

 , Jie Ma 1,2, Xu Zhao 1, Jing Yu 1,*
, Jie Ma 1,2, Xu Zhao 1, Jing Yu 1,*
1 Department of Cardiology, Lanzhou University Second Hospital, 730030 Lanzhou, Gansu, China
2 The Second Clinical Medical College, Lanzhou University, 730030 Lanzhou, Gansu, China
Abstract
Heart failure with preserved ejection fraction (HFpEF) has progressively emerged as the predominant form of heart failure. Thus, studies on the underlying mechanisms of HFpEF have shifted from pathophysiological to molecular factors. Meanwhile, previous studies have primarily focused on inflammation, oxidative stress, metabolic dysregulation, and impaired cardiac compliance (manifesting as ventricular hypertrophy and interstitial fibrosis). In addition to conventional guideline-directed medical therapies, novel therapeutic strategies targeting these aforementioned pathogenic pathways have been investigated. This review aimed to summarize recent progress in HFpEF pathogenesis and emerging treatment approaches, offering insights for developing novel diagnostic and management strategies.
Keywords
- HFpEF
- inflammation
- oxidative stress
- energy metabolism
- therapy
Heart failure with preserved ejection fraction (HFpEF) is a cardiovascular syndrome characterized primarily by left ventricular diastolic dysfunction. As the aging population and the prevalence of metabolic diseases such as hypertension and obesity increase, the incidence of HFpEF continues to increase. HFpEF has surpassed heart failure with reduced ejection fraction (HFrEF) as the predominant form of heart failure. Studies have indicated that HFpEF constitutes approximately 50% of all heart failure cases [1]. Although its age-specific incidence rate shows a declining trend, the magnitude of this decline is significantly lower than that observed in the case of HFrEF [2].
Studies on the pathological mechanisms of HFpEF have mainly focused on macroscopic hemodynamic characteristics, such as left ventricular diastolic dysfunction, left atrial dysfunction, and epicardial factors. However, these studies have not elucidated its pathogenesis at a deeper and fundamental level. In 2013, Paulus and Tschöpe [3] proposed the inflammation hypothesis to link HFpEF with systemic comorbidities (e.g., obesity, diabetes, and hypertension), suggesting that chronic low-grade inflammation and oxidative stress driven by these comorbidities are key molecular mechanisms leading to myocardial cell dysfunction, coronary microvascular dysfunction (CMD), and myocardial interstitial fibrosis. These findings shifted the focus of research from organ-level pathophysiological factors to cellular and molecular factors, substantially improved the understanding of the pathogenesis of HFpEF, and guided the development of novel therapeutic strategies targeting inflammatory and oxidative stress–related pathways.
Despite preserved systolic function, problems such as decreased exercise tolerance, reduced quality of life, and high hospitalization rates are prevalent among patients with HFpEF, and effective treatments remain lacking. Consequently, elucidating the pathological mechanisms of HFpEF and optimizing therapeutic strategies have become major research focuses in recent years. This review aimed to summarize the pathological mechanisms of HFpEF and discuss recent therapeutic advances, providing insights for improving the diagnosis and treatment of HFpEF.
Analysis of different types of heart failure using SOMAscan technology has
revealed that HFpEF exhibits a unique proteomic signature characterized by the
upregulation of inflammation-related proteins, such as interleukin-6 receptor
fraction (IL-6R), indicating that inflammation is the main factor driving HFpEF
pathogenesis (Fig. 1) [4]. Among various inflammatory mediators, the
interleukin-6 (IL-6)/IL-6R signaling pathway plays a key role in the development
of HFpEF. It not only acts as the main regulator of acute-phase responses but
also directly promotes the production of C-reactive protein (CRP) in the liver,
amplifying the systemic inflammatory response, and induces cardiac hypertrophy
and fibrosis through the gp130–JAK–STAT pathway [5]. Therefore, it is
considered the key driver of the inflammatory cascade in HFpEF. Tumor necrosis
factor-alpha (TNF-
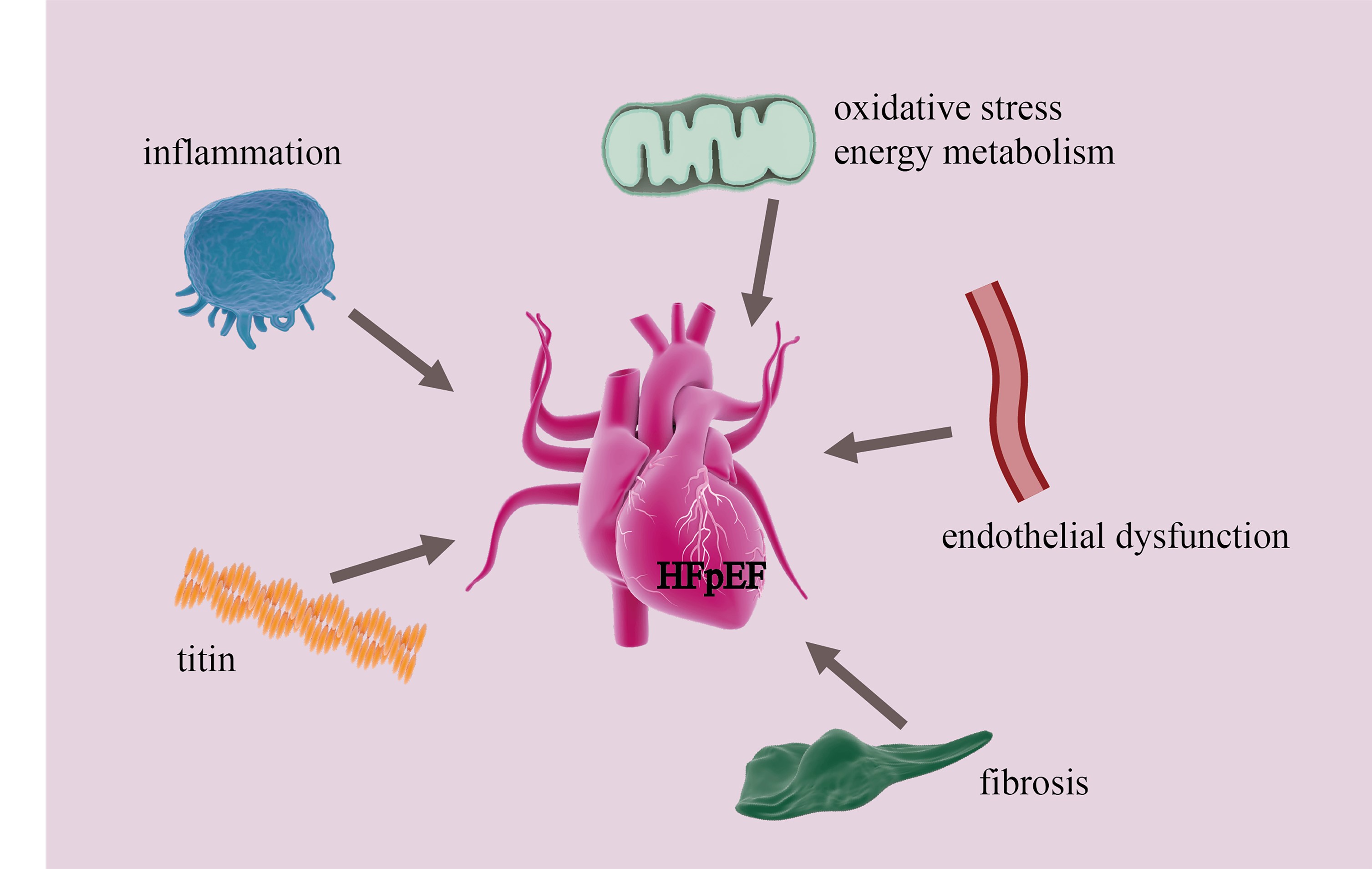 Fig. 1.
Fig. 1.
Major mechanisms underlying cardiac diastolic dysfunction in HFpEF. HFpEF, Heart failure with preserved ejection fraction.
Oxidative stress is a key mechanism underlying myocardial injury in HFpEF, forming a vicious cycle with inflammation (Fig. 1). Superoxide and hydrogen peroxide (H2O2) generated from the activation of NADPH oxidase 1 (NOX1) disrupt calcium (Ca2+) homeostasis through direct post-translational modifications. On the one hand, reactive oxygen species (ROS) oxidize the key thiol group of the ryanodine receptor type 2 (RyR2) protein and activate Ca2+/calmodulin-dependent protein kinase II (CaMKII), resulting in the “hyperopen” state of RyR2 channels. This state leads to Ca2+ leakage and depletion in the sarcoplasmic reticulum during relaxation. On the other hand, ROS directly inhibits sarco/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) pump activity through nitration or glutathione modification, and Ca2+ depletion caused by RyR2 leakage suppresses SERCA2a activity, increasing the consumption of adenosine triphosphate (ATP) and exacerbating Ca2+ recycling disorders [11, 12, 13, 14]. However, the contribution of diastolic Ca2+ dysregulation to injury in HFpEF varies by underlying etiological factors. Ca2+ dysregulation is specifically observed in diabetic HFpEF but not in ischemic or hypertensive HFpEF [15].
Abnormal energy metabolism is common among patients with HFpEF (Fig. 1). Compared with age-matched healthy individuals, elderly patients with HFpEF have a 46% lower skeletal muscle mitochondrial content and a 54% lower expression level of the mitochondrial fusion protein mitofusin 2 (MFN2). These alterations are significantly correlated with reduced peak oxygen consumption (peak VO2) and 6-minute walk distance, indicating that abnormalities in mitochondrial dynamics are a key contributor to exercise intolerance in HFpEF [16]. It is noteworthy that the extent and underlying mechanisms of these abnormalities vary by the phenotype of HFpEF and are strongly associated with specific clusters of comorbidities. Patients with HFpEF with diabetes mellitus can simultaneously exhibit hyperglycemia-induced accumulation of advanced glycation end products (AGEs). These factors aggravate oxidative stress and directly damage mitochondrial DNA and electron transport chain protein complexes through their receptor (RAGE), leading to more severe impairment of ATP production [17]. Therefore, the reduction of MFN2 expression and mitochondrial content is not the result of a single mechanism but the terminal manifestation of different pathogenic pathways (e.g., lipotoxicity, inflammation, glucotoxicity, and oxidative stress). Understanding their association with specific phenotypes is crucial for the development of precise metabolic therapeutic strategies for HFpEF.
Mitochondrial dynamic imbalance driven by inflammation and oxidative stress, especially excessive fission mediated by dynamin-related protein 1 (Drp1), leads to fragmentation of the mitochondrial network. The disordered formation of cristae leads to abnormal assembly of electron transport chain complexes, which significantly suppresses ATP synthesis and causes energy starvation in cardiomyocytes. Furthermore, fragmented mitochondria increase ROS production owing to inefficient electron transport, whereas the accumulation of dysfunctional mitochondrial fragments impairs energy metabolism, forming a vicious cycle [18, 19]. Inhibiting DRP1 or enhancing the expression of mitochondrial fusion proteins (e.g., MFN2) can improve myocardial energy metabolism and diastolic function [20].
Metabolomic analyses of endomyocardial biopsies from patients with HFpEF have revealed significantly decreased levels of glycolytic intermediates, such as glucose-6-phosphate and fructose-1,6-bisphosphate. Exacerbated myocardial oxidative stress is correlated with the activation of inducible nitric oxide synthase (iNOS). iNOS activation inhibits the Akt signaling pathway through S-nitrosylation, leading to insulin resistance and mitochondrial dysfunction. Concurrently, downregulation of mitochondrial pyruvate carrier protein 1 (MPC1) causes pyruvate accumulation, indicating impaired cardiac glucose metabolism in HFpEF [21, 22]. In addition, angiotensin II (Ang II) and norepinephrine reduce glucose oxidation, which is associated with myocardial hypertrophy and diastolic dysfunction. Targeting these pathways is a novel therapeutic approach for HFpEF [23].
The prevalence of endothelial dysfunction is higher in patients with HFpEF than in patients with hypertension and healthy individuals [24]. Inflammatory abnormalities triggered by obesity and diabetes impair vascular endothelial soluble guanylate cyclase (sGC), cyclic guanosine monophosphate (cGMP), and protein kinase G (PKG) signaling. This impairment causes endothelial vascular damage, resulting in CMD that diminishes cardiomyocyte protection [25]. Concurrently, increased NOX activity and uncoupled endothelial nitric oxide synthase (eNOS) elevate myocardial superoxide production, further reducing nitric oxide (NO) bioavailability and impairing endothelium-dependent vasodilation in coronary arterioles [26]. Although the prevalence of CMD is similar between male and female patients with HFpEF, its driving factors differ by sex. For instance, inflammatory CMD phenotypes appear predominantly in men, whereas ventricular remodeling and fibrosis are more common in women [27]. This difference can be attributed to the anti-inflammatory properties of estrogen. In endothelial cells, estrogen activates eNOS by rapid signaling through the Phosphoinositide 3-Kinase/Protein Kinase B (PI3K/Akt) pathway to release NO for vasodilation [28]. Therefore, clinicians should pay attention to inflammation levels in male patients with HFpEF and cardiac structure, especially fibrosis, in female patients, and choose treatment plans according to the observed pathological changes (Fig. 1).
Increased myocardial stiffness is a primary factor leading to diastolic dysfunction (Fig. 1). It is closely associated with extracellular matrix (ECM) fibrosis and abnormalities in the sarcomeric protein titin.
Titin serves as the primary structural determinant of cardiomyocyte stiffness. The two major cardiac titin isoforms are the more flexible N2BA and the stiffer N2B. A negative N2BA/N2B ratio (i.e., increased N2B expression) is consistently observed in both animal models of HFpEF and cardiac biopsies from patients with HFpEF [29]. HFpEF models also exhibit abnormal titin phosphorylation patterns. Hypophosphorylation occurs at I-band phosphoserine residues (Ser3991, Ser4043, and Ser4080) and Ser12884 in the PEVK domain, whereas hyperphosphorylation occurs at Ser12742 in the PEVK domain [30]. Alterations in the expression and phosphorylation patterns of titin isoforms significantly contribute to increased cardiomyocyte stiffness. Therefore, titin regulation is a major research focus. Key kinases modulating cardiomyocyte stiffness include alpha kinase 2, sGC, PKG, and PKA [30, 31, 32]. Genetic inhibition of RBM20 promotes N2BA expression and significantly reduces myocardial passive tension [33]. These findings suggest that titin-targeted gene editing technologies (e.g., CRISPR) and novel biomarkers, such as matrix metalloproteinase-12 (MMP-12)–cleaved titin fragments, should be further investigated [34]. The subtypes and phosphorylation of titin are the main factors determining myocardial stiffness in HFpEF, and the goal of drug therapy is to improve these parameters. Because myocardial biopsy is an invasive procedure, it is not recommended for patients with preserved ejection fraction heart failure who can be diagnosed clearly based on the patient’s condition, common laboratory tests, and imaging examinations. Therapeutic effects should be evaluated not only through biopsy, molecular profiling, and gene editing but also based on laboratory indicators and the symptoms and signs of disease.
In addition to titin, aberrant stabilization of the microtubule network can
contribute to myocardial stiffness. Increased levels of detyrosinated
ECM, deposited between cardiomyocytes, provides structural support (Fig. 1).
Aberrant accumulation of ECM components, particularly elastin and collagen, leads
to myocardial interstitial fibrosis. This fibrosis reduces cardiac tissue
compliance and is a prominent pathological feature observed in endomyocardial
biopsies from 93% of patients with HFpEF [37]. Cardiac fibrosis is closely
associated with neurohumoral regulation abnormalities, inflammation, metabolic
dysregulation, and intracellular molecular pathways. Oxidative stress stimulates
fibroblast activation and pathological ECM remodeling primarily through the
activation of the fibrogenic transforming growth factor-beta (TGF-
Renin–angiotensin system (RAS) inhibitors are important treatment agents for
HFpEF. However, the long-term outcomes of conventional RAS inhibitors in HFpEF
have shown mixed results. For example, in the CHARM-Preserved trial, patients
with chronic heart failure (CHF), New York Heart Association (NYHA) functional
classes II–IV, and left ventricular ejection fraction (LVEF) of
Among RAS inhibitors, the therapeutic effects of angiotensin receptor–neprilysin inhibitors (ARNIs) on hypertension and heart failure have gradually received attention. Sacubitril–valsartan, a representative ARNI, can not only block angiotensin receptors but also inhibit neprilysin. Mechanistic studies have shown that sacubitril–valsartan enhances titin phosphorylation by activating the cgPM–PKG pathway and improves myocardial stiffness in diabetic mice [41]. In addition, it can improve diastolic function by reversing ventricular hypertrophy and reducing the overall strain on the heart in HFpEF [42, 43]. The PARAGON-HF trial showed that sacubitril–valsartan was more effective than valsartan alone in reducing N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels in patients with HFpEF and significantly improved the NYHA class [44, 45]. A meta-analysis indicated that sacubitril–valsartan reduced the incidence of decompensated heart failure and composite decompensated heart failure/all-cause mortality but increased the risk of hypotension [46].
Whether the use of
Jan-Christian Reil et al. [50] found that increased insulin and blood
glucose levels in diabetic mice (db/db) led to increased vascular stiffness,
which was associated with increased heart rate, disturbed ventricular–arterial
coupling, and diastolic dysfunction. At the pathological level, the N2B isoform
of titin was significantly upregulated in the cardiomyocytes of db/db mice, which
was one of the causes of cardiac diastolic dysfunction. When db/db mice were
administered the If channel inhibitor ivabradine, the heart rate decreased, the
ventricular–arterial coupling disorder was corrected, and the expression of N2B
in cardiomyocytes was downregulated, resulting in the recovery of diastolic
function. However, a meta-analysis on the use of ivabradine in human HFpEF failed
to replicate these benefits [51]. This discrepancy in the therapeutic effects of
ivabradine in animal HFpEF models and patients with HFpEF is related to species
differences, comorbidities, and subgroup analysis. Although the mechanisms
underlying cardiac and vascular lesions in diabetic mice overlap with those
underlying HFpEF, they are different from those of human HFpEF. Although db/db
mice showed reduced cardiac diastolic function, they cannot completely mimic the
diastolic dysfunction observed in HFpEF. Therefore, these findings can be used
only as a reference for further clinical research and not as an indication for
the clinical application of ivabradine. In addition, in animal models, the use of
ivabradine is based on a significantly elevated heart rate; however, in clinical
settings, only a few patients with HFpEF have heart rates of
Overactivation of the mineralocorticoid receptor (MR) promotes myocardial
fibrosis, arterial stiffening, and inflammatory responses. Its role in HFpEF has
attracted considerable attention [52]. The FINEARTS-HF trial showed that
finerenone reduced the risk of cardiovascular death and worsening heart failure
in patients with LVEF values of
A post-hoc analysis of the TOPCAT trial indicated that spironolactone can improve clinical outcomes in patients with HFpEF with specific phenotypes, such as those exhibiting increased levels of inflammatory markers or myocardial fibrosis [54]. Kosmala et al. [55] analyzed the effects of spironolactone on exercise capacity in patients with HFpEF and found that 6 months of treatment improved left ventricular untwisting rate and the E/eʹ ratio. Moreover, the improvement in the E/eʹ ratio was independently correlated with increased peak VO2, suggesting that spironolactone functions by suppressing aldosterone-mediated myocardial remodeling. In addition, spironolactone use was associated with a 17% reduction in the hospitalization rate for heart failure [56].
Sodium–glucose cotransporter 2 (SGLT2) inhibitors are established as first-line
drugs for HFpEF. The EMPEROR-Preserved trial showed that empagliflozin
significantly reduced the risk of cardiovascular death or hospitalization for
heart failure in patients with HFpEF [57]. The DELIVER trial indicated that
dapagliflozin reduced the risk of worsening heart failure or cardiovascular
death, with consistent efficacy in patients with LVEF values of
In addition to SGLT2 inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists have shown promise in the treatment of HFpEF. The STEP-HFpEF trial showed that semaglutide significantly improved quality of life in obese patients with HFpEF, evidenced by an increased Kansas City Cardiomyopathy Questionnaire Clinical Summary Score (KCCQ-CSS) and improved 6-minute walk distance [64]. The SUMMIT trial indicated that the dual GIP/GLP-1 receptor agonist tirzepatide improved quality of life and reduced the risk of cardiovascular death or worsening heart failure in patients with HFpEF [65]. In addition, metformin has been shown to improve diastolic function in mice with transverse aortic constriction/deoxycorticosterone acetate (TAC/DOCA)–induced HFpEF by increasing N2B phosphorylation [66].
Angiotensin-converting enzyme 2 (ACE2), a homolog of ACE, is a monocarboxypeptidase that converts Ang II into Ang 1–7. ACE2 and Ang 1–7 negatively regulate RAS at two nodes. JiuChang Zhong et al. [67] infused mice with Ang II to induce HFpEF. They found that elevated Ang II levels induced hypertension, myocardial hypertrophy, fibrosis, and diastolic dysfunction, whereas ACE2 administration restored diastolic function by attenuating the pathological effects of excess Ang II. Furthermore, Ang 1–7 can ameliorate cardiac diastolic dysfunction by improving endothelial function, reducing myocardial fibrosis, and reversing cardiac hypertrophy in db/db mice [68]. These findings indicate that ACE2 and Ang 1–7 are potential treatments for HFpEF. The successful completion of phase I (NCT00886353) and II (NCT01597635) clinical trials of ACE2 has provided key translational evidence for the potential use of race2 as a therapeutic agent.
As mentioned earlier, inflammation is involved in the development of HFpEF. The
D-HART study, inspired by the treatment of rheumatoid arthritis, investigated the
therapeutic value of the IL-1 blocker anakinra in HFpEF [69]. Patients with HFpEF
were randomly divided into anakinra (100 mg) and placebo groups. Results revealed
that anakinra significantly reduced systemic inflammation and improved exercise
capacity in patients with HFpEF with high levels of inflammatory markers. No
major adverse events were observed, except for mild and self-limited injection
site reactions in three patients. These findings suggest that anti-inflammatory
drugs can improve the quality of life of patients with HFpEF with increased
inflammatory marker levels to some extent. However, the sample size of the study
was small, and the follow-up duration was 1 month. Moreover, the long-term
effects of anakinra on HFpEF remain unclear. Therefore, the depth and breadth of
anakinra-related clinical research should be expanded based on these findings.
Miyamoto et al. [72] found that TY1, a synthetic non-coding RNA drug, improved cardiac diastolic function in mice with HFpEF through sustained inhibition of oxidative stress–induced mitogen-activated protein kinase (MAPK) signaling and expression of downstream inflammatory, fibrosis-related, and hypertrophy-related genes in cardiac tissue, with oral and intravenous administration showing comparable effects. Although non-coding RNA therapy is promising, existing studies are limited to animal experiments. Whether it has the same therapeutic effects on human HFpEF remains unclear; therefore, its clinical applicability warrants investigation.
Cardiac systolic and diastolic function are intricately related to energy metabolism. Trimetazidine inhibits long-chain 3-ketoacyl coenzyme A thiolase to shift cardiac energy metabolism from fatty acid oxidation to glucose oxidation, which is more energy efficient and is theoretically favorable for both contraction and relaxation. However, the DoPING-HFpEF study showed that trimetazidine did not improve myocardial energy homeostasis or exercise hemodynamics in patients with HFpEF [73]. Theoretically, phosphodiesterase type 5 inhibitors (PDE5is), such as sildenafil, increase intracellular cGMP concentrations, thereby protecting endothelial function. Animal studies have shown that sildenafil suppresses left ventricular remodeling, hypertrophy, and fibrosis. However, the RELAX trial did not show clinical benefits of phosphodiesterase type 5 (PDE5) inhibition in patients with HFpEF [74]. The mechanisms underlying the development of HFpEF are very complex and not limited to energy metabolism, which may be the reason for the negative results of the DoPING HFpEF and RELAX studies. On the contrary, clinical trials of sGC stimulants have shown more promising results. The DYNAMIC trial showed that the sGC stimulator riociguat improved hemodynamic characteristics but had limited efficacy in alleviating symptoms in patients with HFpEF with pulmonary hypertension [75]. The SOCRATES-PRESERVED trial showed that the sGC stimulator vericiguat improved KCCQ-CSS scores in a dose-dependent manner [76].
Intravenous administration of ferric carboxymaltose (FCM) can reduce the levels of oxidative stress markers (e.g., malondialdehyde) and improve endothelial function [77]. The FAIR-HFpEF trial showed that FCM increased the 6-minute walk distance and reduced the incidence of serious adverse events [78]. A retrospective study showed that FCM improved LVEF and increased right ventricular function normalization rates in patients with HFpEF [79]. Larger-scale trials are warranted to validate the long-term benefits of iron supplementation in HFpEF. Based on the findings of existing studies, FCM should be used in patients with HFpEF after assessing serum iron levels.
In the PIROUETTE trial, oral administration of the antifibrotic drug pirfenidone
significantly reduced myocardial extracellular volume in patients with HFpEF
[80]. In addition to pirfenidone, statins can inhibit fibrosis and inflammation.
For instance, simvastatin can suppress the phosphorylation of Smad2/3 and MAPK
pathways downstream of TGF-
Numerous recent studies have shown that exercise training is a crucial non-pharmacological intervention for improving cardiac diastolic function and exercise capacity in patients with HFpEF [84]. Roeder et al. [85] found significantly reduced left atrial conduit strain in patients with HFpEF, which was strongly correlated with peak VO2. High-intensity exercise training (HIIT) significantly reduces left ventricular myocardial stiffness and increases peak oxygen uptake [86]. Both HIIT (comprising a warm-up of 10 min at moderate intensity, four intervals of 4 min at high intensity, alternating with three intervals, and a 3-min cool-down phase at moderate intensity, totaling 38 min) and moderate-intensity continuous training (moderate-intensity exercise for 47 minutes) can improve the E/eʹ ratio and quality of life, with HIIT leading to greater improvement in VO2 [87]. HIIT rapidly enhances exercise capacity by upregulating the activity of enzymes involved in skeletal muscle energy metabolism and enhancing satellite cell function [88]. Furthermore, low-intensity training improves exercise tolerance in pigs with HFpEF by inhibiting MMP-2 and increasing type III collagen expression, thereby alleviating myocardial fibrosis and enhancing diastolic function [89]. The aforementioned clinical studies on exercise training were performed under the guidance of professional coaches and clinicians. When their condition is stable, each patient with HFpEF should be prescribed a step-by-step exercise program under the guidance of professionals, and this program should be adjusted according to the changes in their condition and the degree of adaptation.
With the advancement of interventional techniques, studies have focused on neuromodulation for treating HFpEF. A study showed that renal denervation (RDN) decreased left ventricular diastolic stiffness, left ventricular filling pressure, and NT-proBNP levels at the 6-month follow-up in patients with HFpEF, indicating a significant effect of RDN on HFpEF [90]. The RDT-PEF trial showed that compared with control individuals, patients with HFpEF undergoing RDN showed greater improvements in peak VO2 and E/eʹ ratio after 3 months [91]. In a study on rats with obesity-induced cardiac dysfunction, early radiofrequency renal denervation (RF-RDN) (at 8 weeks) significantly reduced renal norepinephrine levels, delayed myocardial fibrosis, improved endothelial function, and ameliorated cardiac dysfunction. However, RF-RDN failed to exert beneficial effects when administered to 20-week-old rats with HFpEF [92]. These findings suggest that RDN should be initiated as soon as possible to achieve the best therapeutic effect against HFpEF.
In the REBALANCE-HF trial, splenic artery vasomodulation (SAVM) was performed on 18 patients with HFpEF. At 1 month, SAVM significantly reduced pulmonary capillary wedge pressure (PCWP) during exercise and improved KCCQ-CSS scores [93]. At 12 months, the number of hospitalizations for heart failure, motor function, and health status showed no significant differences between the SAVM and control groups, indicating that SAVM is safe and feasible for the treatment of HFpEF [94].
In another study, thoracoscopic ablation of the right greater splanchnic nerve
was performed on 10 patients with heart failure with ejection fraction (EF)
values of
The REDUCE LAP-HF II trial evaluated the therapeutic efficacy of an atrial shunt
device in patients with HFpEF and HFmrEF. The shunt group showed a 5.65-mL
reduction in left ventricular end-diastolic volume, an increase in right
ventricular volume, and a reversal of ventricular remodeling without the
worsening of right ventricular systolic function within 24 months [98]. These
results suggest that atrial shunt therapy leads to more favorable changes in
cardiac structure/function in patients with HFpEF. Furthermore, pericardiectomy
can relieve pericardial constraint on left ventricular filling. In a porcine
HFpEF model, pericardiectomy decreased the increase in left ventricular
end-diastolic pressure from 13
HFpEF management is primarily based on guideline-directed medical therapy (GDMT). Existing studies have revealed the complex mechanisms underlying HFpEF, particularly the interplay between inflammation, metabolic dysregulation, and cell death pathways, prompting the evaluation of novel treatments such as RNA-based therapies, kinase modulators, and interventional procedures. However, clinical trials remain limited by small sample sizes, high heterogeneity, or suboptimal endpoint designs without achieving translational breakthroughs. In addition, owing to the numerous complications of HFpEF, subgroup analysis of patients with HFpEF should be actively performed, and targeted treatment should be administered based on the results of the subgroup analysis. For instance, subgroup analysis should assess whether antiarrhythmic drugs can offer long-term benefits for patients with abnormally elevated heart rates.
Future studies should prioritize integrating multimodal therapeutic strategies, such as exercise training combined with targeted pharmacotherapy, to enhance synergistic therapeutic efficacy. Combination therapy should be administered after thoroughly examining patients and understanding the underlying etiological factors and complications. For example, SGLT2 inhibitors combined with exercise training can not only control blood glucose levels but also improve the quality of life and reduce the incidence of long-term adverse events in patients with HFpEF with diabetes. However, combination therapy should not be initiated hastily based on theoretical evidence. Further preclinical and clinical studies are warranted to evaluate the clinical applicability, safety, and effectiveness of combination therapy in patients with HFpEF.
The pathogenesis of HFpEF is intricately related to inflammatory activation, oxidative stress, and metabolic dysregulation. In addition to directly damaging vascular and myocardial cells, these mechanisms increase myocardial stiffness and aggravate cardiac interstitial fibrosis. This cascade eventually reduces cardiac compliance and impairs diastolic function.
Although clinical evidence supporting the use of GDMT for HFpEF remains limited, existing management strategies are primarily based on the established treatment regimens for HFrEF. Concurrently, novel therapeutic agents and strategies targeting the underlying pathophysiological mechanisms of HFpEF are being investigated. Although these studies are predominantly at the preclinical stage, they provide substantial support for the development of novel drugs for HFpEF.
XF designed and wrote the manuscript, reviewed the literature, and performed the summarization and organization. JM conducted a literature search and summarization on HFpEF mechanisms. XZ undertook the drafting of Fig. 1. JY contributed the treatment framework for HFpEF, drawing from his clinical expertise. JM was responsible for polishing and XZ was responsible for proofreading the article. JY revised the manuscript and oversaw the organization of the article. All authors contributed to the critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
This study was supported by the National Natural Science Foundation of China (NSFC 81960086). This study was also supported by the Special Fund Project for Doctoral Training of the Lanzhou University Second Hospital (YJS-BD-24) and the International Science and Technology Cooperation Base (PR0124002).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.