



 , Elham Kayvanpour 1,2,3, Benjamin Meder 1,2,3
, Elham Kayvanpour 1,2,3, Benjamin Meder 1,2,31 Department of Cardiology, Angiology and Pneumology, Institut für Cardiomyopathien Heidelberg, University of Heidelberg, 69120 Heidelberg, Germany
2 German Center for Cardiovascular Research (DZHK) Partnerside Heidelberg, 69120 Heidelberg, Germany
3 Precision Digital Health and Informatics for Life, Clinic of Cardiology, Angiology and Pulmonology, University of Heidelberg, 69120 Heidelberg, Germany
Abstract
Hypertrophic cardiomyopathy (HCM) represents the most common inherited cardiac disease and a leading cause of heart failure, arrhythmias, and sudden cardiac death in young individuals. For decades, management of HCM has relied on symptom control with β-blockers, calcium channel blockers, disopyramide, or invasive septal reduction in advanced cases. The identification of pathogenic sarcomere variants and the recognition of hypercontractility as a central disease mechanism have paved the way for cardiac myosin inhibitors (CMIs), the first truly disease-specific pharmacological therapy for HCM. Indeed, CMIs represent a revolutionary therapeutic paradigm that redefines the standard of care by translating molecular discovery into clinical application. This review provides a guide to the mechanistic basis of sarcomere modulation, summarizes the clinical evidence for mavacamten and aficamten, and critically evaluates the evolving roles of both medications in obstructive and non-obstructive HCM.
Keywords
- hypertrophic cardiomyopathy
- myosin inhibitors
- myosin heavy chains
- mavacamten
- aficamten
- precision medicine
Hypertrophic cardiomyopathy (HCM) is the most prevalent inherited cardiovascular
disease, affecting approximately 1 in 200–500 individuals worldwide and
representing a leading cause of heart failure, arrhythmias, and sudden cardiac
death in the young [1, 2]. Despite significant advances in genetic mechanisms
leading to HCM, treatment strategies have for decades remained confined to
symptom control using
The identification of pathogenic variants in sarcomere genes, particularly myosin heavy chain 7 (MYH7) and myosin-binding protein C (MYBPC3), established HCM as a primary disease of the contractile apparatus [5]. This recognition shifted therapeutic aspirations from symptomatic palliation toward disease-specific intervention. Over the past decade, insights into cross-bridge kinetics and actin–myosin interaction have culminated in the development of selective cardiac myosin inhibitors (CMIs). By directly modulating myosin ATPase activity, these agents reduce excessive cross-bridge formation, increase myosin in super-relaxed state (SRX) thereby normalize contractility, and mitigate left ventricular outflow tract (LVOT) obstruction [6]. Mavacamten, the first-in-class myosin inhibitor, provided proof-of-concept that targeted sarcomere modulation could alter the natural history of HCM. Parallel development of aficamten, a next-generation myosin inhibitor with different pharmacokinetics, has reinforced the therapeutic potential of sarcomere modulation [7]. Together, these advances mark a paradigm shift in the management of HCM. For the first time, a pharmacological therapy targets the underlying molecular pathophysiology rather than its downstream consequences. Myosin inhibition has thus emerged as a revolutionary game changer, bridging the gap between genetic discovery and clinical translation (Fig. 1). This review summarizes the historical evolution of myosin inhibitors, synthesizes evidence from pivotal trials, and highlights future directions including use in non-obstructive HCM, pediatric populations, and novel sarcomere-targeting agents.
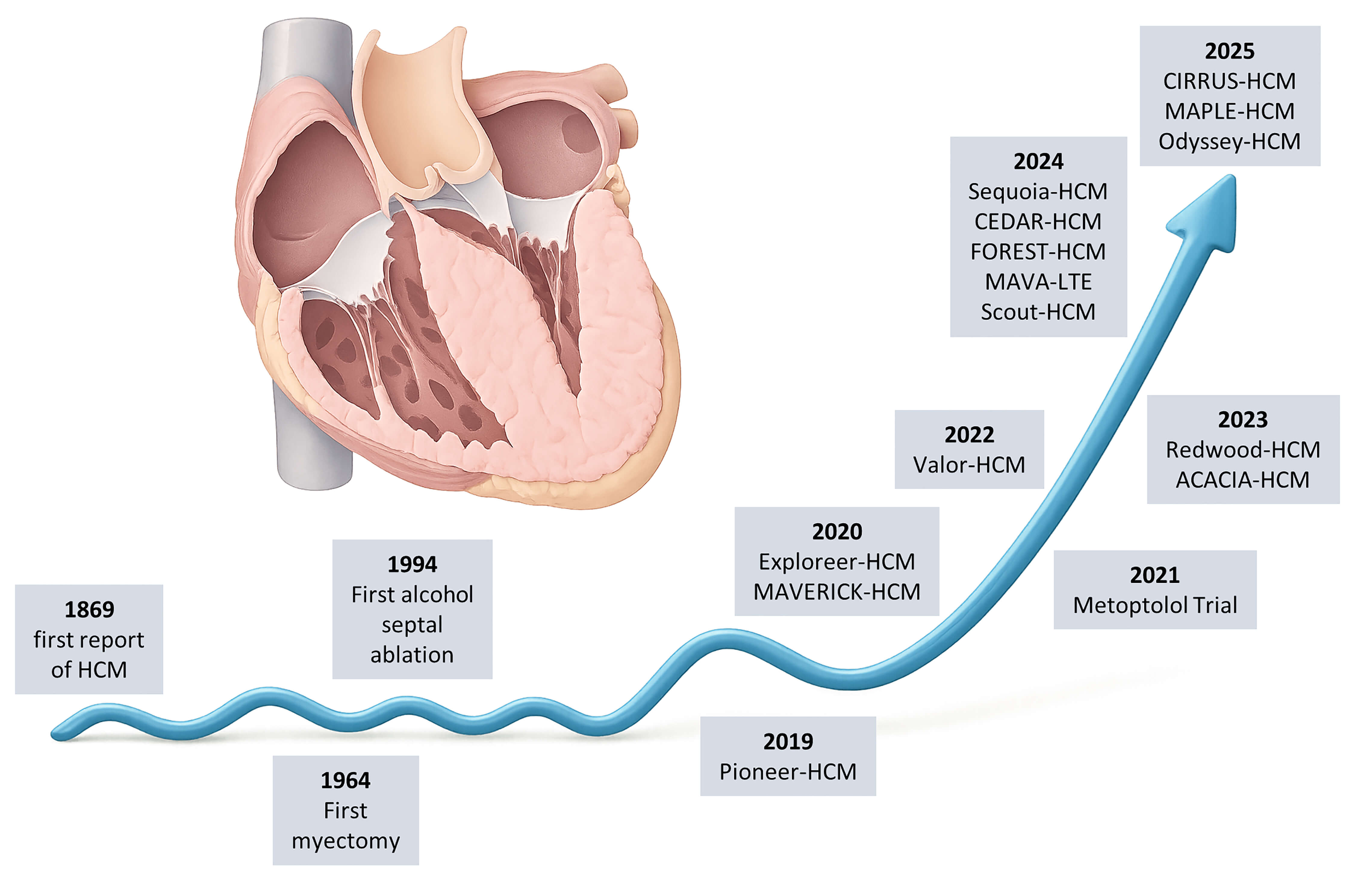 Fig. 1.
Fig. 1.
Timeline of milestones in hypertrophic cardiomyopathy (HCM). Key advances are shown from the first clinical description (1869) and early surgical/interventional therapies (myectomy, 1964; alcohol septal ablation, 1994) to the modern era of cardiac myosin inhibitors, with landmark trials illustrating the paradigm shift from invasive to disease-specific pharmacological therapy. Created in Illustrae (https://illustrae.co/)
The defining hallmark of HCM is sarcomeric hypercontractility, a direct consequence of pathogenic variants in genes encoding thick and thin filament proteins, most notably MYH7 and MYBPC3 [8]. At the cellular level, these variants increase the proportion of myosin heads available for actin interaction, thereby augmenting force generation but impairing diastolic relaxation and myocardial energetics [6, 9]. For mutation elusive obstructive HCM, the mechanism is not as well understood, but response to myosin inhibition seems comparable as shown in recent clinical trials. The resulting hyperdynamic contractile state promotes left ventricular hypertrophy, microvascular ischemia, fibrosis, and ultimately heart failure [10, 11, 12].
Cardiac myosin exists in a continuum of conformational states. In the SRX, myosin heads are sequestered along the thick filament backbone, minimizing adenosine triphosphate (ATP) turnover and conserving energy. In contrast, the disordered relaxed state (DRX) exposes myosin heads, increasing their probability of actin engagement and cross-bridge formation [13, 14]. Genetic variants destabilize the SRX conformation, shifting the balance toward DRX, thereby driving hypercontractility and energetic inefficiency [14, 15].
Selective CMIs, including mavacamten and aficamten, act by stabilizing the SRX
state and reducing the number of myosin heads available for cross-bridge cycling
(Fig. 2). This allosteric modulation of myosin ATPase activity normalizes
contractility, improves diastolic filling, and reduces LVOT gradients [6]. Unlike
traditional negative inotropes, which blunt adrenergic signaling
(
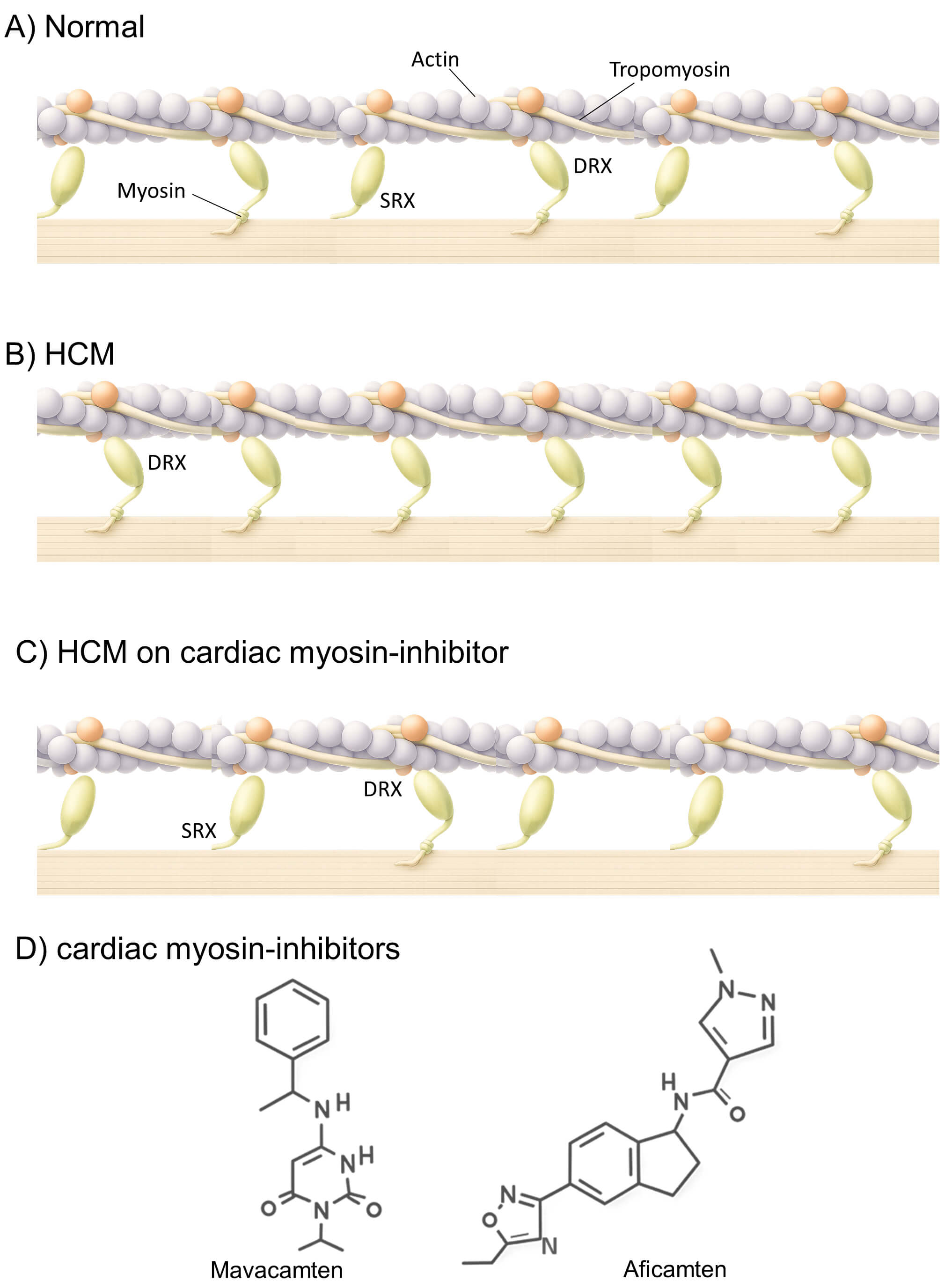 Fig. 2.
Fig. 2.
Mechanism of action of cardiac myosin inhibitors (CMIs). Balanced distribution of myosin heads between the super-relaxed (SRX) and disordered relaxed (DRX) states, permitting normal contractility and efficient relaxation (A). In hypertrophic cardiomyopathy (HCM), destabilization of the SRX state increases myosin head availability for cross-bridge formation, resulting in hypercontractility, diastolic dysfunction, and impaired energetics (B). CMIs such as mavacamten and aficamten stabilize the SRX state, reduce excessive cross-bridge cycling, and restore energetic efficiency (C). Schematic chemical structures of mavacamten and aficamten are shown (D). Created in Illustrae (https://illustrae.co/)
The translation of myosin inhibition from mechanistic insight to clinical application has been remarkable. Within little more than a decade, selective CMIs have advanced from preclinical proof-of-concept to phase 3 trials demonstrating meaningful clinical benefit in obstructive HCM.
The first-in-class inhibitor, mavacamten, was initially evaluated in the PIONEER-HCM trial, an open-label phase 2 study in symptomatic obstructive HCM patients [18, 19]. PIONEER demonstrated for the first time that pharmacological sarcomere modulation could achieve significant reductions in LVOT gradients, improve exercise capacity, and favourably remodel cardiac structure [18]. These results established the feasibility of myosin inhibition as a disease-specific therapeutic strategy. The subsequent EXPLORER-HCM trial, a global, randomized, double-blind, placebo-controlled phase 3 study, confirmed and extended these findings [20]. In EXPLORER, the largest randomized trial on HCM when it was conducted, mavacamten achieved the primary endpoint of improving exercise capacity and symptoms, with 37% of patients reaching the composite endpoint versus 17% in the placebo group. Moreover, nearly three-quarters of patients experienced improvement of at least one New York Heart Association (NYHA) functional class, paralleled by significant reductions in LVOT gradient, N-terminal pro-B-type natriuretic peptide (NT-proBNP), and troponin levels. Importantly, quality of life scores (Kansas City Cardiomyopathy Questionnaire, KCCQ) improved substantially, underscoring the clinical relevance of symptom relief [20]. Beyond symptomatic benefit, the VALOR-HCM trial provided evidence that myosin inhibition may alter established treatment pathways in obstructive HCM. In this randomized phase 3 study of patients referred for septal reduction therapy (SRT), only 18% of those receiving mavacamten remained eligible for SRT at week 16, compared with 77% in the placebo group [21, 22]. This dramatic reduction highlights the capacity of mavacamten to defer or even obviate the need for invasive septal reduction procedures, which have long been the cornerstone of management in advanced obstructive HCM. Importantly, these clinical improvements were accompanied by consistent reductions in resting and Valsalva LVOT gradients, NT-proBNP levels, and troponin concentrations, underscoring both hemodynamic and biomarker evidence of therapeutic benefit [21, 22].
While mavacamten validated the therapeutic concept, its pharmacokinetic profile
necessitates regular echocardiographic monitoring due to its long half-life time,
a relatively narrow therapeutic window and potential for relative overdosing with
consecutive left ventricular systolic dysfunction. Aficamten, a next-generation
CMI, was designed to overcome these limitations (Table 1). In the REDWOOD-HCM
phase II trial, aficamten demonstrated dose-dependent, rapid, and reversible
reductions in LVOT gradients, paralleled by improvements in symptoms and
biomarkers, with an excellent safety profile [23, 24]. The SEQUOIA-HCM phase III
trial has also established aficamten as an effective therapy in symptomatic
obstructive HCM [25]. At 24 weeks, aficamten significantly improved exercise
capacity (between-group difference in peak VO2: +1.7 mL/kg/min), health status
(Kansas City Cardiomyopathy Questionnaire-Clinical Summary Score (KCCQ-CSS): +7
points vs placebo), and NYHA class (58.5% vs 24.3% improved), while achieving
near-complete gradient relief in almost half of treated patients [25]. The
MAPLE-HCM trial provided the first direct, head-to-head comparison of a CMI with
standard-of-care beta-blockade [26, 27]. In this phase III, double-blind study,
175 untreated patients with symptomatic obstructive HCM were randomized to
aficamten (5–20 mg quaque die once daily, titrated) or metoprolol (dosed to
50–200 mg QD) for 24 weeks [27]. Aficamten was superior to metoprolol for the
primary endpoint of peak VO2, and demonstrated consistent benefits across key
secondary endpoints: NYHA class improvement, KCCQ-CSS (+6.9 points), Valsalva
LVOT gradient (–34.9 mmHg), NT-proBNP (ratio 0.19), and left atrial volume index
(-7 mL/m2). Importantly, after a 4-week washout, physiologic effects waned,
consistent with aficamten’s short half-life and pharmacodynamic reversibility.
The safety profile was acceptable, with left ventricular ejection Fraction (LVEF)
| Characteristic | Mavacamten (Camzyos |
Aficamten |
| Molecular class | Small-molecule, allosteric cardiac myosin inhibitor (1st generation) | Small-molecule, allosteric cardiac myosin inhibitor (2nd generation) |
| Binding/Mechanism | Stabilizes super-relaxed state; reduces cross-bridge cycling | Same mechanism, but distinct allosteric binding site; designed for wider therapeutic window |
| Half-life (t½) | ||
| Time to steady state | ||
| Wash-out | Requires |
Faster wash-out due to shorter t½; reversibility within days–weeks |
| Metabolism | Primarily CYP2C19, minor CYP3A4 | Minimal CYP involvement |
| Drug–drug interactions | Contraindicated with strong CYP2C19/CYP3A4 inhibitors or inducers | Low DDI risk reported in trials |
| Dose titration | Tablets 2.5/5/10/15 mg once daily; titration based on LVOT gradient & LVEF | 5–20 mg once daily in trials; echo-guided titration |
| Monitoring requirements | Mandatory REMS program (serial echocardiography) | May require less intensive long-term monitoring, no REMS (not yet approved) |
| Key efficacy (Phase III) | EXPLORER-HCM, VALOR-HCM: Significant improvements in exercise capacity, symptoms, and quality of life; marked LVOT gradient reduction; reduced eligibility for septal reduction therapy | SEQUOIA-HCM: Significant improvements in exercise capacity, symptoms, and quality of life; marked LVOT gradient reduction, comparable to mavacamten |
| Safety (LVEF |
6–14% across trials; all reversible | |
| Regulatory status (2025) | FDA (2022) and EMA (2023) approved for symptomatic oHCM | NDA under FDA review (PDUFA Dec 2025) |
CYP, Cytochrome P450; DDI, drug–drug interaction; EMA, European Medicines Agency; FDA, U.S. Food and Drug Administration; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract; NDA, New Drug Application; oHCM, obstructive hypertrophic cardiomyopathy; PDUFA, Prescription Drug User Fee Act date; REMS, Risk Evaluation and Mitigation Strategy.
While most drug development has centered on obstructive HCM, nearly one-third of
patients present with non-obstructive HCM. These patients experience substantial
symptom burden and progressive functional limitation, yet no disease-specific
therapies are currently approved. Conventional agents such as
The introduction of CMIs into clinical practice has been accompanied by close
attention to safety, tolerability, and monitoring requirements. While both
mavacamten and aficamten demonstrate consistent efficacy across pivotal trials,
their pharmacokinetic properties, potential for left ventricle (LV) systolic
dysfunction, and regulatory frameworks necessitate careful implementation. A key
safety consideration is the potential for transient reductions in LVEF,
reflecting the intended negative inotropic mechanism. In EXPLORER-HCM, VALOR-HCM,
and MAVA-LTE, between 5% and 14% of mavacamten-treated patients experienced a
reduction in LVEF
Mavacamten is metabolized primarily via Cytochrome P450 2C19 (CYP2C19) and CYP3A4, creating potential for drug-drug interactions with agents such as antiarrhythmics, antifungals, and proton-pump inhibitors [33]. Accordingly, comprehensive medication reconciliation is essential prior to and during therapy. In a recent analysis of real-world data of a post-market approval registry in the US, 99% of patients did not show any clinically relevant interacting medication before initiation of mavacamten. Aficamten, while not yet approved, undergoes minimal CYP-mediated metabolism, which may improve safety in elderly patients and those with polypharmacy [33].
Overall, CMIs have shown a favorable tolerability profile. The most frequently
reported adverse events include dizziness, fatigue, and transient reductions in
ejection fraction, with no evidence of excess arrhythmias or proarrhythmic risk
compared with placebo. Importantly, treatment discontinuation due to adverse
events remains low (
Despite their promise, several important uncertainties and challenges remain in
the clinical implementation of CMIs. While MAVA-LTE and FOREST-HCM provide
reassuring data over 2–3 years, the long-term consequences of chronic sarcomere
inhibition are unknown. Whether lifelong therapy in young patients alters
survival, sudden cardiac death risk, or progression to end-stage heart failure
remains unanswered. In patients otherwise eligible for SRT, whether CMIs should
be offered as first-line alternatives or as a bridge remains debated. VALOR-HCM
demonstrated a striking reduction in SRT eligibility, but head-to-head
comparisons with surgical myectomy or alcohol septal ablation are lacking [22].
CMI use after acute myocardial infarction raises concern: transient negative
inotropy in a myocardium already compromised by ischemic injury could exacerbate
pump failure. No study has systematically evaluated post-myocardial infarction
(MI) use, and this remains a relative contraindication until further evidence
emerges. Similarly, in overdose scenarios, the predictable risk is profound
systolic dysfunction; while reversible, optimal management strategies (temporary
mechanical support, pharmacological reversal) remain theoretical and require
guideline development. Not all HCM patients harbor sarcomere mutations, and
treatment response may differ by genotype. Preliminary analyses suggest that
sarcomere-positive patients may derive greater structural reverse remodeling, but
robust genotype–phenotype–treatment interaction data are limited. Precision
cardiology approaches incorporating genetics, imaging, and biomarkers will be
essential to refine patient selection. CMIs can be used safely in patients with
mild-to-moderate renal impairment (estimated Glomerular Filtration Rate, eGFR
The therapeutic horizon for cardiac myosin inhibition is rapidly expanding. Ongoing studies will determine whether the symptomatic and biomarker improvements observed in early-phase studies translate into robust functional and quality-of-life benefits in non-obstructive HCM. Parallel pediatric and adolescent studies (e.g., CEDAR, SCOUT) are evaluating safety and efficacy in younger patients, where early intervention may prevent maladaptive remodeling and alter lifetime disease trajectory. Beyond classical HCM, CMIs may also have a role in other conditions of hypercontractility. Early exploratory work suggests potential application in heart failure with preserved ejection fraction (HFpEF), where sarcomeric hypercontractility and diastolic dysfunction contribute to pathophysiology [36]. Further investigation will determine whether CMIs can extend benefit into this broader heart failure population.
Integration of CMIs with genetic testing and precision cardiology frameworks could ultimately enable targeted therapy for at-risk mutation carriers before overt disease manifests, opening the door to disease prevention. In parallel, real-world registries and long-term extension studies will be essential to address questions of durability, late safety signals, and cost-effectiveness, and to define their impact on hard outcomes such as progression to heart failure, arrhythmia burden, and mortality.
The positioning of CMIs within current treatment algorithms is becoming
increasingly relevant.
In conclusion, cardiac myosin inhibitors represent the first truly disease-specific pharmacological therapy for hypertrophic cardiomyopathy. By directly modulating sarcomeric hypercontractility, CMIs achieve outcomes previously attainable only through invasive septal reduction procedures: relief of obstruction, symptomatic improvement, and evidence of structural reverse remodeling. As development extends to non-obstructive disease, pediatric populations, and next-generation molecules, CMIs are poised to fundamentally redefine the standard of care in HCM. While challenges remain—including uncertainties about long-term outcomes, equitable access, and patient selection—myosin inhibition stands as a revolutionary game changer, bridging decades of genetic discovery with tangible clinical translation.
FSH conceived the review, performed the literature search, interpreted the data, drafted the manuscript, and critically revised the final version. EK supported data acquisition and contributed to the critical revision of the manuscript. BM substantial contributions to the conception of the work and critically reviewed the manuscript for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
FBS has received advisory honoraria as well as speaker and travel honoraria from Bristol Myers Squibb. EK has received speaker honoraria from Bristol Myers. BM reports honoraria for scientific advisory activities, speaking engagements, and travel support from Bristol Myers Squibb and Cytokinetics. Farbod Sedaghat-Hamedani is serving as Guest Editor of this journal. Farbod Sedaghat-Hamedani had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Theodoros Karamitsos.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.