




 , Andrea Segreti 1,2,*
, Andrea Segreti 1,2,* , Francesco Piccirillo 1,2, Michele Pelullo 1,2, Simone Pasquale Crispino 1,2, Martina Ciancio 1,2, Gian Paolo Ussia 1,2, Francesco Grigioni 1,2
, Francesco Piccirillo 1,2, Michele Pelullo 1,2, Simone Pasquale Crispino 1,2, Martina Ciancio 1,2, Gian Paolo Ussia 1,2, Francesco Grigioni 1,21 Cardiology Unit, Fondazione Policlinico Universitario Campus Bio-Medico, 00128 Roma, Italy
2 Research Unit of Cardiovascular Science, Department of Medicine and Surgery, Università Campus Bio-Medico di Roma, 00128 Roma, Italy
Abstract
Heart failure (HF) is a multifaceted clinical syndrome that frequently precipitates disturbances in perfusion, ventilation, and metabolic regulation, all of which are rapidly detectable through arterial blood gas (ABG) analysis. Meanwhile, clinical markers such as lactate, arterial pH, arterial partial pressure of carbon dioxide (PaCO2), arterial partial pressure of oxygen (PaO2), bicarbonate, and electrolyte concentrations provide dynamic insight into the pathophysiologic status of patients and can serve as early indicators of decompensation. This review evaluates the clinical significance of key ABG and electrolyte parameters in both acute and chronic HF, emphasizing the prognostic value of the analyses, contribution to risk stratification, and utility in guiding therapy. In acute HF and cardiogenic shock, hyperlactatemia and acidosis are associated with increased mortality and the need for hemodynamic or ventilatory support. Furthermore, electrolyte abnormalities, particularly those involving sodium and potassium, are common and driven by neurohormonal activation, pharmacological therapies, and volume shifts. Therefore, integrating ABG and electrolyte monitoring into routine HF management can enhance diagnostic precision and support timely, targeted interventions. This narrative review synthesizes current evidence and proposes a practical framework for interpreting ABG results in the context of contemporary HF care.
Keywords
- acute heart failure
- chronic heart failure
- arterial blood gas
- lactate
- electrolyte imbalance
Heart failure (HF) is a complex clinical syndrome characterized by structural or
functional cardiac impairment, leading to elevated intracardiac pressures and/or
reduced cardiac output, either at rest or during exertion [1]. This hemodynamic
dysfunction manifests with cardinal symptoms such as breathlessness, fatigue, and
ankle swelling, often accompanied by signs including elevated jugular venous
pressure, pulmonary rales, and peripheral edema. The diagnosis of chronic HF
requires the presence of symptoms and/or signs of HF and objective evidence of
cardiac dysfunction. The etiology of HF is heterogeneous and often
multifactorial, with ischemic heart disease and hypertension as predominant
causes globally [1, 2]. HF is an emerging public health priority, driven
primarily by demographic aging and the rising incidence of risk factors,
including hypertension, obesity, and diabetes. The estimated lifetime risk of
developing HF is 20–25%, with one in four individuals likely to experience the
condition during their lifetime [3, 4]. Despite therapeutic advances, HF
continues to be associated with high mortality with 1-year and 5-year mortality
rates approximately of 20% and up to 50%, respectively. Among older patients
(
Arterial blood gas (ABG) offers rapid insight into the respiratory, metabolic, and perfusion status of patients with HF. Parameters such as lactate, pH, partial pressure of carbon dioxide (PaCO2) and oxygen (PaO2), the PaO2/FiO2 ratio, bicarbonate concentration (HCO3–), and electrolytes provide rapid insights into tissue perfusion, acid-base status, and cardiorenal-respiratory interplay [9, 10].
Due to its rapid turnaround, bedside availability, and favorable safety profile, ABG analysis is widely used in both emergency and inpatient settings to support immediate clinical decision-making. Emerging evidence suggests that specific ABG variables at presentation are significantly associated with critical clinical outcomes in acute HF, including mortality, need for mechanical ventilation, ICU admission, and prolonged hospitalization [11, 12].
This narrative review explores the diagnostic and prognostic utility of ABG including electrolytes in patients with HF, focusing on their clinical relevance, pathophysiological implications, and role in contemporary HF management.
Lactate is a key biomarker of anaerobic metabolism and tissue hypoperfusion.
Under conditions of impaired oxygen delivery, pyruvate generated from glycolysis
is reduced to lactate by L-lactate dehydrogenase. Lactate is then transported to
the liver, where it is oxidized back to pyruvate, and undergoes gluconeogenesis
or enters the Krebs cycle as acetyl-CoA [13]. Approximately 70–75% of
circulating lactate is metabolized by the liver; the kidneys contribute the
remaining clearance. Hyperlactatemia (
During shock, lactate becomes a primary energy substrate for the heart. Hyperlactatemia reflects a stress response characterized by increased metabolic rate, sympathetic activation, accelerated glycolysis, and altered bioenergetic pathways [15]. While mild lactate elevations may occur in chronic HF, levels are often significantly higher in acute HF (AHF). The underlying mechanism can differ between the conditions. In chronic HF, hyperlactatemia is thought to result from increased glycolysis due to chronic metabolic dysregulation, sustained myocardial injury, and persistent sympathetic nervous system activation, which together enhance lactate production and efflux from cells [16].
In contrast, acute HF is typically associated with systemic hypotension, hypoxia, and hypoperfusion, which shift metabolism toward anaerobic pathways and markedly increase lactate levels. In advanced stages, multiorgan dysfunction, including hepatic and renal impairment, further limits lactate clearance, exacerbating hyperlactatemia [17].
Multiple studies, as shown in Table 1 (Ref. [18, 19, 20, 21, 22, 23, 24, 25, 26, 27]) and Fig. 1, have demonstrated the prognostic value of elevated lactate levels in AHF, even in the absence of overt clinical signs of hypoperfusion [18]. High lactate correlates with in-hospital mortality, the need for circulatory support and ICU admission [19]. Conversely, normalization of lactate levels, or evidence of lactate clearance, is a favorable prognostic sign. Lactate thus serves as both a prognostic indicator and a therapeutic target in the acute management of HF [20, 28, 29]. In cardiogenic shock, lactate is integral to the Society for Cardiovascaular Angiography and Interventions (SCAI) staging system, helping identify patients who may benefit from advanced therapies [30].
| Study (year) | Type of study | Sample size | HF type | Lactate threshold | Outcomes | OR (95% CI) | p-value |
| Kawase et al. (2015) [19] | Retrospective single-center observational study | 754 | Acute HF in ICU | In-hospital all-cause death | 2.14 (1.10–4.21) | 0.03 | |
| Gjesdal et al. (2018) [22] | Retrospective single-center observational study | 1260 | Patients with AMI underwent PCI and with signs of mild to moderate heart failure (Killip class II–III) | 30-day mortality | 5.94 (1.23–28.64) | ||
| Zymliński et al. (2018) [18] | Retrospective single-center observational study | 237 | Patients with AHF without overt clinical evidence of peripheral hypoperfusion | All-cause 1-year mortality | 2.7 (1.6–4.5) | ||
| Biegus et al. (2019) [23] | Retrospective single-center observational study | 89 | Hospitalized patients with AHF | One-year mortality | 3.4 (1.3–8.7) | 0.009 | |
| Biegus et al. (2019) [24] | Prospective single-center observational study | 222 | Hospitalized patients with Acute HF | Persistent hyperlactataemia within the first 24 h of hospitalization is a predictor of a worse outcome in AHF and is related to higher rates of in-hospital adverse events and one-year mortality | 2.5 (1.5–4.3) | ||
| Bosso et al. (2021) [25] | Prospective single-center observational study | 96 | AHF presenting to Emergency | In-hospital composite outcome (need for ICU admission, LOS |
1.51 (1.24–1.84) | ||
| Uyar et al. (2020) [26] | Prospective single-center observational study | 85 | AHF admitted to the hospital | Composite of cardiovascular death and HF hospitalizations at 6 months | 5.35 (1.243–23.093) | 0.024 | |
| Lindholm et al. (2020) [27] | Post hoc analysis of CardShock study | 217 | AMI-CS | 30-day all-cause mortality | 1.20 (1.14–1.27) | ||
| Marbach et al. (2022) [20] | Post-hoc analysis of the DOREMI trial | 142 | All-cause cardiogenic shock (SCAI stages B–E) | Lactate clearance at 24 hours | In-hospital survival | 5.44 (2.14–13.8) | |
| Hu et al. (2018) [21] | Retrospective single-center observational study | 7558 | Acute HF admitted in ICU | 2.3–4.3 mmol/L | In-hospital all-cause mortality was gradually increased with lactic acid levels increasing | 2.36 (1.73–3.22) |
Abbreviations: AMI, acute myocardial infarction; PCI, percutaneous coronary intervention; HF, heart failure; SCAI, Society for Cardiovascular Angiography and Interventions; LOS, length of stay; AHF, acute HF; OR, odds ratio; CI, confidence interval.
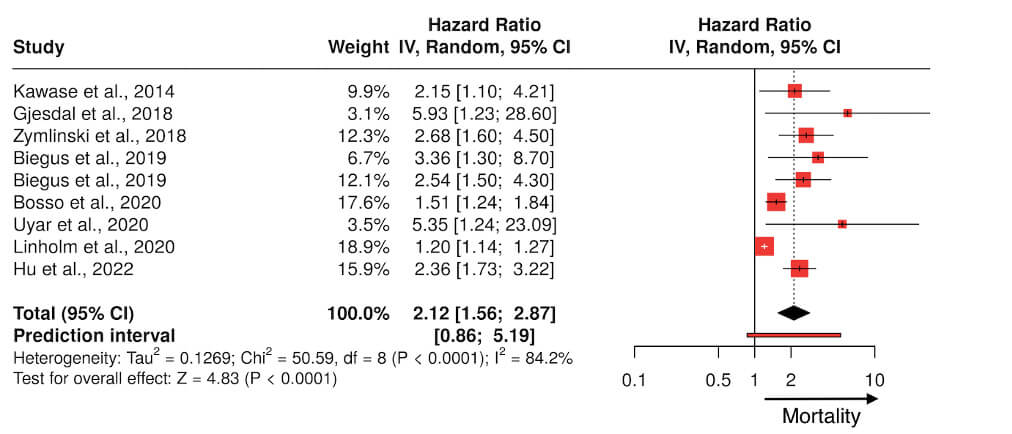 Fig. 1.
Fig. 1.
Forest plot summarizing the clinical studies investigating lactate as a prognostic marker in patients with heart failure. IV, intravenour.
The lactate/albumin (L/A) ratio has been linked with poorer outcomes in adults suffering from a variety of conditions such as sepsis, trauma and heart failure. Some studies suggest that the L/A ratio may correlate with poor prognosis [31, 32].
Furthermore, studies have shown that lactate accumulation in acute HF is closely related to cardiac index, with mixed venous oxygen saturation, heart rate, and systemic vascular resistance emerging as the strongest determinants [33].
In clinical practice, a serum lactate
From a practical standpoint, the interpretation of lactate in HF should move
beyond a single admission value and incorporate both the underlying HF phenotype
and serial trends over time. Patients with cardiogenic shock, mixed
cardiogenic–septic shock, or advanced chronic HF with multiorgan dysfunction may
all exhibit elevated lactate, but for different pathophysiological reasons,
including impaired tissue perfusion, adrenergic stimulation, hepatic and renal
dysfunction, or
Arterial pH reflects the balance between the respiratory (PaCO2) and metabolic (HCO3–) systems, components of acid-base homeostasis. In red blood cells, carbon dioxide (CO2) combines with water under the action of carbonic anhydrase to form carbonic acid, which rapidly dissociates into bicarbonate (HCO3–) and hydrogen ions (H+). CO2 crosses cell membranes via simple diffusion, dissolves in the blood, and is primarily eliminated through pulmonary exhalation. This process is modulated by the rate and depth of respiration. The level of bicarbonate in the blood is controlled through the renal system, where it is filtered and then reabsorbed in the proximal convoluted tubule [34].
In a normal ABG analysis, HCO3– and PaCO2 typically shift in the same direction as part of a compensatory mechanism (Fig. 2); however, renal compensation is generally slower than respiratory compensation. When both pH and HCO3– change in the same directions, a primary metabolic disorder is likely. Conversely, when pH and PaCO2 move in opposite direction, the primary disorder is respiratory [35]. In contrast, a mixed disorder is characterized by HCO3– and PaCO2 moving in opposite directions, a pattern that is not expected in isolated disturbances, and pH may be either normal or abnormal.
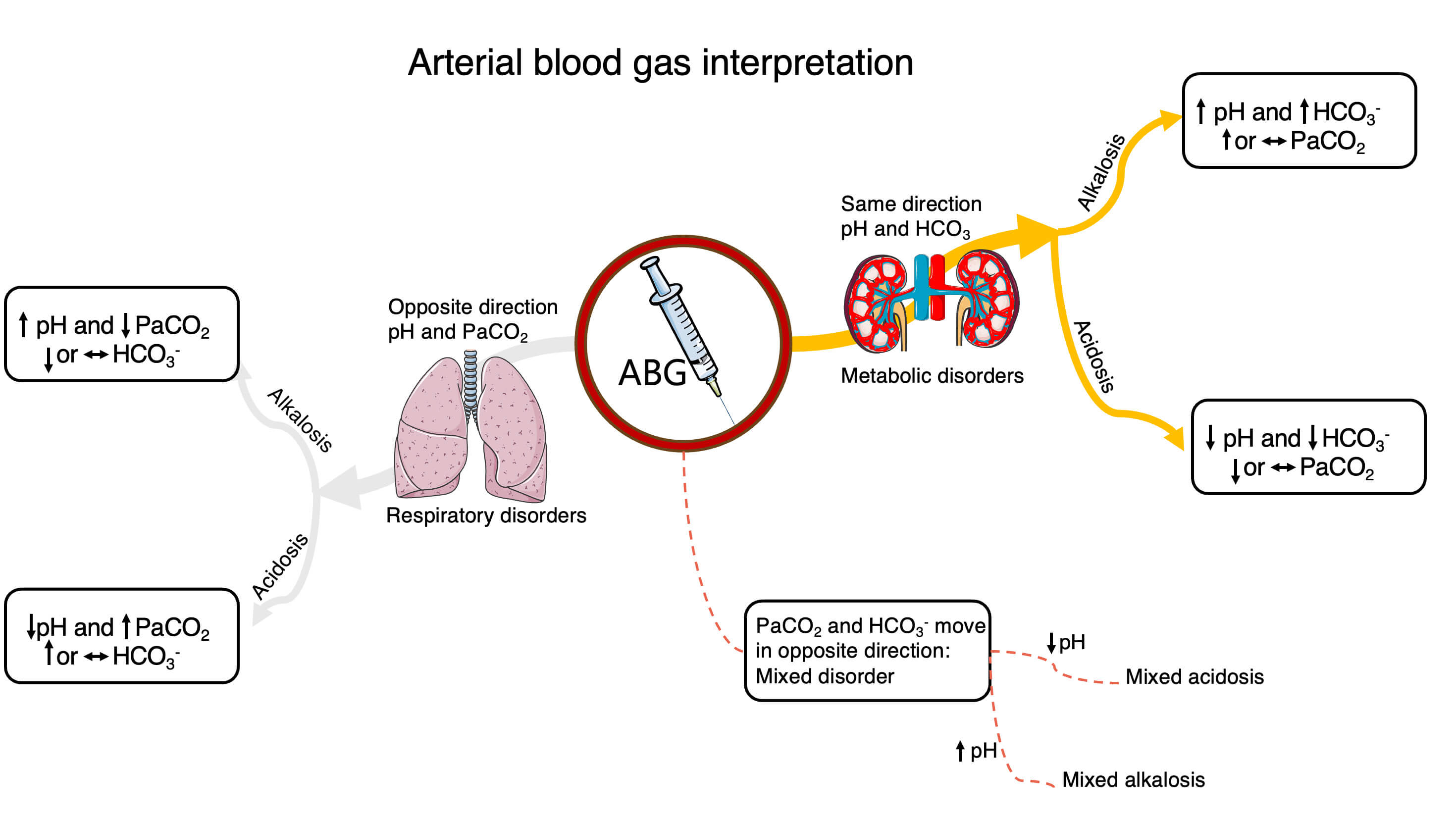 Fig. 2.
Fig. 2.
An example of ABG interpretation. Abbreviations: ABG, arterial
blood gas; PaO2, arterial partial pressure of oxygen; PaCO2, arterial
partial pressure of carbon dioxide; HCO3, carbonate ion;
In HF, acidemia (pH
Also, lactate can acutely stimulate carotid chemoreceptors, much like hypoxia and is able to stimulate carotid body sensory activity in the absence of other hypoxic signals, increasing respiratory rate [38].
Several studies have identified acidosis at admission as a predictor of mortality in acute HF [37, 39]; however, little evidence has yielded conflicting results [40]. These discrepancies may be attributed to substantial differences in the characteristics of the study populations. A large multinational registry of 1982 AHF patients (KorHF Registry) stratified patients by admission pH. Acidosis was present in roughly 19% on arrival, primarily metabolic or mixed-type acidosis. In adjusted Cox analysis, admission acidosis emerged as an independent predictor of mortality (hazard ratio 1.9, 95% CI 1.27–2.93). The largest group had respiratory alkalosis, whereas only 7% had metabolic alkalosis. Notably, alkalosis was not associated with increased mortality in that cohort [9].
An arterial pH
Acid-base status also exerts a powerful influence on electrolyte distribution, particularly for potassium and chloride, and this interaction is frequently encountered in HF. In metabolic or respiratory acidosis, hydrogen ions move into cells and potassium shifts to the extracellular space, leading to hyperkalemia even when total body potassium is normal or reduced. Conversely, acute respiratory or metabolic alkalosis promotes intracellular potassium uptake, predisposing to hypokalemia and increasing the risk of ventricular arrhythmias in patients already vulnerable due to structural heart disease. Chloride, together with sodium and bicarbonate, is a key determinant of the strong ion difference: hyperchloremia from chloride-rich fluids can narrow the strong ion difference and drive a non-anion gap metabolic acidosis, whereas chloride loss from vomiting or loop and thiazide diuretics widens it and contributes to metabolic alkalosis. In HF, these mechanisms often coexist—for example, a decompensated patient with hypercapnic acidosis on non-invasive ventilation may develop rising potassium levels despite stable renal function, while another on high-dose loop diuretics may present with metabolic alkalosis, hypochloremia, and hypokalemia. Recognizing these linked patterns on ABG and electrolyte panels is essential for targeted correction and for avoiding oversimplified interpretations such as attributing all abnormalities solely to “renal failure” or “diuretic therapy”.
The arterial partial pressure of oxygen (PaO2) in arterial blood is a key component of ABG analysis. The PaO2/FiO2 ratio (arterial oxygen partial pressure divided by the inspired oxygen fraction) is a well-established index of oxygen perfusion. It is used to characterize acute respiratory distress syndrome (ARDS) severity as per the Berlin definition, but it is equally applicable to cardiogenic pulmonary edema and correlates with the need for ventilatory support and ICU care [42, 43].
In acute HF, the rapid accumulation of fluid within the interstitial and alveolar spaces due to elevated cardiac filling pressure, leading to acute cardiogenic pulmonary edema. Edema reduces pulmonary compliance, promotes alveolar and small airway collapse, gas exchange can be severely impaired, leading to hypoxemia and significant dyspnea. A low PaO2/FiO2 ratio in AHF indicates significant intrapulmonary shunting or diffusion impairment due to fluid-filled alveoli. The early application of positive end-expiratory pressure (PEEP) keeps the airway open, counteracting alveolar collapse and improving gas exchange [44].
Oxygen delivery (DO2) is directly proportional to hemoglobin concentration, arterial oxygen saturation (SaO2), and cardiac output (Fig. 3). Cardiac output, in turn, is determined by heart rate and stroke volume. The primary determinants of stroke volume include preload, afterload, and myocardial contractility. Reduction in preload or contractility typically diminishes stroke volume, whereas elevated afterload may impede ventricular ejection, thereby reducing stroke volume. A comprehensive understanding of these hemodynamic relationships is essential for the assessment and management of cardiovascular conditions such as heart failure and circulatory shock [45].
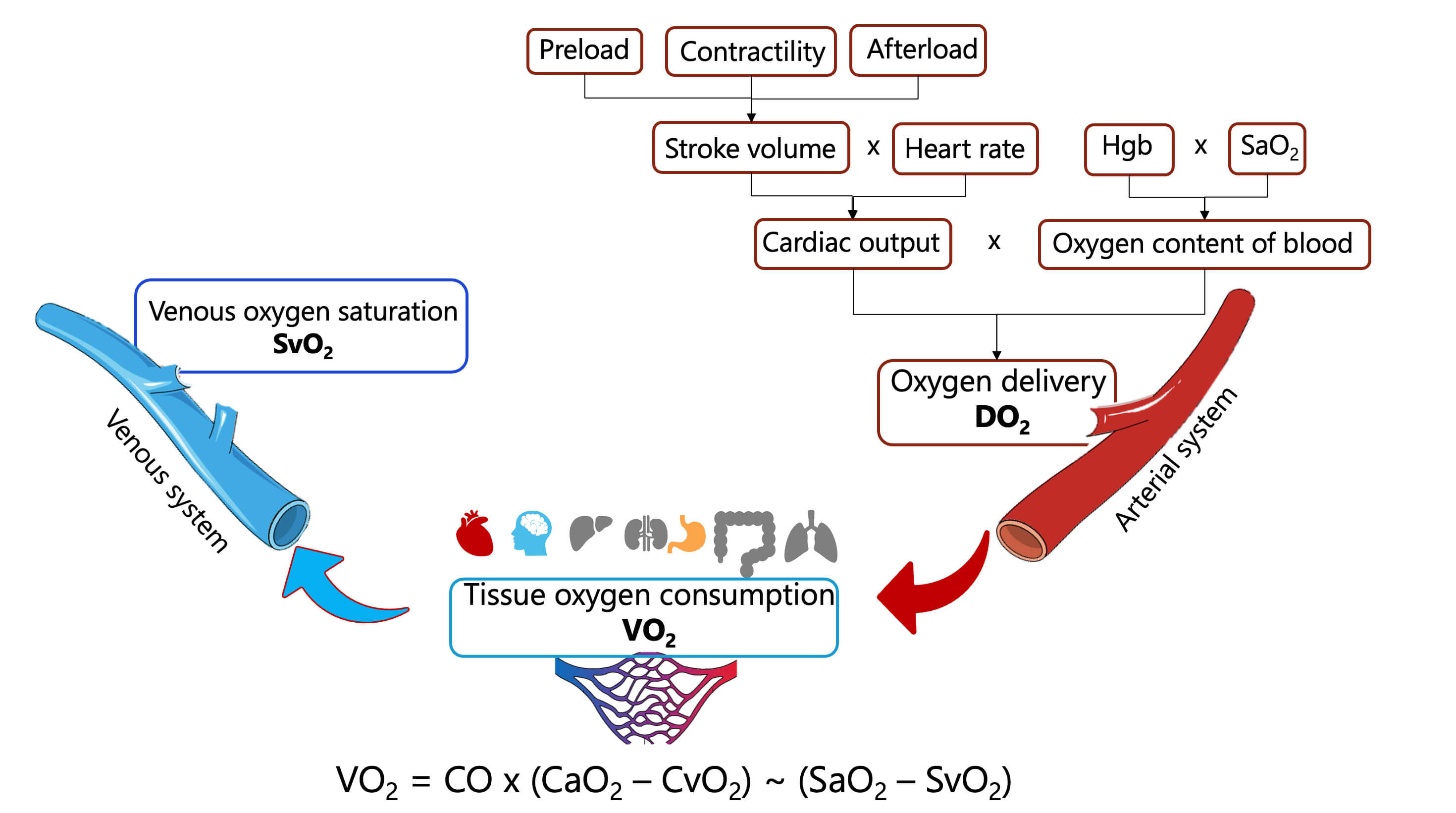 Fig. 3.
Fig. 3.
Determinants of systemic oxygen delivery and tissue oxygen consumption. Abbreviations: DO2, systemic oxygen delivery; VO2, tissue oxygen consumption; SaO2, arterial oxygen saturation; SvO2, venous oxygen saturation; Hgb, hemoglobin; CO, cardiac output; CaO2, arterial oxygen content of blood; CvO2, venous oxygen content of blood.
Central venous oxygen saturation (ScvO2) represents the percentage of hemoglobin saturated with oxygen in venous blood in the right heart via a central venous catheter. It serves as a surrogate marker for the global balance between supply (DO2) and tissue oxygen consumption (VO2). The total rate of DO2 is usually around 15 mL/kg/min, and the normal range for VO2 is approximately 3.5–4.0 mL O2/kg/min relative to body mass. Under physiological conditions, ScvO2 values are between 70 and 75%. A decrease in ScvO2 suggests either increased oxygen consumption (VO2) or DO2, as in cardiogenic shock [46].
Patients with chronic HF may be adapted to a low venous oxygen saturation (SvO2) due to chronic tissue hypoxia. An acute drop in SvO2 is an indication of cardiac dysfunction. On the other hand, SvO2 improving following cardiopulmonary resuscitation is a marker for the return of spontaneous circulation [46, 47]. Thus, in such settings, it is useful to monitor SvO2 [48]. Furthermore, the Surviving Sepsis Campaign guidelines recommend the use of ScvO2 or mixed venous oxygen saturation to assess the balance of tissue oxygen delivery and consumption in sepsis [49].
In cardiogenic shock, the veno-arterial difference in partial pressure of carbon
dioxide (PCO2 gap or
In VA-ECMO, ABG interpretation becomes more complex because measured PaO2
and PaCO2 depend on cannulation configuration, native cardiac output,
circuit flow, and the degree of mixing between oxygenated extracorporeal blood
and desaturated native cardiac output. Samples drawn from the right radial
artery, femoral artery, or post-oxygenator line may yield substantially different
results, particularly in the presence of “north-south” (Harlequin) syndrome
with preserved left ventricular ejection and severe pulmonary dysfunction. In
this setting, a normal PaO2 in a post-oxygenator sample can coexist with
cerebral or myocardial hypoxia, while extracorporeal CO2 removal may mask
ongoing tissue hypoperfusion if lactate and
ABG-derived indices such as lactate, PaO2/FiO2, PaCO2, and ScvO2 have the potential to complement existing HF and cardiogenic shock risk scores. Lactate is already incorporated into several shock staging systems and reflects the severity of systemic hypoperfusion, while PaO2/FiO2 captures the burden of respiratory failure and pulmonary congestion and ScvO2 provides a dynamic estimate of the balance between oxygen delivery and consumption. Integrating these parameters into multiparametric scores that also include clinical signs, biomarkers, and imaging findings could improve discrimination and reclassification for key outcomes such as the need for mechanical circulatory support, ICU admission, or short-term mortality. In practice, serial changes in lactate, PaO2/FiO2 and ScvO2 under therapy may be more informative than single measurements at admission, helping to identify patients with a “failing trajectory” who warrant early escalation of support despite apparently stable vital signs.
Chronic HF is characterized by progressive neurohormonal and hemodynamic
dysregulation that triggers compensatory mechanisms aimed at maintaining
perfusion. Among these, sympathetic nervous system hyperactivity initially
supports circulatory homeostasis but eventually leads to
In parallel, activation of the renin-angiotensin-aldosterone system (RAAS) constitutes a cornerstone maladaptive response in chronic HF. Reduced cardiac output and renal perfusion stimulate renin release from juxtaglomerular cells, initiating a cascade culminating in angiotensin II and aldosterone secretion. Angiotensin II promotes vasoconstriction and adverse ventricular remodeling, while aldosterone induces renal sodium and water retention, potassium excretion, and myocardial fibrosis. Antidiuretic hormone (ADH) release is concurrently stimulated, compounding sodium-free water retention and contributing to dilutional hyponatremia and volume overload. Although natriuretic peptides such as BNP and ANP are released in response to myocardial stretch, their compensatory effects are often overwhelmed in advanced disease stages [57]. The pathologic consequences of sustained RAAS activation are central to the progression of heart failure, as shown in Fig. 4. Angiotensin II and aldosterone drive myocardial fibrosis, hypertrophy, endothelial dysfunction, and adverse remodeling—amplifying both systolic and diastolic impairment [58, 59].
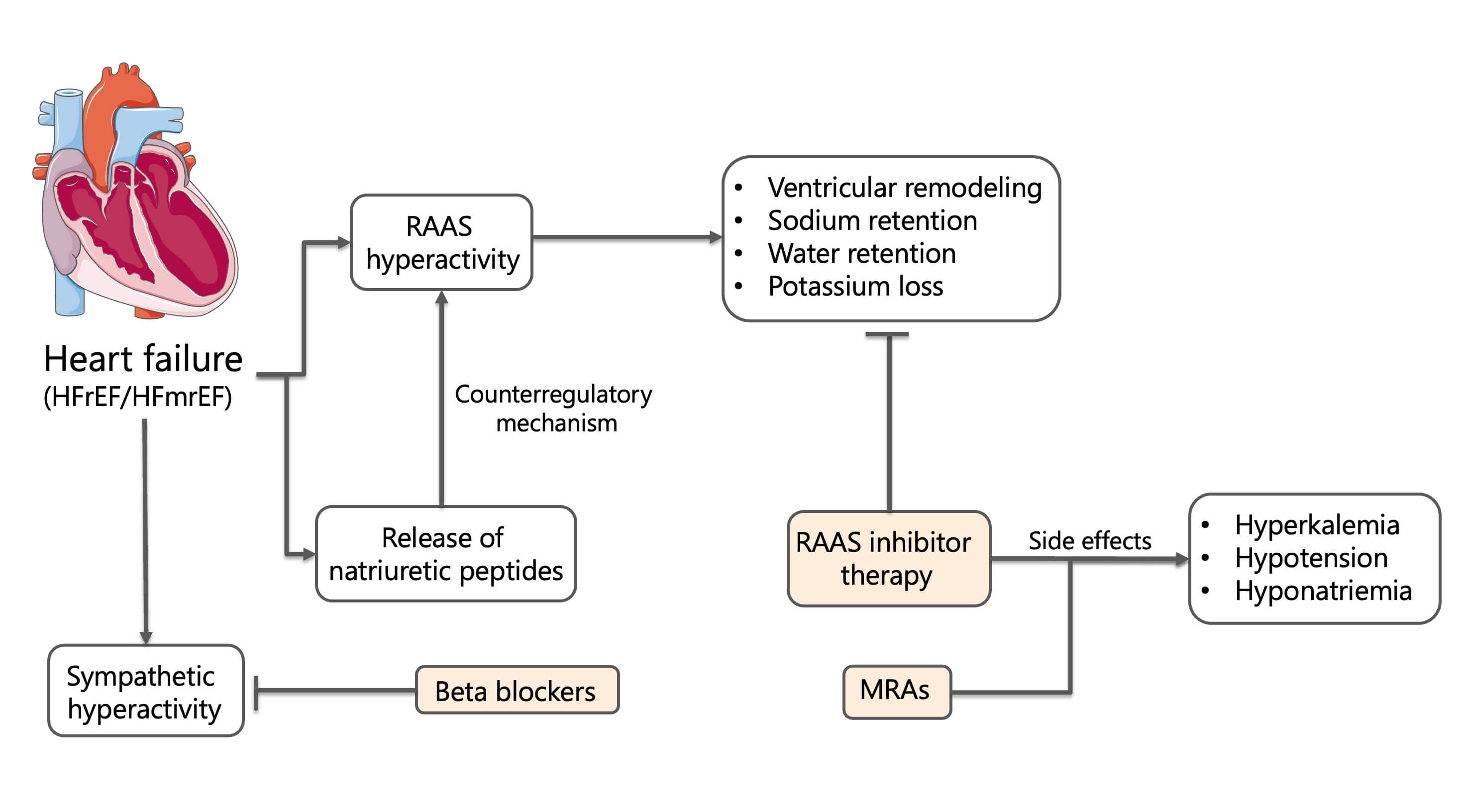 Fig. 4.
Fig. 4.
Neurohormonal activation and pharmacological modulation in heart failure. Abbreviations: HFrEF, heart failure with reduced ejection
fraction (
These neurohormonal and hemodynamic adaptations not only modulate sodium and water balance but also generate characteristic acid-base and electrolyte patterns that are readily captured by ABG analysis. For example, RAAS activation and aldosterone excess favor renal potassium loss and metabolic alkalosis, particularly when combined with loop or thiazide diuretics, whereas advanced renal dysfunction, RAAS inhibition, and acidosis shift the balance toward hyperkalemia and mixed metabolic disturbances. Thus, the electrolyte profile of a patient with HF is best interpreted in parallel with ABG-derived pH, PaCO2, and bicarbonate, viewing both as different facets of the same pathophysiological process rather than isolated laboratory domains.
The therapeutic goal in HF is to reduce mortality, mitigate symptoms, prevent
hospitalization, and improve quality of life. ESC guidelines endorse four classes
of disease-modifying therapies for HFrEF and HF mildly reduced ejection fraction
(HFmrEF): (1) RAAS inhibitors, including angiotensin converting enzyme (ACE)
inhibitors, angiotensin receptor blockers (ARBs), and angiotensin
receptor-neprilysin inhibitors (ARNIs); (2)
While RAAS inhibition improves outcomes, it may provoke clinically significant electrolyte disturbances. Suppressed aldosterone activity can lead to hyponatremia, hypotension, and volume depletion—particularly in patients on loop diuretics or sodium-restricted diets [60]. Simultaneously, impaired potassium excretion predisposes to hyperkalemia, especially in individuals with chronic kidney disease (CKD) or those receiving potassium-sparing agents [61]. Thus, the therapeutic target needed in HF patients is often not reached because of these side-effects. Management of these side-effects is pivotal to maximize the use of renin-angiotensin-aldosterone system inhibitors (RAASi) in HFrEF, particularly in high-risk patients.
Furthermore, SGLT2i, the latest foundational therapy in HFrEF, exert its primary action by promoting glucosuria and natriuresis in the proximal renal tubule. These agents offer a unique hemodynamic and electrolyte-sparing profile, although, due to osmotic diuresis, they can occasionally lead to hypotension and mild hyponatriemia, particularly in elderly or volume-depleted patients [62].
Beyond its traditional application in acid-base analysis, ABG testing now serves as a rapid, point-of-care tool for assessing electrolytes and guiding real-time management in critically ill heart failure patients. ABG analyzers provide concurrent measurements of sodium, potassium, chloride, bicarbonate, and ionized calcium—electrolytes integral to maintaining cardiac electrophysiology, volume status, and acid-base balance.
The advantage of ABG-based electrolyte assessment in HF is the ability to interpret these values in the immediate context of pH, PaCO2, bicarbonate, and lactate. For example, a mild elevation in potassium will carry different clinical implications in a patient with metabolic alkalosis and total body potassium depletion than in one with metabolic or respiratory acidosis on the verge of malignant arrhythmias. Similarly, the combination of rising chloride and falling bicarbonate on ABG may uncover iatrogenic hyperchloremic acidosis from saline administration, whereas low chloride with elevated bicarbonate suggests diuretic-induced metabolic alkalosis and neurohormonal activation. This integrated, physiology-based interpretation is more informative than viewing electrolyte values in isolation from the concurrent ABG profile.
Sodium is the predominant extracellular cation, essential for maintaining osmolality, intravascular volume and hemodynamic stability. It also contributes to acid-base balance through the calculation of the anion gap: AG = [Na+] – ([Cl–] + [HCO3–]).
An elevated anion gap suggests the accumulation of unmeasured anions (e.g., lactate, ketones), characteristic of high anion gap metabolic acidosis [63]. In Chronic HF (CHF), hyponatremia is independently associated with poor prognosis, including increased mortality and rehospitalization rates [64]. Sodium derangements may be further exacerbated by overly aggressive sodium restriction, highlighting the need for individualized dietary and pharmacologic strategies [60].
Potassium is the primary intracellular cation and is crucial in maintaining cellular membrane potential, myocardial conduction, and skeletal muscle function. Approximately 98% of total body potassium is located intracellularly, with only a small fraction circulating in the extracellular space. This distribution is tightly regulated by the Na+/K+ ATPase pump and influenced by acid-base status, insulin, and adrenergic activity. In acidosis, potassium shifts extracellularly in exchange for hydrogen ions, resulting in hyperkalemia; alkalosis produces the opposite effect. Importantly, extracellular potassium may not accurately reflect total body stores. Normokalemia or even hyperkalemia may mask underlying depletion [65].
Hyperkalemia (
| Serum K+ (mmol/L)* | ECG/clinical risk | Acute management | HF-specific medium/long-term management |
Mild 5.1–5.5 |
Usually no ECG changes | - Confirm true hyperkalemia (exclude hemolysis; repeat K+, ABG, check renal function and acid–base status). | - Optimize guideline-directed HF therapy: maintain RAASi/MRA if possible; consider small dose reduction rather than withdrawal. |
| - Review medications: RAASi/MRA, K+-sparing diuretics, NSAIDs, trimethoprim, heparin. | - Advise moderate dietary K+ restriction (avoid high-K+ foods and salt substitutes). | ||
| - No need for emergency K+ lowering if asymptomatic and ECG normal. | - Intensify loop or thiazide diuretic if volume overloaded. | ||
| - In recurrent hyperkalemia or CKD, consider chronic K+ binders (patiromer or sodium zirconium cyclosilicate) to allow continuation/up-titration of RAASi/MRA. | |||
Moderate 5.6–6.0 |
May be asymptomatic or show subtle ECG changes (peaked T waves); higher arrhythmic risk in HFrEF, or ischemic cardiomyopathy |
- Repeat ABG and K+ to confirm. - 12-lead ECG and continuous monitoring if K+ - If no ECG changes and hemodynamically stable: consider loop diuretic IV or PO, adjust RAASi/MRA dose, and start a K+ binder early. - Address triggers (dehydration, AKI, high-K+ diet, metabolic acidosis). |
- Reassess need/dose of ACEi/ARBs/ARNIs and MRA; try to maintain life-saving drugs using K+ binders and diuretics rather than stopping them outright. |
| - Schedule close K+ and creatinine monitoring (e.g., within 48–72 h after any medication change). | |||
| - Educate patient on diet, over-the-counter drugs (NSAIDs), and sick-day rules. | |||
Severe |
Very high risk of malignant arrhythmias and cardiac arrest | 1. Stabilize myocardium: IV calcium gluconate or calcium chloride (according to local protocol) if ECG changes. Avoid calcium if digoxin toxicity is suspected. 2. Shift K+ intracellularly: - IV insulin + glucose (avoid excessive volume in HF; use concentrated dextrose and monitor glycemia). - Nebulized - IV sodium bicarbonate only if significant metabolic acidosis and appropriate volume status. 3. Remove K+ from body: - High-dose loop diuretic IV if euvolemic or congested and kidneys responsive. - Potassium binders (patiromer or sodium zirconium cyclosilicate) as soon as feasible. - Urgent dialysis in refractory or life-threatening hyperkalemia, especially in advanced CKD/AKI. |
- After stabilization, reassess RAASi/MRA strategy: avoid permanent discontinuation if possible; restart/down-titrate under K+ binder and close monitoring. - Optimize diuretic regimen to prevent recurrence (consider adding thiazide in resistant edema). - Address underlying triggers (e.g., dehydration, infection, AKI, excessive K+ intake). - Define a target K+ range in HFrEF (typically 4.0–5.0 mmol/L) and individualized follow-up plan. |
Abbreviations: HFrEF, heart failure with reduce ejection fraction; ECG, electrocardiogram; PO, per os; K+, potassium; CKD, chronic kidney disease; AKI, acute kidney injury; RAASi, renin-angiotensin-aldosterone system inhibitors; MRA, mineralocorticoid receptor antagonist; NSAIDs, nonsteroidal anti-inflammatory drugs; ACEi, angiotensin converting enzyme inhibitors; ARBs, angiotensin receptor blockers; ARNIs, angiotensin receptor-neprilysin inhibitors; *Approximate thresholds; should be interpreted in the context of local laboratory reference ranges, ECG findings, comorbidities (e.g., CKD, diabetes, COPD), and overall clinical status.
| Serum K+ (mmol/L)* | ECG/clinical risk | Acute management | HF-specific medium/long-term management |
Mild 3.0–3.5 |
Often asymptomatic; increased arrhythmic risk in HFrEF, LV hypertrophy, or QT-prolonging drugs | - Confirm with repeat K+ and ABG. - Oral KCl supplementation if no contraindications. - Assess for concurrent hypomagnesemia and replace Mg2+ if low. |
- Review diuretic regimen; consider reducing loop/thiazide dose or adding a K+-sparing agent/MRA (if not already on and no contraindication). |
| - Ensure adequate RAASi/MRA and | |||
| - Target K+ 4.0–5.0 mmol/L in HFrEF to reduce arrhythmic risk. | |||
Moderate 2.5–3.0 |
Increased risk of ventricular ectopy, especially with digoxin, ischemia, or LV dysfunction | - Oral KCl (divided doses) if GI tract usable and no severe symptoms. - If symptomatic, unable to take PO, or multiple risk factors for arrhythmia: slow IV KCl via peripheral or central line according to local protocols, with ECG monitoring. |
- Reassess diuretic and RAASi/MRA balance: reduce loop/thiazide dose, maximize MRA if tolerated. |
| - Review medications that lower K+ (steroids, high-dose | |||
| - Correct hypomagnesemia (IV MgSO4 if needed). | - Arrange short-interval K+ checks after any change. | ||
Severe |
High risk of malignant ventricular arrhythmias, especially in HFrEF and ischemic heart disease | - Continuous ECG monitoring and admission to monitored setting. - Prompt IV KCl replacement with strict rate limits and central line if high concentration is required; avoid dextrose-only fluids which can worsen hypokalemia. - Correct Mg2+ deficiency aggressively. - Temporarily reduce or hold digoxin and QT-prolonging drugs if feasible. - ABG monitoring to assess concomitant metabolic alkalosis or respiratory derangements. |
- Once stabilized, adjust chronic therapy: lower loop/thiazide doses, up-titrate MRA/ACEi/ARBs/ARNIs if blood pressure and renal function allow. - Educate patient on maintaining adequate dietary K+ intake (unless contraindicated by prior hyperkalemia or CKD). - Define individualized K+ target and follow-up frequency based on HF phenotype, LV function, arrhythmic history, and comorbidities. |
Abbreviations: *Approximate thresholds; should be interpreted in the context of local laboratory reference ranges, ECG findings, comorbidities (e.g., CKD, diabetes, COPD), and overall clinical status.
Chloride plays a pivotal role in acid-base homeostasis by its inverse relationship with bicarbonate. Hyperchloremia—often induced by excessive saline administration—can cause a non-anion gap metabolic acidosis and has been linked to worse renal outcomes [67]. Conversely, hypochloremia, may accompany metabolic alkalosis due to vomiting or diuretic use [68].
Ionized calcium represents the physiologically active fraction of calcium and is critical in coagulation, myocardial contractility, and vascular tone. It is influenced by pH: acidosis increases ionized calcium due to reduced protein binding, while alkalosis reduces it [21].
While this review provides a comprehensive synthesis of available evidence on ABG and electrolyte analysis in heart failure, several limitations should be acknowledged. First, the discussion is based primarily on observational studies, registry data, and retrospective analyses, which may be subject to selection bias and confounding. As such, the prognostic associations identified between ABG parameters and clinical outcomes do not imply causality. Second, most of the evidence pertains to acute heart failure and cardiogenic shock, with fewer robust data available for stable chronic HF populations. Third, the variability in ABG measurement techniques, timing of sampling, and institutional protocols may limit the generalizability of findings across different clinical settings. Furthermore, this narrative review did not employ a systematic methodology for study selection, which may introduce publication bias. Although key references from major guidelines and high-quality studies were prioritized, the absence of formal quality assessment or meta-analytic techniques may limit the reproducibility of conclusions.
ABG analysis has emerged as a vital tool in the management of HF, enabling rapid assessment of acid-base balance, respiratory efficiency, tissue perfusion, and electrolyte status at the bedside. In both acute and chronic HF, ABG parameters such as lactate, arterial pH, PaCO2, PaO2, and bicarbonate concentrations yield essential prognostic information and offer a window into the complex interplay between cardiac, pulmonary, and renal systems [69].
The integration of ABG and electrolyte monitoring in HF care not only facilitates early detection of decompensation but also supports tailored therapeutic decision-making, including the initiation of inotropic support, adjustment of diuretic regimens, or escalation to respiratory or circulatory support.
The interaction between electrolytes, ABG patterns, and HF is modified by sex and age. Women with HF often present with lower body weight, higher prevalence of HFpEF, and more frequent use of thiazides for hypertension, all of which increase the risk of diuretic-induced hyponatremia and hypokalemia. In post-menopausal women, changes in sex hormone profile may further alter renal sodium handling and vasopressin sensitivity, predisposing to dilutional hyponatremia. By contrast, men more commonly present with HFrEF, larger ischemic burden, and more severe neurohormonal activation, which are associated with higher rates of hyperkalemia when RAAS-inhibiting therapies are optimized.
Age also acts as a major effect modifier. Older patients have reduced GFR, diminished renal acid excretion, and lower respiratory reserve, which favor chronic hypercapnia and chronic metabolic compensation. Consequently, the same diuretic dose may produce more pronounced hypochloremia and metabolic alkalosis in an elderly patient than in a younger adult, and superimposed lactic acidosis may be partially masked on ABG. In younger patients, preserved respiratory drive and renal function often allow a more “pure” respiratory alkalosis or metabolic acidosis pattern in acute decompensation, with fewer mixed disorders.
Beyond sex and age, several comorbidities systematically influence electrolyte and ABG profiles in HF, including chronic kidney disease, COPD/OSA, obesity, and diabetes, as well as therapies such as SGLT2 inhibitors, MRAs, and acetazolamide. A careful interpretation of ABGs in HF should therefore integrate patient age, sex, comorbidity profile, and current pharmacotherapy to avoid misattributing an acid–base disturbance solely to HF decompensation.
As demonstrated in this review, hyperlactatemia and acidemia are robust markers of impaired perfusion and predict adverse outcomes [70, 71], while derangements in sodium and potassium are associated with increased morbidity and mortality [72, 73, 74, 75].
Moreover, serial ABG measurements provide dynamic feedback on treatment efficacy and can guide ongoing hemodynamic optimization. The ability to rapidly detect mixed or evolving acid-base disturbances, particularly in critically ill patients, underscores the value of ABG as a real-time, physiologic monitor.
Looking ahead, the expanding capabilities of point-of-care testing and integrated clinical decision support systems may enhance the precision and utility of ABG analysis in HF management. Future research should aim to standardize ABG-guided risk stratification algorithms and explore its integration with biomarker-based and imaging modalities to refine prognostic models.
Miniaturized devices capable of measuring pH, PaO2, PaCO2, lactate, and key electrolytes at the bedside, in ambulatory clinics, or even in home-based care models could allow earlier detection of decompensation and more agile titration of diuretics, RAAS inhibitors, and SGLT2 inhibitors. When combined with non-invasive hemodynamic monitoring and weight, blood pressure, and symptom data, serial ABG-like measurements may contribute to a richer “digital phenotype” of HF that captures both congestion and perfusion status.
In parallel, machine learning–based risk models offer an opportunity to integrate ABG parameters, electrolytes, biomarkers, imaging, and comorbidity profiles into dynamic prognostic and decision-support tools. Such models could support clinicians in identifying patients at high risk of deterioration shortly after presentation, refining the selection of candidates for intensive monitoring, non-invasive or invasive ventilation, or early mechanical circulatory support. However, these approaches require rigorous prospective validation, attention to data quality and calibration, and transparent, interpretable algorithms to ensure that they augment rather than complicate bedside decision-making.
In summary, ABG analysis, when interpreted in the appropriate clinical and pathophysiological context, represents a cornerstone of personalized, physiology-guided care in heart failure. By integrating information on acid-base status, oxygenation, ventilation, tissue perfusion, and electrolyte balance, ABG supports earlier recognition of decompensation, more precise risk stratification, and timely escalation or de-escalation of therapy. Emphasizing its systematic use in routine practice, and exploring its incorporation into structured risk scores and digital decision-support tools, may help reduce treatment delays and ultimately improve outcomes for patients across the spectrum of HF.
AS, FG, NT, GPU: Conceptualization, methodology, software; AS, FG, GPU: validation; NT, AS: formal analysis; NT: investigation, writing—review and editing; AS, NT, FP, SPC, MC, MP: data curation; FG, GPU: supervision; All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by the Fondazione Policlinico Universitario Campus Bio-Medico.
The authors declare no conflict of interest. This research was funded by the Fondazione Policlinico Universitario Campus Bio-Medico, the institution where several authors are affiliated. The authors declare that the funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.