3]. Although factors such as hypertension, connective tissue disorders, and medial degeneration have been implicated [4, 5], the molecular mechanisms driving AD progression remain incompletely defined, limiting the development of effective targeted therapies.
Pyroptosis is an inflammatory form of programmed cell death marked by membrane
pore formation, cytoplasmic swelling, and release of pro-inflammatory cytokines
such as interleukin-1 beta (IL-1
Pyruvate kinase M2 (PKM2), a key glycolytic enzyme, has emerged as a multifunctional protein involved in metabolic reprogramming, inflammation, and regulation of cell death [12, 13, 14]. PKM2 has been shown to promote pyroptotic cell death in several diseases, yet its role in AD is unknown [15, 16, 17]. Of particular interest is whether PKM2 is linked to changes associated with pyroptosis in VSMCs during the progression of AD.
In this study, using integrative bioinformatics analyses and experimental validation, we identified zinc finger protein 460 (ZNF460), a zinc finger transcription factor, as a novel upstream regulator of PKM2. We demonstrate that ZNF460 directly binds the PKM2 promoter, activating PKM2 expression, which in turn promotes GSDME cleavage and pyroptosis. This ZNF460–PKM2–GSDME axis accelerates VSMC destruction and exacerbates AD progression in vivo. These findings uncover a previously unrecognized transcriptional mechanism driving pyroptosis in AD and suggest potential therapeutic targets for this lethal disease.
We analyzed gene expression data from the GSE52093 and GSE153434 public databases to identify differentially expressed genes (DEGs) between AD patients and healthy controls. We performed a Venn analysis to intersect DEGs with pyroptosis-related genes (from GeneCards).
The aortic wall specimens from AD patients were obtained. Control samples were collected from the macroscopically intact, non-dissected portion of the ascending aorta in the same patients. After resection, some specimens were rapidly frozen in liquid nitrogen and stored at –80 °C, while others were fixed in 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) and then preserved in paraffin. All surgeries were performed at Fujian Medical University Union Hospital, and all patients were informed of the purpose of the study and provided written informed consent. This study was approved by the Fujian Medical University Union Hospital Ethics Committee (2024KY180). The study adhered to the Declaration of Helsinki guidelines.
Male C57BL/6 mice, aged 3 weeks and weighing 10–12 g, were used in these
experiments. They were housed in a specific pathogen-free environment with a
temperature of 22–28 °C and relative humidity of 50–60%. The animal experiments were approved by the Ethics Committee of Fujian Medical University Union Hospital (Approval Number: IACUC FJMU 2024-0275). All procedures were performed in accordance with the animal welfare guidelines and regulations. Mice (male C57BL/6, 3 weeks old) were housed under specific pathogen-free conditions with controlled temperature and humidity, and all efforts were made to minimize suffering. Mice were randomly
divided into six groups: control, model, model + si-NC, model + si-PKM2,
model + vector, and model + PKM2, with 6 mice per group included in the
final analysis. Adenovirus-associated virus (AAV) vectors were used to deliver
siRNA or overexpression constructs targeting PKM2. Specifically, the AAV9
serotype was chosen due to its high tropism for VSMCs, ensuring efficient gene
transfer to the target population. Mice were injected via the tail vein with a
total of 1
After routine dehydration, fixation, paraffin embedding, and serial sectioning, the paraffin sections were placed in a 65 °C oven for 1 h. The sections were dewaxed with three changes of xylene, rehydrated through a graded alcohol series, and rinsed with distilled water. Antigen retrieval was performed in a citrate buffer in a microwave oven, and the sections were incubated with 3% hydrogen peroxide (MACKLIN, Shanghai, China) for 20 min. After three washes with PBS (MACKLIN, Shanghai, China), the sections were blocked with 8% goat serum (Solarbio Life Sciences, Beijing, China) at room temperature for 1 h. The primary antibody was incubated overnight at 4 °C. The next day, the sections were washed three times with PBS, incubated with the secondary antibody for 30 min, and washed three more times with PBS. DAB staining was then performed, and the sections were photographed and examined under an optical microscope.
In AD and normal aortic wall tissue samples, we used TRIzol reagent (Invitrogen, Carlsbad, CA, USA) to extract RNA from the tissue through phenol-chloroform extraction. We transcribed the RNA into cDNA using oligo (dT) primers and the Transcriptor First Strand cDNA Synthesis Kit (4896866001, Roche, Basel, Switzerland). GAPDH was used as an internal reference, and RT-PCR was performed on a Bio-Rad RT-PCR instrument (CFX96, Hercules, CA, USA) to amplify the target gene and the reference gene. We calculated and compared the relative expression levels in each tissue based on the Ct values. The PCR reaction system was 20 µL, using a two-step method, 5 min at 95 °C followed by 40 cycles, each cycle consisting of 5 sec at 95 °C and 30 sec at 60 °C.
In 100 mg of frozen aortic wall tissue, and after cutting and grinding, we extracted total protein from the aortic wall tissue using RIPA lysis buffer (P0013B, Beyotime Biotechnology, Shanghai, China). We measured the protein concentration using a BCA protein quantification kit (23225, ThermoFisher Scientific, Rockford, IL, USA). We performed SDS-PAGE electrophoresis with 24 µg of protein and transferred it onto a PVDF membrane (IPVH00010, Millipore, Bedford, MA, USA). The membrane was blocked with 5% non-fat milk at room temperature for 1 h, then incubated the membrane with the appropriate primary antibody dilution solution at 4 °C overnight. The next day, the membrane was washed 3 times with TBST (TBS containing 0.1% Tween 20, P0231, Beyotime Biotechnology, Shanghai, China), 5 min each time. Then, we added the corresponding alkaline phosphatase-labeled secondary antibody and incubated the tissue at room temperature for 1 h. We used a ChemiDocTM XRS+ imaging system (Bio-Rad, Hercules, CA, USA) to detect the protein signal after adding the developing solution.
After sectioning the paraffin blocks, we baked the slices at 65 °C for 1 h, then sequentially hydrated them through xylene, 100%, 95%, and 70% ethanol, and stained them using hematoxylin (5 min), 1% ethanol hydrochloride (1 s), Scott’s solution (1 min), and eosin (1 min). Finally, the samples were dehydrated through graded ethanol solutions (70%, 95%, and 100%), cleared in xylene, and mounted. The basic pathological changes were compared between AD and normal aortic wall samples under a microscope (CKX41, Olympus, Tokyo, Japan).
The levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), IL-1
We used immunofluorescence to detect the expression of phenotype transformation
markers, Alpha-smooth muscle actin (
Primary mouse aortic VSMCs were purchased from SUNNCELL (SNL-521, Wuhan, China). Mouse aortic VSMCs were cultured in DMEM medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in a 5% CO2 incubator. Cells were cultured according to the manufacturer’s instructions and used at passages 3–5 for all experiments. When the cell density reached 70% under the microscope, we starved the cells by treating them with serum-free medium for 12 h, and then divided the cells into 8 groups for treatment: si-NC, si-PKM2, Ang II + si-NC, Ang II + si-PKM2, as well as vector, PKM2, Ang II + vector, Ang II + PKM2.
The binding of ZNF460 to the PKM2 promoter was predicted using the Just Another Simple Array Retrieval/Simple API for Repository JASPAR and University of California, Santa Cruz UCSC Genome Browser Home databases. The promoter sequence of PKM2 and the ZNF460 binding site were obtained from the JASPAR website. This promoter sequence and the mutant promoter sequence were cloned into the pGL4-Luc-Report vector (Promega, Shanghai, China). The constructed plasmids were then transfected into VSMCs. After transfection, the cells were divided into two groups: one group was transfected with the vector alone; the other group was transfected with pcDNA-ZNF460. The cells were cultured for 48 h after treatment, and luciferase activity was determined using a dual-luciferase reporter assay kit (Cat. No. E1910, Promega, Madison, WI, USA).
Cells were seeded in 100 mm dishes and cultured until the cell density exceeded 90%. Based on the volume of the medium, 37.5% formaldehyde was added to each dish to achieve a final concentration of 0.75%. The mixture was then incubated on a rocker at room temperature for 10 minutes to allow cross-linking. Next, 1.5 mL of 2.5 M glycine was added to the mixture, which was further incubated on a rocker at room temperature for 5 minutes to stop the cross-linking reaction. The cells were collected and resuspended in 750 µL of FA lysis buffer. Chromatin was fragmented by sonication, generating fragments ranging from 100 to 1000 base pairs. 25 µg of the cell lysate was incubated with 4 µg of primary antibody and 50 µL of pre-treated Protein G agarose beads for immunoprecipitation (IP). The mixture was rotated and incubated overnight at 4 °C. The beads were washed three times with wash buffer and once with elution buffer. 200 µL of elution buffer was added to the IP samples and rotated at room temperature for 15 min. The IP samples were then treated with RNase A and proteinase K, and DNA was extracted using the phenol-chloroform method. PCR was used to confirm the binding of ZNF460 to the DNA region of the PKM2 promoter.
All experimental data were statistically analyzed using SPSS 22.0 (IBM Corp.,
Armonk, NY, USA), and the results were expressed as mean
Integrated analysis of two Gene Expression Omnibus (GEO) datasets (GSE52093, GSE153434) identified 161 differentially expressed genes in AD tissues compared with healthy controls. Intersection with pyroptosis-related genes yielded four candidates (PLAUR, MLKL, PKM2, CXCL8), among which PKM2 displayed the most pronounced and specific upregulation (Fig. 1A). IHC confirmed elevated PKM2 protein in AD aortic walls, particularly localized in vascular smooth muscle cells (VSMCs) (Fig. 1B). RT-qPCR further validated significantly higher PKM2 mRNA expression in AD tissues (Fig. 1C), and Western Blotting confirmed increased PKM2 protein levels (Fig. 1D,E). These data established PKM2 as a pyroptosis-associated molecule selectively upregulated in AD, especially within VSMCs.
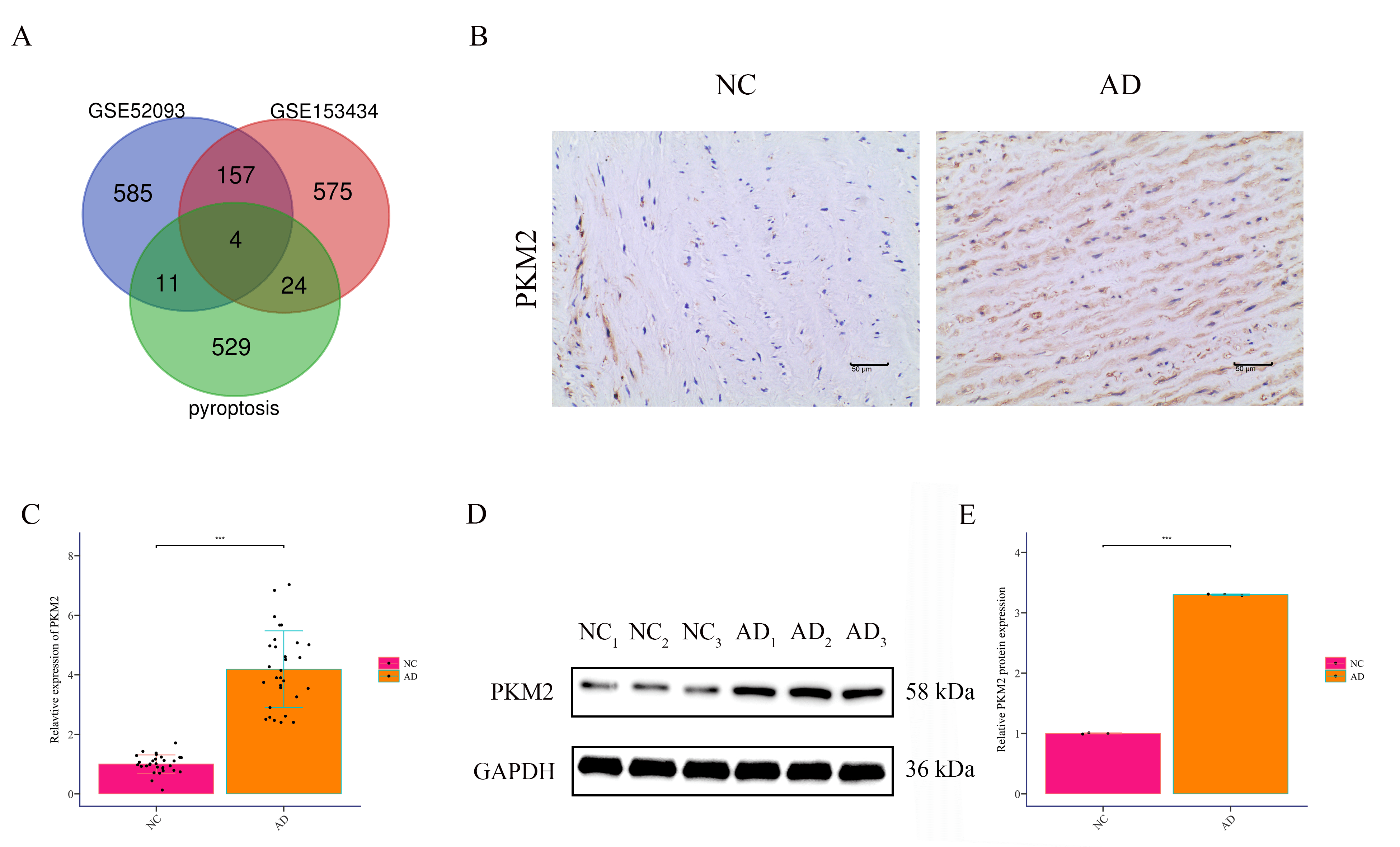 Fig. 1.
Fig. 1.
PKM2 is highly expressed in the aortic wall tissues of
patients with AD. (A) Differential genes in AD patients and healthy controls
from datasets GSE52093 and GSE153434 were analyzed and intersected with
pyroptosis-related genes via Venn analysis. (B) Immunohistochemistry detected
PKM2 expression in the aortic wall tissues of AD patients (AD) and non-dissected
aortic tissues from the same patients (NC) (200
In the murine AD model, HE staining and Masson staining showed marked intimal
tearing and intramural hematomas, which were mitigated by Pkm2 knockdown
(si-Pkm2) but exacerbated by Pkm2 overexpression (Fig. 2A–E).
Serum biochemical assays revealed increased glucose, total cholesterol, and
triglycerides in AD mice, all attenuated by Pkm2 silencing and
aggravated by Pkm2 overexpression (Fig. 2F–H). Western Blotting
demonstrated elevated PKM2, GSDME, GSDME-N, and cleaved caspase3 in AD mice,
reduced by Pkm2 silencing and increased by overexpression (Fig. 3A–F).
Loss of VSMC contractile markers
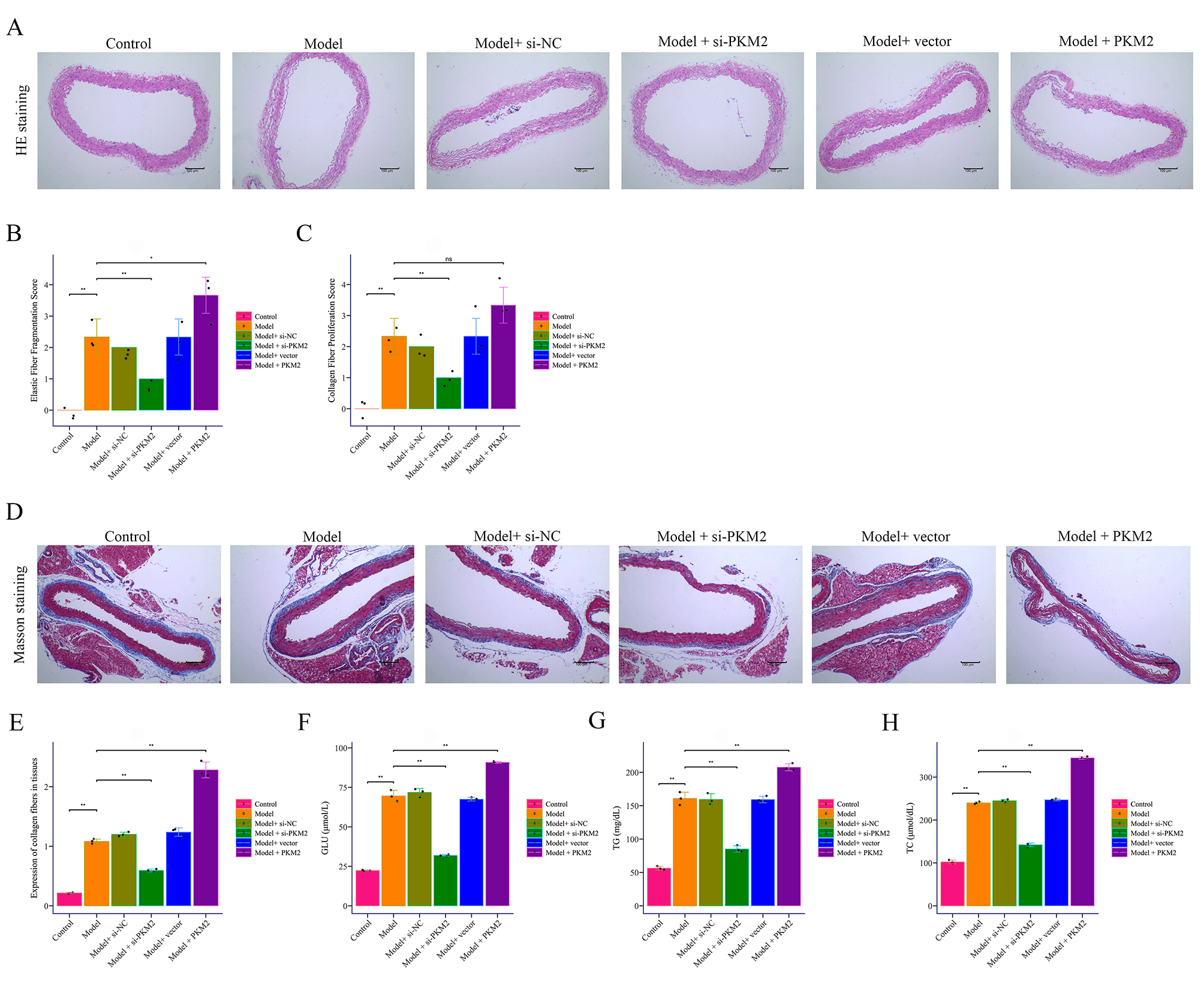 Fig. 2.
Fig. 2.
The effect of PKM2 on BAPN combined with Ang II-induced
mouse AD. (A–E) HE staining and Masson staining to detect pathological changes
in AD (100
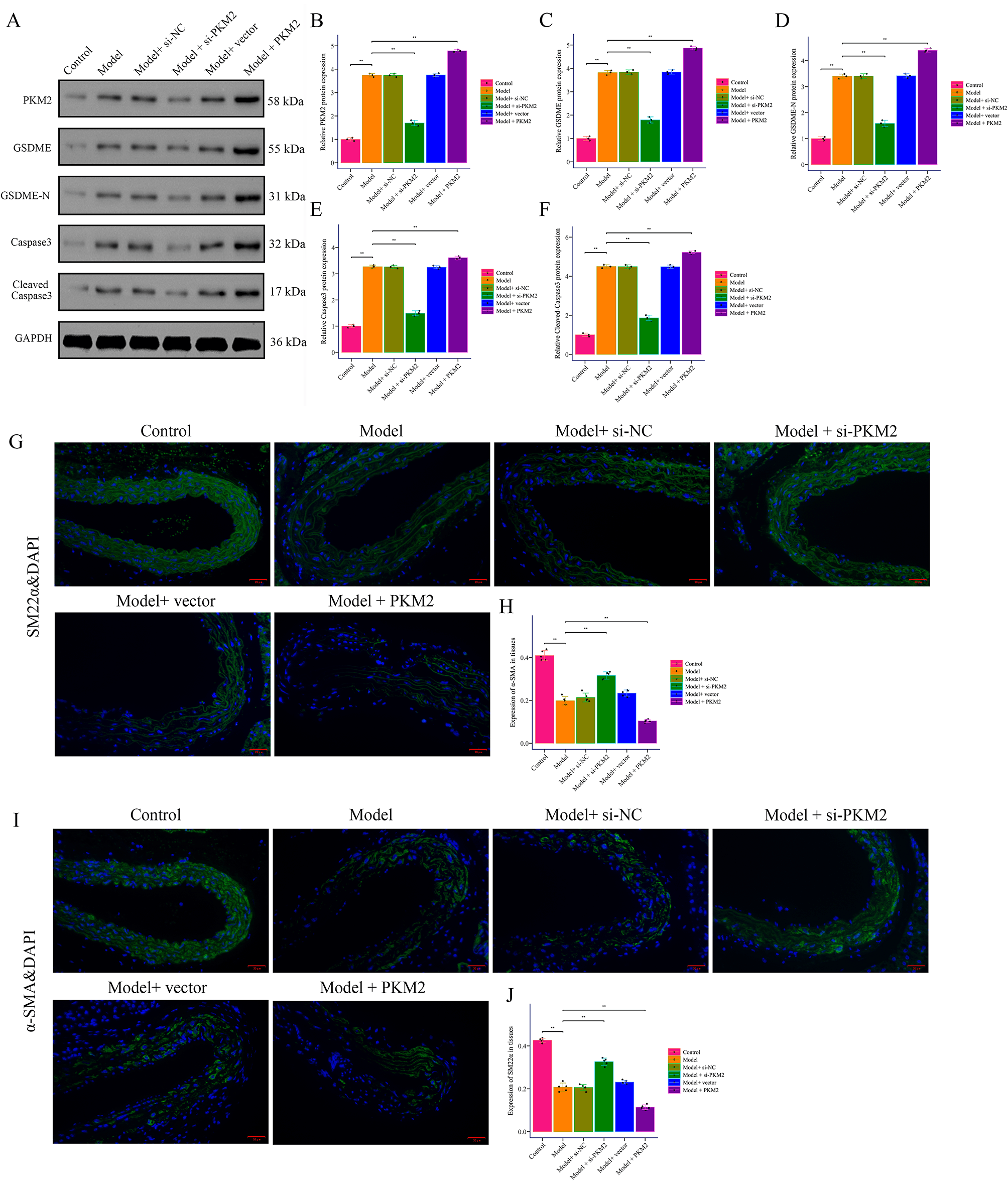 Fig. 3.
Fig. 3.
PKM2 promotes pyroptosis in AD induced by BAPN combined
with Ang II in mice. (A–F) Western Blot was used to detect the expression of
PKM2, GSDME, GSDME-N, caspase3, and cleaved caspase3 in the aortic wall tissue of
mice with AD induced by BAPN combined with Ang II. (G–J) Immunofluorescence was
used to detect the expression of
The results showed that PKM2 expression was significantly increased in the Ang
II treatment group, while si-PKM2 treatment significantly decreased PKM2
expression (Fig. 4A), and pcDNA-PKM2 treatment significantly increased PKM2
expression (Fig. 4B). Based on Western Blot results, the expression of GSDME,
GSDME-N, and Cleaved-Caspase3 was significantly increased in the Ang II treatment
group, while si-PKM2 treatment significantly reduced the expression of these
proteins, and pcDNA-PKM2 treatment significantly increased the expression of
these proteins (Fig. 4C–L). We measured lactate dehydrogenase (LDH) release in
the supernatant of VSMCs under different treatment conditions. LDH release is a
well-established indicator of membrane rupture and cell lysis, which are
characteristic features of pyroptosis. Our results showed a significant increase
in LDH release in the Ang II-treated group compared to the control group
(Supplementary Fig. 1A). Notably, this increase was mitigated by PKM2
knockdown, suggesting that PKM2 plays a crucial role in Ang II-induced pyroptosis
(Supplementary Fig. 1A). Using scanning electron microscopy (SEM) or
high-magnification microscopy, we observed characteristic morphological changes
in VSMCs treated with Ang II. Specifically, we detected membrane blebs and
swelling, which are hallmark features of pyroptotic cell death
(Supplementary Fig. 1B). These morphological changes were significantly
reduced in cells where PKM2 was knocked down, further supporting the involvement
of PKM2 in Ang II-induced pyroptosis (Supplementary Fig. 1B). The
results of immunofluorescence staining indicated that SM22
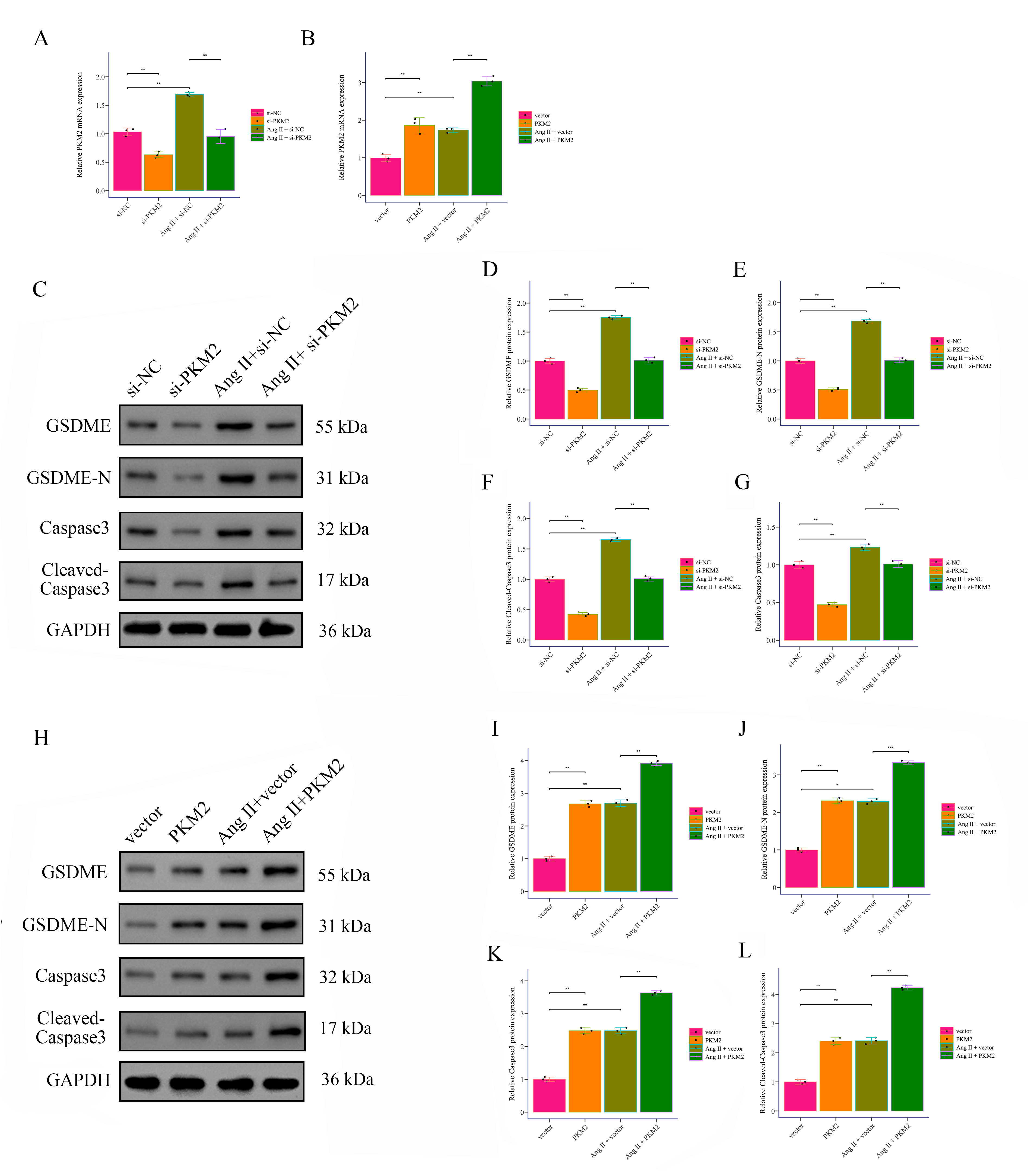 Fig. 4.
Fig. 4.
PKM2 promotes Ang II-induced pyroptosis in VSMCs. (A,B) The effects of PKM2 downregulation and upregulation on PKM2
expression in Ang II-induced and non-induced VSMCs were detected by RT-PCR. (C–L) The expression of pyroptosis markers GSDME, GSDME-N, caspase3, and cleaved
caspase3 in Ang II-induced and non-induced VSMCs was detected by Western Blot.
*p
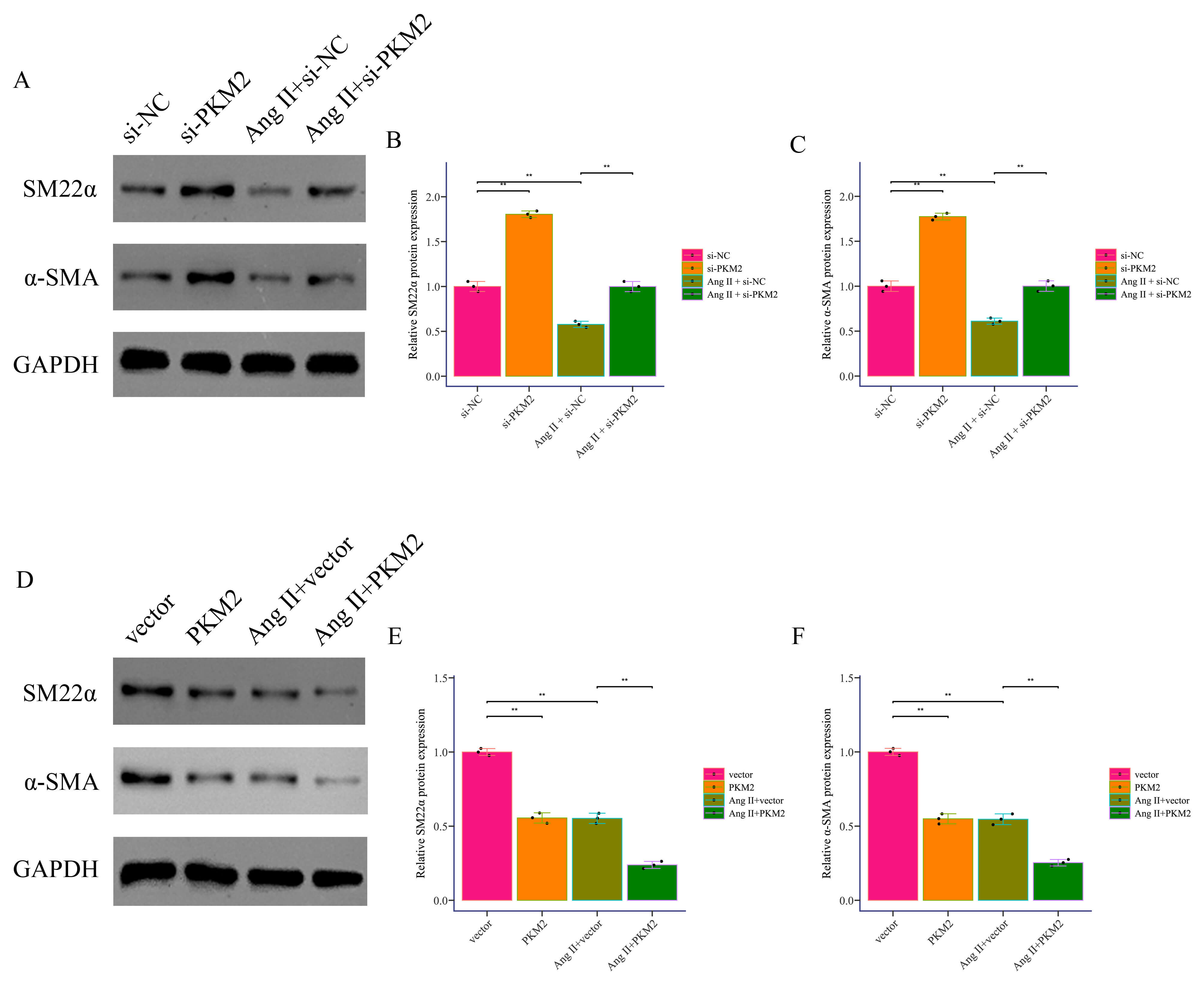 Fig. 5.
Fig. 5.
PKM2 promotes Ang II-induced phenotypic
transformation. (A–C) The expression of SM22
The intersection of PKM2 targets and key pyroptosis proteins was
analyzed by Venn analysis. The blue circle represents PKM2 targets, and
the red circle represents key pyroptosis genes. The overlapping part is the
intersecting gene GSDME (Fig. 6A). In vitro, Ang II treatment
significantly increased the expression of GSDME, while si-PKM2
treatment significantly decreased the expression of GSDME, and
overexpression of GSDME partially restored this effect (Fig. 6B).
Western Blot analysis results showed that Ang II treatment significantly
increased the expression of GSDME and GSDME-N, si-PKM2 treatment
significantly decreased the expression of these proteins, and overexpression of
GSDME partially restored these changes (Fig. 6C–E). As Fig. 6F–H, the results
demonstrated that compared to the Ang II group alone, si-PKM2 treatment
significantly increased SM22
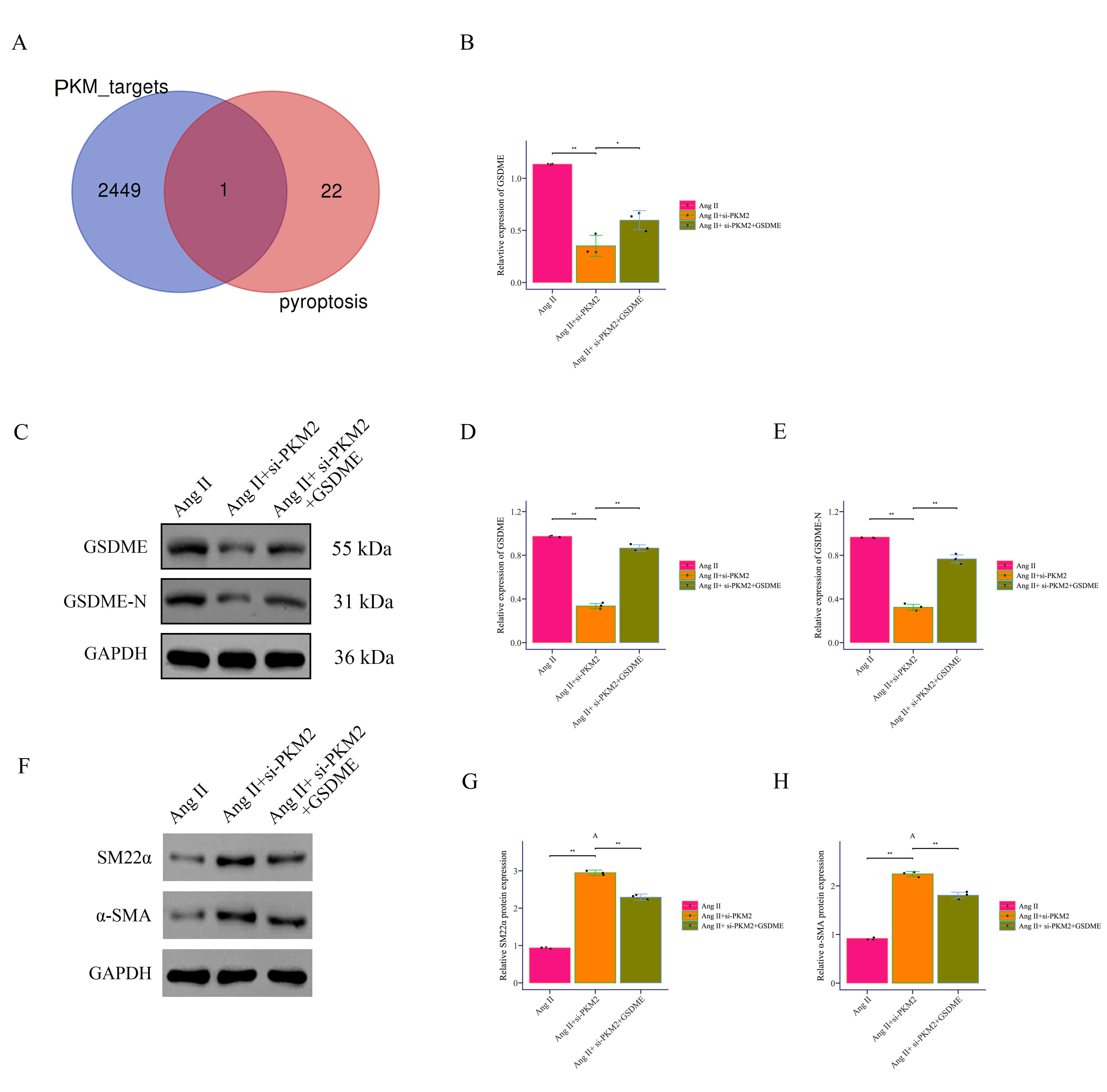 Fig. 6.
Fig. 6.
PKM2 promotes phenotypic transformation and pyroptosis
in Ang II-induced VSMCs by binding to GSDME. (A) Venn diagram analysis
was used to identify the intersection of PKM2 targets and pyroptosis
targets. (B) GSDME expression was detected by RT-PCR. (C–E) GSDME and GSDME-N
expression was detected by Western Blot. (F–H) The expression of SM22
Promoter analysis revealed putative ZNF460 binding sites in the
PKM2 promoter (Fig. 7A,B). Luciferase assays demonstrated that
ZNF460 increased wild-type PKM2 promoter activity but not
mutant constructs (Fig. 7C). ChIP assays confirmed direct binding of
ZNF460 to the PKM2 promoter in VSMCs (Fig. 7D). To validate the
clinical relevance of ZNF460 as a key upstream regulator in aortic
dissection, we conducted additional experiments using human tissue samples. We
performed quantitative real-time polymerase chain reaction (qRT-PCR) and IHC staining to assess the expression levels of
ZNF460 in both AD tissues and non-dissected control tissues. The results of
qRT-PCR demonstrated that ZNF460 mRNA was significantly upregulated in
AD tissues compared to non-dissected controls (Supplementary Fig. 2A).
Consistent with the qRT-PCR data, IHC analysis revealed a marked increase in
ZNF460 protein expression in AD tissues (Supplementary Fig. 2B).
ZNF460 silencing reduced GSDME, GSDME-N, caspase3, and cleaved
caspase3 expression, whereas PKM2 overexpression partially restored them
(Fig. 7E–I). Correspondingly, Ang II–treated cells with ZNF460
knockdown showed reduced serum ALT, AST, CRP, and IL-1
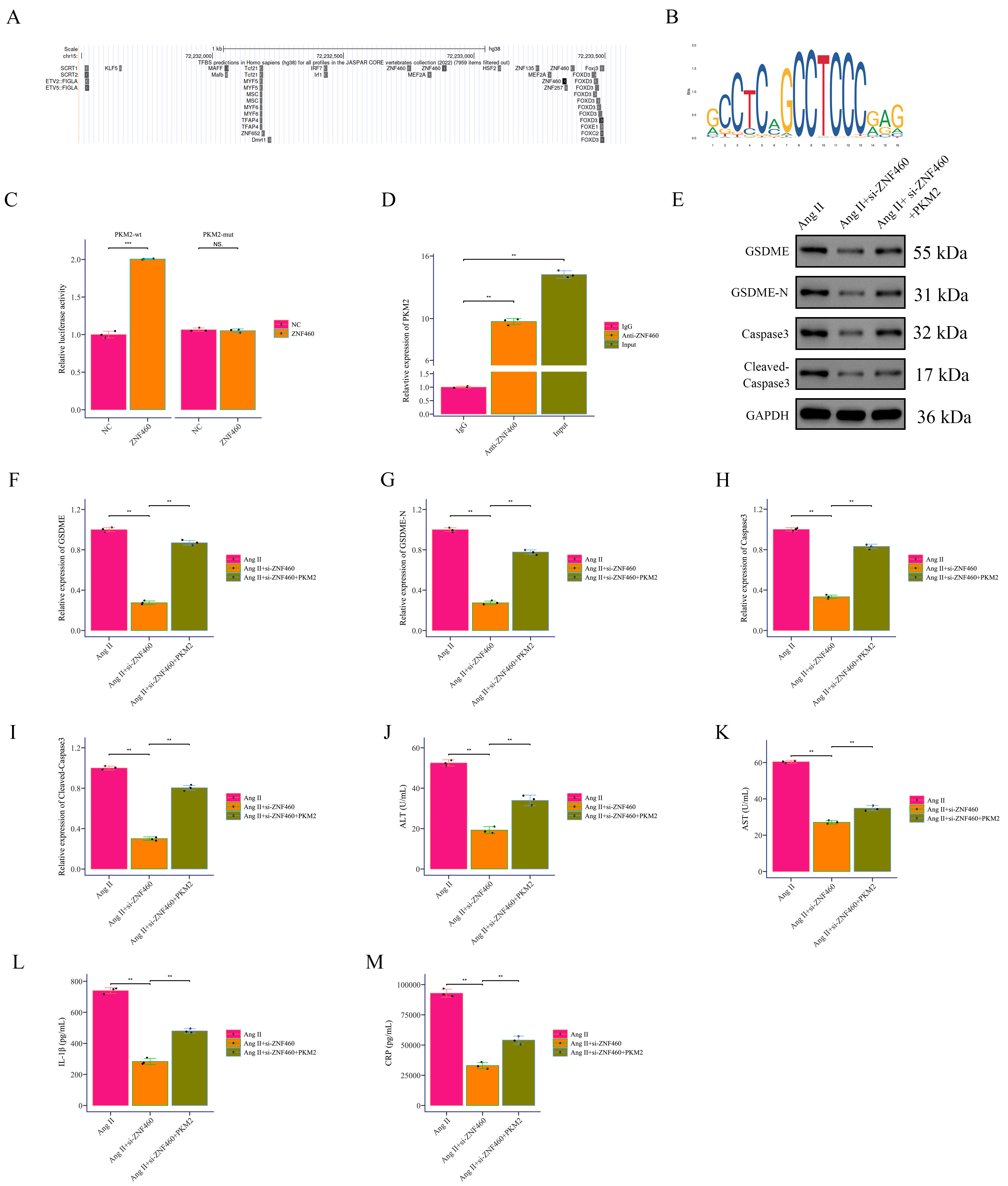 Fig. 7.
Fig. 7.
ZNF460 promotes PKM2 expression at the
transcriptional level. (A) Prediction of transcription factors near the
PKM2 promoter using the UCSC database. (B) Sequence logo of
ZNF460 DNA-binding motif obtained from the JASPAR database. The height
of letters at each position indicates the nucleotide frequency in binding sites.
(C) Luciferase reporter assay showing the effect of transcription factor
ZNF460 on PKM2 expression. (D) ChIP-qPCR analysis of
ZNF460 binding to the PKM2 promoter. (E–I) Western Blot
detection of pyroptosis markers GSDME, GSDME-N, caspase3, and cleaved caspase3.
(J–M) ELISA measurement of inflammatory factor markers ALT, AST, CRP, and
IL-1
In this study, we identified and characterized a previously unrecognized
transcriptional axis, ZNF460–PKM2–GSDME, as a driver
of VSMC pyroptosis and the progression of AD. Using integrated bioinformatics,
in vitro experiments, and in vivo murine models, we
demonstrated that ZNF460 directly binds the PKM2 promoter,
enhancing its transcriptional activity. Elevated PKM2 expression
promoted caspase3 activation, GSDME cleavage, and pyroptotic cell death,
resulting in the release of IL-1
Pyroptosis has emerged as a crucial link between cellular stress responses and inflammatory vascular injury. Pyroptosis, classically mediated by the gasdermin family proteins, is characterized by pore formation in the plasma membrane and efflux of pro-inflammatory cytokines, which amplify local immune activation [18]. In cardiovascular pathology, pyroptosis contributes to atherosclerotic plaque instability [19], myocardial ischemia-reperfusion injury [20], and abdominal aortic aneurysm (AAA) [21]. Our findings extend this paradigm to AD, revealing that GSDME is the principal driver of pyroptosis in VSMCs, consistent with evidence that GSDME is preferentially cleaved by caspase3 and bridges apoptotic and pyroptotic signaling [22]. The dominance of caspase3–dependent pyroptosis in our model suggests that classical apoptotic stimuli in AD, such as oxidative stress and biomechanical injury, may be rerouted towards inflammatory cell death, thereby increasing medial damage. Compared with macrophage or neutrophil-driven pyroptosis, VSMC pyroptosis directly undermines the structural integrity of the aortic wall, making its pathological consequences potentially more irreversible.
PKM2 is increasingly recognized as a multifunctional metabolic enzyme
that integrates glycolytic flux with transcriptional regulation of inflammatory
mediators [23]. In immune cells, PKM2 can dimerize and translocate to
the nucleus, acting as a co-activator of hypoxia-inducible factor-1 alpha (HIF-1
While pyroptosis in macrophages has been extensively studied in vascular pathology [25], our results underscore that VSMCs themselves are highly susceptible to pyroptosis via the ZNF460–PKM2–GSDME axis. The destruction of contractile VSMCs not only weakens the aortic wall mechanically but also alters the extracellular matrix environment, potentially facilitating infiltration of inflammatory cells that undergo their own pyroptotic death. This creates a feed-forward loop of inflammation and tissue damage. Therapeutically, selectively modulating pyroptosis in VSMCs while preserving immune surveillance could be a sophisticated strategy for AD management, especially during the acute phase when rapid wall destabilization is imminent.
Beyond vascular disorders, GSDME-mediated pyroptosis has been implicated in chemotherapy-induced tissue injury, neurodegeneration, and septic organ damage in sepsis [26, 27, 28]. The recurrent theme across these contexts is that GSDME links apoptotic executioner caspase3 to the inflammatory outcomes of pyroptosis, particularly in non-immune parenchymal cells. Our findings suggest that the same coupling exists in VSMCs during AD, highlighting GSDME as a pivotal molecular switch that converts apoptotic cues into inflammatory cell death. Given ongoing efforts to develop inhibitors of GSDME cleavage or pore formation in oncology, there is an opportunity to accelerate their use for cardiovascular indications.
ZNF460 is a zinc finger transcription factor with limited characterization in cardiovascular biology. Previous studies have implicated ZNFs in regulating inflammatory and apoptotic signaling [29], but a direct role in pyroptosis has not been reported. We demonstrate that ZNF460 binds directly to the PKM2 promoter and enhances its transcriptional activity. This positions ZNF460 as a novel upstream driver of pyroptosis in VSMCs, expanding the functional repertoire of zinc finger proteins in vascular pathology. Given that transcription factors are generally considered challenging drug targets, future studies should explore indirect inhibition strategies, such as interfering peptides or small molecules disrupting ZNF460–DNA binding.
It is plausible that the ZNF460–PKM2–GSDME axis
interacts with known inflammatory signaling hubs such as NLRP3 inflammasomes,
MAPK cascades, or nuclear factor kappa B (NF‑
The dual function of PKM2, as both a metabolic and a signaling regulator, underscores its therapeutic potential. Small‑molecule PKM2 inhibitors (e.g., shikonin, compound 3K) have demonstrated anti‑inflammatory and anti‑tumor activities in preclinical settings [31, 32]. Disrupting GSDME cleavage or modulating caspase‑3 activation could mitigate pyroptotic damage without abolishing vital cell death pathways. Although transcription factors like ZNF460 have traditionally been considered “undruggable”, advances in targeting protein-DNA interactions and RNA therapeutics make indirect modulation increasingly feasible. Moreover, PKM2 expression in aortic tissue or potentially in circulating vesicles could serve as a biomarker for disease activity and response to therapy.
There are several limitations to this study. While our model recapitulates acute AD-like medial rupture, the chronic degenerative aspects seen in human AD may not be fully represented. It is important to acknowledge a limitation regarding the interpretation of PKM2 upregulation in human AD tissues. Since samples were obtained post-operatively, it is difficult to distinguish whether the increase in PKM2 was a primary driver of the dissection or a secondary response to the acute inflammatory storm following rupture. However, our in vitro data demonstrating that PKM2 knockdown mitigates Ang II-induced injury suggest that PKM2 plays a functional role in the pathogenesis, rather than being a mere consequence. We propose that PKM2 may be involved in a positive feedback loop where the initial vascular injury upregulates PKM2, which subsequently amplifies inflammatory responses and vascular smooth muscle cell death, further aggravating the disease. The transcriptional network of ZNF460 is incompletely mapped; other pyroptosis- or metabolism-related target genes may exist. Co‑culture systems, including VSMCs and immune cells, or vessel‑on‑a‑chip models, could further clarify intercellular pyroptosis crosstalk. Future studies will also need to validate these findings in larger patient cohorts and across multiple centers to strengthen their translational potential. Lastly, the safety and efficacy of targeting the ZNF460–PKM2–GSDME axis should be established in large‑animal studies before clinical translation. Another limitation of this study is that we did not distinguish between VSMC dedifferentiation and cell death in the analysis of marker expression. Future studies using co-staining of contractile markers with pyroptosis markers are needed to clarify the temporal relationship between these events. These adjacent tissues may still exhibit subtle pathological changes compared to healthy aortas, and thus our comparisons reflect differences between diseased and less-affected tissues rather than healthy versus diseased states.
In summary, this study identifies a critical regulatory axis involving ZNF460–PKM2–GSDME in the pathogenesis of AD. PKM2 promotes VSMC phenotypic switching and pyroptosis, accelerating disease progression, while GSDME exacerbates the inflammatory response. The modulation of PKM2 or GSDME, or the inhibition of ZNF460, could offer promising therapeutic avenues for the treatment of AD and other vascular diseases driven by pyroptosis. Future studies are required to explore the translational potential of targeting this axis in preclinical and clinical settings.
AD, aortic dissection; VSMCs, vascular smooth muscle cells; PKM2, pyruvate kinase M2; ZNF460, zinc finger protein 460; GSDME, Gasdermin E; AAA, abdominal aortic aneurysm; DEGs, differentially expressed genes.
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
BH, XD, and QY conceived and designed the study. BH, XD, QY, and ZZ conducted the research and analyzed the data. QY, XS, ML, CX, and BC collected the experimental data. QY drafted the manuscript. GT, BH, and XD interpreted the data and critically revised the manuscript. All authors contributed to the critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All surgeries were performed at Fujian Medical University Union Hospital, and all patients were informed of the purpose of the specimen usage and signed the informed consent form. This study was approved by the Fujian Medical University Union Hospital Ethics Committee (2024KY180). The study has adhered to the Declaration of Helsinki guidelines. The animal experiments were approved by the Ethics Committee of Fujian Medical University Union Hospital (Approval Number: IACUC FJMU 2024-0275). All animal experiments were conducted in strict compliance with the ARRIVE 2.0 guidelines and relevant animal welfare regulations (e.g., the 3Rs principles – Replacement, Reduction, and Refinement). The study design, housing conditions, experimental procedures, and reporting adhere to the standards set forth by ARRIVE 2.0 to ensure transparency, reproducibility, and ethical rigor.
Not applicable.
This study was supported by the Foundation of Key Laboratory of Cardio-Thoracic Surgery (Fujian Medical University), Fujian Province University (No. XHZDSYS202406), awarded to the first author, Quanlin Yang, the National Natural Science Foundation of China (No. 82070476) and the Joint Funds for the innovation of science and Technology, Fujian province (No. 2023Y9219), awarded to the corresponding author, Xiaofu Dai. The National Natural Science Foundation of China (No. 82300543).
The authors declare no conflict of interest. Guowei Tu is serving as an Editorial Board member and Guest Editor of this journal. We declare that Guowei Tu had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Yong Peng and Karol E. Watson.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/RCM48463.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.