


1 Department of Cardiovascular Medicine, The Second Affiliated Hospital, Jiangxi Medical College, Nanchang University, 330006 Nanchang, Jiangxi, China
2 Department of Gastroenterology, Jiangxi Provincial Hospital of Traditional Chinese Medicine, 330006 Nanchang, Jiangxi, China
3 Department of Cardiovascular Medicine, Jingdezhen First People’s Hospital, 333000 Jingdezhen, Jiangxi, China
†These authors contributed equally.
Abstract
Dystrophin deficiency is the core pathological feature of Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD). Indeed, a deficiency in dystrophin results in the progressive degeneration of skeletal muscle and severely compromises the structure and function of cardiomyocytes, eventually leading to dilated cardiomyopathy and heart failure. Thus, this review provides an in-depth analysis of the molecular mechanisms underlying dystrophin-deficient cardiomyopathy, including membrane instability, calcium dysregulation, mitochondrial dysfunction, and fibrosis. The role of inflammatory responses in disease progression is also discussed. In addition, we evaluate current and emerging therapeutic strategies, including gene therapy, pharmacological interventions, and regenerative medicine approaches, and highlight recent preclinical and clinical trial data. Finally, we explore future directions in precision medicine, novel biomarkers for early detection, and combination treatment regimens, to provide a comprehensive resource for clinicians and researchers working in this challenging field.
Keywords
- dystrophin
- Duchenne muscular dystrophy
- heart failure
- gene therapy
Dystrophin is a crucial protein that maintains the structural integrity of muscle cells by linking the cytoskeleton to the extracellular matrix [1]. Its absence or deficiency, as seen in Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD), leads to the progressive degeneration of both skeletal and cardiac muscles [2]. While DMD is more commonly associated with skeletal muscle weakness, cardiac involvement becomes a major concern as the disease progresses, particularly in the form of dystrophin-deficient cardiomyopathy (DDCM), which is responsible for much of the morbidity and mortality in these patients [3].
Cardiac complications, such as dilated cardiomyopathy (DCM), often emerge in DMD and BMD patients in their teens or early twenties, leading to reduced ejection fraction, arrhythmias, and ultimately heart failure [4]. This makes the heart a critical organ in dystrophinopathies, necessitating a deeper understanding of the molecular mechanisms underlying cardiac dysfunction. Key factors contributing to cardiac dysfunction include membrane instability, calcium dysregulation, mitochondrial dysfunction, and fibrosis [5]. In the absence of dystrophin, mechanical stress leads to membrane damage and calcium influx, triggering cellular damage, inflammation, and fibrosis, all of which contribute to a decline in cardiac function.
Despite recent advances in understanding the molecular basis of DCM, effective therapies for this condition in DMD and BMD patients are limited. However, emerging treatments like gene therapy, stem cell-based therapies, and targeted pharmacological interventions hold great promise for slowing or halting disease progression [6]. This review aims to provide a concise synthesis of our current understanding of the molecular mechanisms underlying DDCM. We also explore the potential of emerging therapeutic strategies for DCM.
Dystrophin deficiency in cardiomyocytes severely compromises the integrity of the sarcolemma, which is the cell membrane of cardiac muscle cells [7]. The absence of this key structural protein prevents proper linking of the actin cytoskeleton to the extracellular matrix (ECM), leading to significant vulnerability of the heart muscle to mechanical stress during contraction. In DMD patients, repetitive mechanical injury to the sarcolemma causes localized membrane tears that allow the influx of calcium and other ions into the cell, disrupting cellular homeostasis [7]. This disruption is associated with immediate damage and also triggers a cascade of events that contribute to cell death and muscle degeneration. Additionally, the compromised ability of the sarcolemma to repair itself exacerbates this process, making the myocardium prone to progressive dysfunction [8].
Recent studies have highlighted that dystrophin-deficient hearts exhibit a distinct pattern of membrane damage and cell death compared to hearts with traditional forms of heart failure [7, 9]. This specific pattern underscores the uniqueness of dystrophinopathies and suggests that targeted therapies should consider the mechanistic role of membrane instability as a critical therapeutic target.
Under normal physiological conditions, calcium is tightly regulated in heart cells to maintain proper contraction and relaxation cycles [10]. However, in the absence of dystrophin, the sarcolemma’s fragility leads to calcium influx, causing pathological elevation of intracellular calcium levels. This abnormal calcium influx disrupts normal cellular signaling and triggers a cascade of downstream events that compromise cellular function.
Intracellular calcium overload activates proteolytic enzymes, such as calpains, which degrade structural proteins, leading to the breakdown of essential cellular structures. This contributes to the deterioration of sarcomeres, the contractile units of muscle fibers, and ultimately leads to myocardial dysfunction [11]. Mitochondria, which are responsible for generating energy in the form of Adenosine Triphosphate (ATP), are overwhelmed by calcium and begin to produce reactive oxygen species (ROS). ROS damage cellular components, including lipids, proteins, and DNA, contributing to the overall oxidative stress within cells [12]. This vicious cycle of calcium overload, mitochondrial dysfunction, and oxidative stress exacerbates the damage to cardiomyocytes and accelerates heart failure. Importantly, mitochondrial dysfunction further impairs the ability of the sarcolemma to repair itself, as mitochondria play a role in providing the energy required for membrane stabilization [13].
The intertwined relationship between calcium dysregulation and mitochondrial damage suggests that therapies aimed at normalizing calcium levels or protecting mitochondria could provide substantial benefits for patients with dystrophin-deficient heart failure.
Dystrophin-deficient hearts undergo significant inflammatory responses due to
the constant cycle of muscle damage and repair [14]. In the early stages of
injury, dying cardiomyocytes release pro-inflammatory cytokines such as Tumor
Necrosis Factor-alpha (TNF-
Persistent cytokine signaling (e.g., TNF-
Recent findings have highlighted the significant role of metabolic dysfunction in DDCM. In addition to calcium dysregulation and mitochondrial dysfunction, alterations in lipid metabolism and reduced mitochondrial fatty acid oxidation contribute significantly to disease progression [21]. These metabolic changes increase oxidative stress and disrupt energy production, further impairing mitochondrial function. The failure to efficiently utilize fatty acids for energy exacerbates the pathological processes already present due to mitochondrial dysfunction. Additionally, altered nitric oxide signaling has been observed in dystrophin-deficient hearts, which may compromise vascular function and myocardial perfusion [22]. Abnormal nitric oxide metabolism, combined with mitochondrial dysfunction, creates a vicious cycle that accelerates cardiac dysfunction.
The contribution of these metabolic changes to disease progression underscores the importance of considering metabolic therapy as a potential intervention for dystrophinopathies. The investigation of pharmacological agents that could restore normal metabolic processes in cardiomyocytes is a promising direction for future research.
The cardiac cytoskeleton plays an essential role in maintaining the shape and mechanical integrity of cardiomyocytes [23]. Dystrophin is an integral component of this structure, functioning to link the actin cytoskeleton to the ECM through the dystrophin-glycoprotein complex. This structural framework is compromised in the absence of dystrophin, making the myocardium more susceptible to mechanical stress [24]. The structural integrity of cardiomyocytes is crucial for efficient contractile function, and its loss contributes significantly to the progression of cardiomyopathy in DMD and BMD patients.
Disruption of the cytoskeletal network in dystrophin-deficient hearts has been shown to lead to a series of mechanical and biochemical abnormalities, including altered sarcomere structure and impaired force transmission during contraction [25]. The inability to withstand mechanical stress causes progressive myocardial damage, resulting in further degeneration of cardiomyocytes. Moreover, the loss of dystrophin also impairs the mechanical coupling of cardiomyocytes, which affects the contractile force and overall efficiency of the heart [26].
This phenomenon further emphasizes the need to develop therapeutic strategies that restore the structural integrity of the cardiac cell membrane. Strategies that focus on strengthening the cytoskeletal network or targeting the dystrophin-glycoprotein complex could potentially halt the progression of cardiomyopathy in dystrophinopathies [27].
Autophagy is the process by which cells degrade and recycle their components. It plays a crucial role in maintaining cellular homeostasis and in the cell response to stress [28]. Autophagic processes are often impaired in dystrophin-deficient hearts, leading to the accumulation of dysfunctional mitochondria and other damaged cellular components. The failure of cellular repair mechanisms compounds the oxidative stress and mitochondrial dysfunction already present, further accelerating cardiomyocyte death and myocardial degeneration. Studies have shown that enhancing autophagy may have beneficial effects on cellular survival, tissue repair, and mitochondrial function in dystrophin-deficient hearts [29].
The targeting of autophagy-related pathways could provide a novel therapeutic angle. Recent research has indicated that pharmacological modulation of autophagy could improve cellular health by promoting the clearance of damaged cellular components, thus potentially mitigating the adverse effects of dystrophin deficiency [30].
The interplay between these pathological mechanisms ultimately leads to progressive cardiomyocyte loss, fibrosis, and heart failure in DDCM (Fig. 1). Understanding these interconnected pathways provides valuable insights into potential therapeutic targets. Approaches aimed at stabilizing the sarcolemma, restoring calcium homeostasis, reducing fibrosis, and improving metabolic function hold promise for mitigating disease progression.
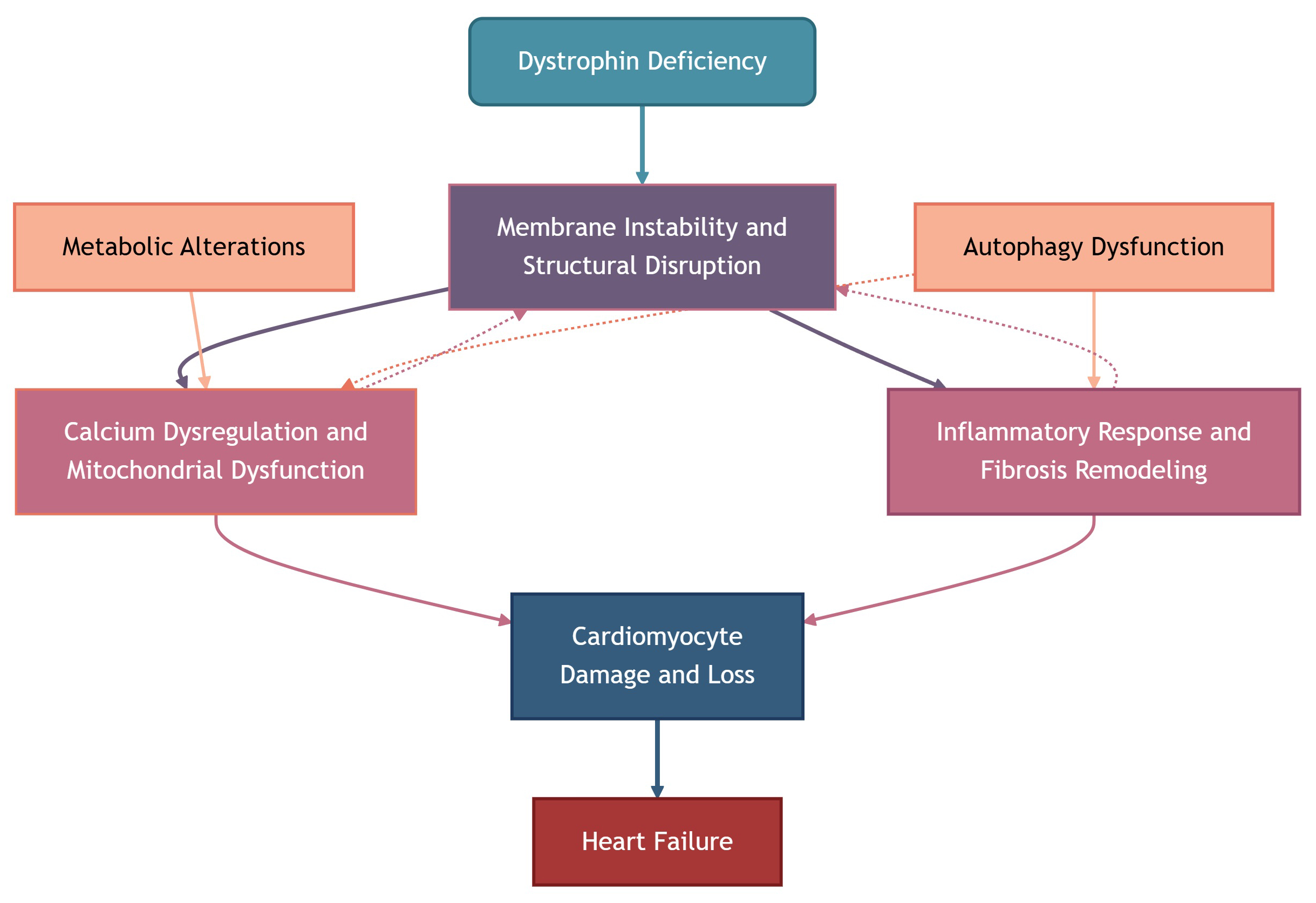 Fig. 1.
Fig. 1.
Molecular mechanisms underlying dystrophin-deficient cardiomyopathy. Solid arrows indicate direct causal relationships, while dashed arrows represent exacerbating or feedback effects.
As previously described, cardiac dysfunction in patients with DMD and BMD typically begins to manifest in adolescence. The progressive nature of heart failure in these individuals often leads to significant delays in diagnosis, as skeletal muscle degeneration generally precedes cardiac symptoms [31]. However, the onset of cardiac symptoms is insidious and can be masked by the concurrent skeletal muscle weakness. In the early stages, left ventricular dilation and reduced ejection fraction are commonly observed with echocardiography [32].
Given the high variability in disease progression, clinical practitioners face challenges in predicting the exact timeline of heart failure in dystrophin-deficient patients. Early biomarkers, such as elevated levels of cardiac troponin I or other myocardial proteins, are critical for detecting subclinical cardiac dysfunction. Additionally, advanced imaging techniques like cardiac magnetic resonance (CMR) imaging, which offer more detailed insights into myocardial fibrosis and structure, have become essential tools for monitoring disease progression [33].
Arrhythmias are a major cause of mortality in dystrophin-deficient patients. Structural changes, including fibrosis and abnormal electrical conduction pathways, contribute to the development of arrhythmias, often leading to sudden cardiac death [34]. Prolonged exposure to arrhythmic events can significantly impair quality of life, especially in adolescent and young adult patients. To better manage these risks, continuous cardiac monitoring and proactive interventions are critical.
Greater awareness of the arrhythmogenic substrate in dystrophinopathies has prompted the exploration of new therapies aimed at stabilizing myocardial electrical activity [35]. The use of antiarrhythmic medications, implantable defibrillators, and pacemakers is becoming more common in clinical practice. Nevertheless, the benefits of these interventions are often limited, underscoring the need for therapies that target the underlying molecular causes of arrhythmia.
Traditional imaging modalities such as echocardiography and CMR imaging are useful in evaluating structural and functional abnormalities [36]. However, these tools often detect changes only after significant damage has occurred. To facilitate earlier intervention, research is increasingly focused on the development of sensitive biomarkers. Circulating biomarkers such as cardiac troponin I, various microRNAs (miRNAs), and other myocardial-specific proteins are under investigation as potential early indicators of cardiac involvement [37]. Table 1 (Ref. [38, 39, 40, 41, 42, 43, 44, 45]) provides a comprehensive overview of these emerging biomarkers and their detection methods, performance, and clinical significance.
| Biomarker | Detection method | Performance | Clinical significance | Key references |
| Cardiac troponin I (high-sensitivity assays) | Serum/plasma | Quantitative sensitivity/specificity for DMD cardiac involvement are not uniformly reported; longitudinal hs-cTnI correlates with cardiac involvement and is recommended for a prospective study. | Candidate early myocardial injury marker in DMD/BMD; useful for longitudinal monitoring, but thresholds and prognostic cutoffs are still under study. | Spurney et al., 2021 [38]; Yamaguchi et al., 2022 [42] |
| miR-1, miR-133a/b | Plasma/serum qPCR or sequencing | Multiple DMD cohorts report high AUCs distinguishing DMD vs controls (ROC/AUC reported in some studies; e.g., many myomiRs show AUCs |
Robust circulating markers of muscle injury; some (miR-1, miR-133) associate with cardiac involvement in cohort studies—promising for early detection but need cardiac-focused prospective validation. | Zaharieva et al., 2013 [43]; Li et al., 2014 [44]; Meng et al., 2022 [39] |
| miR-208a/b, miR-499 (cardiac myomiRs) | Plasma/serum qPCR | Reported dysregulation in DMD; in non-DMD cardiology literature, these have high specificity for cardiac injury. Cohort studies in DMD report correlations with fibrosis/CMR changes. | More cardiac-specific than skeletal myomiRs; candidate markers for myocardial injury/fibrosis in dystrophinopathies. | Jeanson-Leh et al., 2014 [40]; Liu et al., 2015 [41] |
| Exosome/EV-associated miRNAs and proteins | Plasma/serum exosome profiling (RNA-seq, MS) | Emerging studies report altered EV-cargo in DMD; sensitivity/specificity not yet standardized across platforms. | Exosomal cargo may improve tissue-specificity and stability versus free miRNA; technical standardization and large prospective validation are needed. | Yedigaryan and Sampaolesi, 2023 [45] |
AUC, area under the ROC curve; BMD, Becker muscular dystrophy; CMR, cardiac magnetic resonance; cTnI, cardiac troponin I; DMD, Duchenne muscular dystrophy; EV, extracellular vesicle; hs-cTnI, high-sensitivity cardiac troponin I; miR-1, MicroRNA 1; miR-133a/b, MicroRNA 133a/b; miR-208a/b, MicroRNA 208a/b; miR-499, MicroRNA 499; miRNA, microRNA; myomiR, muscle-enriched microRNA; MS, mass spectrometry; qPCR, quantitative polymerase chain reaction; RNA-seq, RNA sequencing; ROC, receiver operating characteristic.
Among these, the detection of cardiac troponin I with high-sensitivity assays is a plausible early indicator of myocardial injury and has shown longitudinal associations with cardiac involvement in dystrophinopathies [38]. Muscle-specific miRNAs released into the circulation following myocyte damage, such as miR-1 and miR-133a/b, have also shown promise. Significantly elevated serum levels of miR-1, miR-133a, and miR-133b were found in DMD patients compared to healthy controls, and these levels were higher in patients with documented cardiac involvement [39]. Although much of this work pertains to skeletal muscle pathology, several studies have suggested potential applicability to cardiac manifestations of dystrophinopathy. Another review noted that miR-1 and miR-133 were widely investigated in cardiovascular diseases as biomarkers of myocardial injury and fibrosis [46], although prospective, cardiac-endpoint–anchored validation and standardized clinical cutoff levels are still lacking [39]. Cardiac-biased myomiRs (miR-208a/b, miR-499) show greater cardiac specificity and have been linked to fibrosis or CMR changes in smaller cohorts. However, their lower circulating levels and the heterogeneity of assays limit their immediate clinical translation [40, 41]. Extracellular vesicles (EV) such as exosomes can carry miRNAs (including miR-1, miR-133) and may provide enhanced specificity for cardiac tissue injury. A recent review of exosomal miRNAs in cardiovascular disease highlights their value as non-invasive biomarkers and identifies miR-1 and miR-133 among those most frequently detected [47]. Nonetheless, while these biomarkers are promising, their specificity for cardiac vs skeletal muscle pathology in DMD/BMD patients remains to be firmly established, and large-scale clinical validation is still lacking [48].
Advances in molecular imaging techniques that target specific cellular and metabolic processes may also provide earlier detection of pathological changes.
While DMD and BMD are X-linked recessive disorders, gender differences have been observed in disease presentation [49]. For example, female carriers of DMD may exhibit variable degrees of cardiac involvement, with some experiencing progressive heart failure despite their relatively normal skeletal muscle function [50]. This is particularly evident in female carriers with skewed X-inactivation patterns, where the defective X chromosome is preferentially silenced, leading to lower levels of dystrophin expression in cardiac tissue.
In addition to the traditional male phenotype, female carriers of dystrophinopathies may show a more subtle onset of cardiomyopathy, often going undiagnosed until later in life. The occurrence of heart failure in these female carriers has attracted attention, as it suggests the need for more tailored screening and monitoring strategies [51]. Research is increasingly focused on understanding the underlying genetic and epigenetic factors that contribute to the variability in disease progression. This could lead to more personalized approaches in the diagnosis and management of DDCM [52].
Recent advancements in non-invasive imaging technologies are helping to detect early changes in the myocardium that may not yet be clinically apparent with conventional methods. For example, CMR imaging can identify myocardial fibrosis, a hallmark of heart failure, long before the onset of overt symptoms [53]. This ability to detect early pathological changes opens the door for interventions that could slow or halt the progression of heart failure.
Similarly, speckle tracking echocardiography is an emerging technique that assesses myocardial strain and can detect subclinical myocardial dysfunction, even in the absence of significant structural changes [54]. This technique holds promise as a diagnostic tool for the early identification of cardiac involvement in patients with dystrophinopathies. As these advanced imaging methods become more widely available, they may revolutionize the timing of interventions, allowing clinicians to initiate therapies before the occurrence of irreversible damage [55].
Effective therapeutic strategies are crucial for managing heart failure related to dystrophin deficiency. This section provides an overview of current and emerging approaches, including gene therapy, pharmacological interventions, and regenerative medicine. Table 2 presents a comprehensive summary of these strategies, detailing their mechanisms, current status, advantages, and challenges.
| Therapeutic strategy | Mechanism of action | Key examples | Current status | Advantages | Challenges |
| Gene therapy | Partially restore dystrophin expression | Micro-dystrophin gene therapy | Clinical trial (Phase III) | Addresses root cause, reduces membrane instability | Immune responses to viral vectors, optimal delivery route |
| Anti-fibrotic agents | modulate the fibrotic response | TGF- |
Preclinical | Reduces fibrosis, improves cardiac function | Lack of clinical research |
| Calcium-regulating agents | Restore calcium homeostasis, modulate calcium flux | Ryanodine receptor stabilizers | Preclinical | Stabilizes Ca2+ levels, protects cardiomyocytes | Lack of clinical research |
| Regenerative medicine | Repair damaged myocardium, tissue regeneration | iPSC-derived cardiomyocytes | Preclinical | Integrate with host tissue, improve contractile function | Survival, maturation, Immune responses, Safety concerns |
| Combination therapies | Target multiple pathological pathways simultaneously | Gene therapy and pharmacological agents | Preclinical | Synergistic benefits, comprehensive approach | Requires carefully designed clinical trials, complexity |
| Gene editing | Directly correct genetic mutations for dystrophin deficiency | CRISPR/Cas9 | Preclinical | Potential for long-term, curative treatment | Minimizing off-target effects, long-term stability, and ethical concerns |
iPSC, induced pluripotent stem cells; CRISPR/Cas9, Clustered Regularly
Interspaced Short Palindromic Repeats/CRISPR-associated protein 9; TGF-
Gene therapy has emerged as one of the most promising approaches for treating dystrophin deficiency. Recent advances in viral vector technologies have enabled the delivery of micro-dystrophin constructs that can partially restore dystrophin expression in affected tissues [56]. Micro-dystrophin gene therapy has been shown to reduce membrane instability, stabilize calcium levels, and prevent further cardiomyocyte death in a preclinical model [57].
Building on encouraging preclinical results, clinical trials of gene therapy for DMD-related heart failure have advanced considerably, including the EMBARK Phase 3 randomized trial (NCT05096221) [57]. Continued advances in gene delivery systems and micro-dystrophin therapy hold great promise as components of a comprehensive treatment strategy. Nonetheless, significant challenges remain, including immune responses to viral vectors, optimal delivery route, and the long-term stability of dystrophin expression [58].
While current pharmacological therapies for heart failure (e.g.,
angiotensin-converting enzyme [ACE] inhibitors, beta-blockers) have shown some
efficacy in slowing disease progression, they do not target the primary molecular
defects [59]. Recent research has focused on agents that modulate the fibrotic
response and restore calcium homeostasis. TGF-
In addition to calcium channel blockers, other therapeutic agents that target calcium handling, such as ryanodine receptor stabilizers currently in preclinical investigation, are being investigated. These drugs act on intracellular calcium release channels to prevent excessive calcium release from the sarcoplasmic reticulum, a major source of intracellular calcium overload [64, 65]. By stabilizing calcium homeostasis, these agents may offer a complementary treatment to gene therapy or stem cell-based therapies in dystrophin-deficient heart failure.
Regenerative medicine holds promise for repairing damaged myocardium in dystrophin-deficient hearts. The use of induced pluripotent stem cells (iPSCs) to generate patient-specific cardiomyocytes is one of the most exciting developments in this field [66]. Experimental studies have shown that iPSC-derived cardiomyocytes can integrate with host tissue, improve contractile function, and even contribute to the regeneration of damaged myocardium [67]. However, significant challenges remain regarding the survival, maturation, and long-term functionality of these transplanted cells. Further research into optimizing cell delivery methods and ensuring proper integration with the host’s cardiac tissue is ongoing.
Translational challenges include:
• Cell survival and integration: The survival of transplanted cells within the damaged myocardium remains a major issue. Host immune responses may lead to cell rejection, and the harsh ischemic and inflammatory environment of the heart poses a challenge for cell integration. A recent study highlighted the need for optimized immunosuppressive strategies to reduce immune rejection and support graft persistence [68].
• Cell maturation: iPSC-derived cardiomyocytes do not always mature to a functional adult-like state, thus affecting their ability to contribute to long-term heart function. The development of strategies to promote proper maturation and integration into the host tissue is crucial. Current research has shown promise in using small molecules and engineered scaffolds to enhance cell maturation [69].
• Immune responses: Although iPSC-derived cells are patient-specific, immune activation still poses a risk, particularly in cases where non-autologous sources of stem cells are used, or if the cells undergo genetic modifications. The use of immunomodulatory agents or the development of immune-tolerant iPSCs may help to mitigate adverse responses [70].
• Safety concerns: Long-term safety remains a concern, particularly the risk of tumorigenicity from undifferentiated iPSCs. Effective strategies for controlling the differentiation of these cells and avoiding tumor formation are critical. Ongoing work in the field has focused on purifying differentiated cells and ensuring that only fully mature cardiomyocytes are transplanted [71].
While significant progress has been made in improving the delivery methods and optimizing cell survival, additional research is needed to address these challenges. Clinical trials must focus on ensuring the long-term efficacy and safety of these therapies, with special attention paid to minimizing immune responses and improving cell function. In combination with gene therapy or pharmacological agents, stem cell therapies may ultimately hold the key to reversing or halting the progression of heart failure in dystrophin-deficient patients.
Given the multifactorial nature of DDCM, a single therapeutic modality may not be sufficient to halt disease progression. Combination therapies that target multiple pathological pathways simultaneously are being explored [72]. For example, integrating gene therapy with pharmacological agents that reduce fibrosis and oxidative stress could offer synergistic benefits. Similarly, combining regenerative medicine approaches with anti-inflammatory treatments might enhance the survival and integration of transplanted cells. This type of strategy remains in the experimental stage, and rigorous clinical trials are needed to assess its effectiveness and safety [73, 74].
Gene editing technologies, particularly Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/Cas9), have emerged as powerful tools for directly correcting the mutations responsible for dystrophin deficiency. Recent advances in genome editing have demonstrated that dystrophin expression can be restored in animal models of DMD through precise edits of the dystrophin gene [75]. These promising results have prompted ongoing efforts to adapt CRISPR technology for clinical applications, with the potential to correct genetic mutations in human patients [76]. While still in the preclinical stages, the use of CRISPR to correct dystrophin mutations holds significant promise for providing a long-term, potentially curative treatment for DMD and BMD patients [77].
However, several translational challenges still hinder the widespread clinical application of these therapies:
• Delivery systems: One of the key challenges in genome editing is the efficient delivery of CRISPR/Cas9 components to the target cells. Non-viral delivery systems, such as nanoparticles, are promising but often suffer from low efficiency and poor targeting. Viral vectors, while more efficient, carry risks of immune responses and off-target effects [78].
• Immune responses: The use of CRISPR/Cas9 may trigger immune reactions, particularly when viral vectors are used for delivery. These immune responses can reduce the effectiveness of treatment and increase the risk of side effects. A key area of ongoing research is the development of CRISPR systems that minimize immune activation [79].
• Off-target effects: Although the precision of CRISPR/Cas9 technology has improved significantly, off-target mutations remain a concern. These unintended genetic changes could potentially lead to adverse outcomes, such as the activation of oncogenes or the disruption of other important genes. Researchers are now focused on improving the specificity of CRISPR systems by using more refined versions of Cas9, such as high-fidelity Cas9 variants [80].
• Long-term stability: Another challenge is ensuring the long-term stability of dystrophin expression. Even if initial therapeutic effects are achieved, maintaining stable gene expression over time in a patient’s heart and muscle tissue is critical. Concerns remain about the durability of CRISPR-based edits, particularly after the treated cells divide [81].
• Ethical concerns: Beyond the technical challenges, gene editing for therapeutic purposes raises ethical questions, particularly regarding germline editing or the potential for unintended consequences in the genetic code. Regulatory frameworks will need to address these concerns as clinical trials move forward [82].
Despite these challenges, CRISPR-based therapies hold significant promise for treating dystrophinopathies. As research progresses, attention must be paid to minimizing off-target effects and optimizing delivery systems to ensure safe and effective gene editing in human patients [83]. Once such issues are addressed, gene editing could offer a long-term, potentially curative solution for DMD and BMD patients.
Animal models have played a crucial role in advancing our understanding of dystrophin-deficient cardiomyopathy. The mdx mouse is a widely used model of DMD that recapitulates many features of the human disease, including progressive skeletal muscle weakness and cardiac dysfunction [84]. This model has been instrumental in testing new therapies, ranging from gene editing to pharmacological intervention [1].
However, despite their value, animal models often fail to fully replicate the complexity of the human condition, particularly with regard to disease heterogeneity and long-term therapeutic outcomes. Consequently, there is an ongoing need for more sophisticated models that better mirror human disease and provide more predictive data for clinical trials. The development of large animal models, such as pigs and non-human primates, could provide more accurate representations of the human cardiovascular system and enable better testing of new treatments.
The advent of genome editing tools, particularly CRISPR/Cas9, has opened new avenues for directly correcting the genetic mutations responsible for dystrophin deficiency. Recent studies have demonstrated that CRISPR-mediated approaches can restore dystrophin expression in animal models, leading to improvements in both skeletal and cardiac muscle function [75]. Although still in the experimental phase, these technologies represent a promising strategy for future therapeutic interventions that could potentially offer a permanent cure [85].
While preclinical models have provided invaluable insights, several challenges remain in translating these findings to the clinic. Significant hurdles remain in terms of the heterogeneity of the patient population, issues related to immune responses, and the need for long-term efficacy data, as detailed in Section 4 [86]. Future research must focus on bridging the gap between bench and bedside through well-designed clinical trials, the development of robust biomarkers for early detection, and the refinement of therapeutic delivery systems.
In recent years, the concept of precision medicine has gained significant momentum in the treatment of complex diseases, including dystrophin-deficient cardiomyopathy. Advances in genomic profiling technologies, such as next-generation sequencing, have led to a deeper understanding of the genetic mutations that contribute to dystrophinopathies [87]. These technologies can also identify genetic variants that modulate disease severity, thus providing valuable insights into how individual patients may respond to different therapeutic strategies.
Precision medicine in the context of dystrophinopathies goes beyond simply correcting the underlying dystrophin deficiency. By considering individual genetic factors, clinicians can tailor interventions to minimize adverse side effects and maximize therapeutic efficacy [88]. For example, by identifying specific mutations or epigenetic modifications that influence the course of disease, therapies could be personalized to more effectively target both the underlying genetic cause and downstream pathological processes such as fibrosis, inflammation, and mitochondrial dysfunction.
Furthermore, the incorporation of precision medicine can enhance the development of novel therapeutic strategies. Pharmacogenomic approaches, which consider how genetic variations affect drug responses, can optimize the use of existing medications and improve outcomes in patients with DMD and BMD [89]. Tailored therapies could also involve the use of gene editing technologies or RNA-based therapies that are customized according to the specific genetic profile of each patient, ultimately improving their clinical management and outcomes.
Chronic inflammation plays a pivotal role in the progression of DDCM, contributing to both myocardial fibrosis and the ongoing cycle of muscle degeneration. Therefore, immunomodulatory therapies that target inflammatory pathways have gained considerable attention as potential therapeutic options for dystrophinopathies [90]. The goal of immunomodulation in this context is to reduce chronic inflammation without compromising the body’s ability to fight infections and other diseases.
Several cytokines and immune signaling pathways, including TNF-
Regulatory T cell therapies, which aim to enhance the body’s natural anti-inflammatory response, are also being investigated as a potential treatment strategy to mitigate inflammation without exacerbating immune suppression. When combined with other therapeutic modalities such as gene therapy or stem cell therapy, these approaches could help to control the inflammatory process that drives myocardial damage in dystrophinopathies.
Several clinical trials are currently underway to evaluate novel therapies for
DDCM. Early-phase trials of micro-dystrophin gene therapy have reported
encouraging preliminary results, with improvements in cardiac function and safety
profiles that support further investigation [57]. Similarly, trials investigating
the efficacy of Sacubitril/Valsartan and TGF-
Despite these advances, significant challenges remain in the clinical translation of experimental therapies. One of the major hurdles is the variability in disease progression and treatment response among patients [93]. In addition, ensuring the long-term safety and efficacy of gene and cell-based therapies remains a concern. Multidisciplinary collaboration and well-designed, large-scale clinical trials are essential to overcome these obstacles and bring effective therapies to the clinic [94, 95].
The future management of DDCM lies in the integration of precision medicine, advanced biomarker development, and combination therapeutic strategies. Ongoing research should focus on:
• Refining gene editing and gene therapy techniques to maximize efficacy and minimize immune reactions.
• Expanding the use of iPSC-derived cardiomyocytes and improving their long-term integration into host tissue.
• Developing robust, non-invasive biomarkers for early detection and real-time monitoring of disease progression.
• Designing combination therapies that simultaneously target multiple pathological pathways, such as inflammation, fibrosis, and metabolic dysfunction.
• Conducting comprehensive longitudinal studies to evaluate the long-term outcomes of novel therapies.
Dystrophin deficiency initiates a cascade of molecular and cellular events that culminate in the development of cardiomyopathy and heart failure in patients with DMD and BMD. The multifactorial nature of this disease, encompassing membrane instability, calcium dysregulation, mitochondrial dysfunction, inflammatory responses, and metabolic alterations, necessitates a comprehensive, multi-targeted therapeutic approach. Emerging treatments, including gene therapy, pharmacological agents that target fibrosis and calcium handling, and regenerative medicine, have shown promise in preclinical and early clinical trials. However, challenges related to early diagnosis, treatment variability, and long-term efficacy remain.
Future research efforts should focus on integrating precision medicine approaches, developing novel biomarkers, and exploring combination therapies that address the complex interplay of pathogenic mechanisms. With continued advances in genomic technologies, immunomodulatory strategies, and regenerative medicine, there is optimism that more effective treatments will become available to improve the quality of life and survival of patients with DDCM.
ACE, Angiotensin-Converting Enzyme; ATP, Adenosine Triphosphate; AUC, Area Under the ROC Curve; BMD, Becker muscular dystrophy; CMR, Cardiac Magnetic Resonance; CRISPR/Cas9, Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9; cTnI, Cardiac Troponin I; DCM, dilated cardiomyopathy; DDCM, dystrophin-deficient cardiomyopathy; DMD, Duchenne muscular dystrophy; ECM, Extracellular Matrix; EV, Extracellular Vesicle; hs-cTnI, High-Sensitivity Cardiac Troponin I; IL-6, Interleukin-6; iPSCs, Induced Pluripotent Stem Cells; miR-1, MicroRNA 1; miR 133a/b, MicroRNA 133a/b; miR-208a/b, MicroRNA 208a/b; miR-499, MicroRNA 499; miRNAs, MicroRNAs; MS, Mass Spectrometry; myomiR, Muscle-Enriched MicroRNA; qPCR, Quantitative Polymerase Chain Reaction; RNA-seq, RNA Sequencing; ROC, Receiver Operating Characteristic; ROS, Reactive Oxygen Species; TGF-
PL conceived the review topic and developed the overall study framework and critically revised the manuscript. WQY and WW performed the literature search and selection, synthesized and interpreted the findings from the included literature, and drafted the initial manuscript. YBZ and FL screened relevant articles, extracted and collated key information, and contributed to the critical revision of the manuscript. QHW contributed to the interpretation of the literature and provided critical editing and substantive intellectual input during manuscript revision. All authors contributed to the critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript and agree to be accountable for all aspects of the work. All authors confirm that they meet the four ICMJE authorship criteria.
Not applicable.
The authors thank the teacher in the Cardiovascular Department of the Second Affiliated Hospital of Nanchang University.
This work was supported by Innovative Research Group Project of the National Natural Science Foundation of China (No.82160093 and No.81960098), the incubation project of the National Natural Science Foundation of the Second Affiliated Hospital of Nanchang University (No.2021YNFY2021), the Jiangxi Provincial Health Commission Project (No.202210663), and the Nature Science Foundation of Jiangxi Province (No. 20232BAB206013, No. 20232BAB216009 and No. 20224BAB216018).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.