



 , Li Liu 2,†, Guang-Gui Zeng 3, Jinrong He 4, Huiqin Liu 1, Dandan Ma 4, Zixin Yang 4, Xiangyan Ma 4, Yunxiang Cao 4, Chunyan Xu 4
, Li Liu 2,†, Guang-Gui Zeng 3, Jinrong He 4, Huiqin Liu 1, Dandan Ma 4, Zixin Yang 4, Xiangyan Ma 4, Yunxiang Cao 4, Chunyan Xu 41 Department of Intensive Care Medicine, Hunan University of Medicine General Hospital, 418000 Huaihua, Hunan, China
2 Department of Nursing, Hunan University of Medicine General Hospital, 418000 Huaihua, Hunan, China
3 School of Medicine, Shanghai Jiao Tong University, 200127 Shanghai, China
4 Department of Cardiology, Hunan University of Medicine General Hospital, 418000 Huaihua, Hunan, China
†These authors contributed equally.
Abstract
Atherosclerosis, a lipid-driven chronic inflammatory disease, is the primary pathological basis of cardiovascular diseases, characterized by endothelial injury, lipid deposition, immune cell infiltration, and chronic inflammation. The NOD-like Receptor Pyrin Domain-Containing 3 (NLRP3) inflammasome has emerged as a crucial mediator of inflammation in atherosclerosis, with caspase recruitment domain family member 8 (CARD8) acting as a key regulatory component. Indeed, CARD8, a member of the caspase recruitment domain family, regulates immune responses by modulating inflammasome activity, particularly NLRP3. Recent studies suggest that CARD8 influences various aspects of atherosclerotic development, including lipid accumulation, oxidative stress, vascular inflammation, smooth muscle cell proliferation, and plaque instability. Thus, this review summarizes the latest findings on the role of CARD8 in the pathogenesis of atherosclerosis, with a focus on the regulatory effects of this component on immune cells and inflammatory pathways. We also discuss the potential of targeting CARD8 as a therapeutic strategy for atherosclerosis, exploring the current preclinical and clinical evidence.
Keywords
- CARD8
- atherosclerosis
- cardiovascular disease
- therapeutic potential
- molecular mechanisms
Atherosclerosis is a complex, multifactorial disease that is characterized by the chronic accumulation of lipids, inflammatory cells, and extracellular matrix within the arterial wall [1]. This disease is a leading cause of cardiovascular morbidity and mortality worldwide, contributing to major conditions such as coronary artery disease (CAD), stroke, and peripheral artery disease [2, 3]. The pathogenesis of atherosclerosis involves a series of processes, including endothelial injury, lipid accumulation, smooth muscle cell migration, and the formation of atherosclerotic plaques [4, 5]. Despite the widespread use of statins and other lipid-lowering therapies, the incidence of cardiovascular events remains high, and many patients still experience recurrent cardiovascular events even after treatment, underscoring the need for more effective and targeted therapies to address the underlying disease processes. Historically, atherosclerosis was primarily considered a disease of lipid accumulation; however, recent research has revealed that immune cells play a pivotal role in the development and progression of the disease [6]. The immune response in atherosclerosis is initiated by the retention of low-density lipoprotein (LDL) particles in the arterial intima, which undergo oxidative modifications [7]. These modified lipoproteins are recognized by immune cells, primarily macrophages, which engulf them to form foam cells [8]. Foam cell formation, in turn, triggers a cascade of inflammatory events that contribute to plaque instability [9, 10, 11, 12]. Furthermore, T lymphocytes and dendritic cells, which infiltrate the atherosclerotic lesions, release pro-inflammatory cytokines and chemokines, thus perpetuating the inflammatory response and contributing to lesion progression [13, 14, 15, 16, 17, 18]. It is now well-established that inflammation within the arterial wall is a central driver of atherosclerosis and that modulation of the immune response may offer potential therapeutic avenues for treatment [19]. Chronic low-grade inflammation in atherosclerosis is not only responsible for plaque formation but also for the destabilization of plaques, leading to acute cardiovascular events such as myocardial infarction and stroke [20]. Therefore, understanding the mechanisms that regulate immune responses within atherosclerotic plaques is critical for the development of novel therapies aimed at controlling inflammation and stabilizing plaques.
Inflammasomes are multi-protein complexes that are formed in response to
cellular stress and injury [21, 22, 23, 24]. These complexes play a crucial role in innate
immunity by sensing pathogens, danger signals, and cellular damage. The NOD-like
Receptor Pyrin Domain-Containing 3 (NLRP3) inflammasome, one of the
best-studied inflammasomes, is composed of the sensor protein NLRP3, the
adapter protein ASC, and the effector protein caspase-1 [25, 26, 27, 28]. Upon activation,
NLRP3 inflammasome triggers the processing and release of
pro-inflammatory cytokines, particularly Interleukin (IL)-1
Caspase Recruitment Domain Family Member 8 (CARD8) is a recently identified member of the caspase recruitment domain (CARD) family of proteins that has been shown to play an important role in the regulation of inflammasome activation [39, 40, 41]. CARD8 is highly expressed in immune cells such as macrophages, dendritic cells, and neutrophils, where it interacts with the NLRP3 inflammasome to modulate its activation [42, 43, 44]. Unlike other inflammasome components that promote inflammasome activation, CARD8 acts as a negative regulator by inhibiting the excessive activation of NLRP3 [45, 46, 47]. CARD8 achieves this through interactions with NLRP3 and caspase-1, preventing the activation of the inflammasome under basal conditions. In addition to its role in inflammasome regulation, CARD8 is also involved in the modulation of other immune pathways, such as apoptosis and the regulation of cytokine production [42, 48]. The dual role of CARD8 in immune regulation has made it an attractive candidate for further investigation in the context of atherosclerosis. Some studies suggest that CARD8 expression is upregulated in macrophages within atherosclerotic lesions, where it helps control the inflammatory response by inhibiting excessive inflammasome activation [49, 50, 51, 52]. However, the precise role of CARD8 in atherosclerosis remains unclear, as some studies have also suggested that CARD8 may have pro-inflammatory effects under certain conditions, particularly in the context of macrophage activation and foam cell formation. Given these conflicting findings, further research is needed to elucidate the full spectrum of CARD8’s function in atherosclerotic disease and its potential as a therapeutic target.
In recent years, the exploration of CARD8 as a therapeutic target in atherosclerosis has gained attention. Targeting CARD8 could potentially modulate inflammasome activation, reduce inflammation, and stabilize atherosclerotic plaques [43, 51, 53, 54]. The development of small molecules or biologics that can selectively target CARD8 expression or its interactions with inflammasome components holds great promise for therapeutic intervention in atherosclerosis. However, the clinical translation of CARD8-based therapies faces several challenges. For instance, the specificity and selectivity of CARD8-targeting agents need to be carefully evaluated to avoid unintended effects on other immune pathways. Additionally, the potential for off-target effects and the long-term safety of such therapies must be addressed before clinical application. Despite these challenges, the promise of CARD8 as a therapeutic target in atherosclerosis underscores the need for further research to refine and optimize strategies that target this protein. This review aims to provide a comprehensive overview of the current understanding of CARD8’s role in atherosclerosis, with a focus on its function in regulating the NLRP3 inflammasome and its impact on the inflammatory processes driving atherosclerotic disease. We will examine the molecular mechanisms by which CARD8 influences immune cell activation and cytokine production, as well as its potential as a therapeutic target in atherosclerosis. Additionally, we will discuss current research on CARD8-targeted therapies and explore the challenges and opportunities for translating these findings into clinical practice.
Web of Science is a globally authoritative retrieval system operated by Clarivate, widely cited and utilized in the academic community. It boasts an extensive subject coverage, encompassing 256 disciplines across diverse fields such as science and technology, social sciences, arts, and humanities, serving as a comprehensive resource library. Its literature resources are remarkably abundant: as of December 2024, the Web of Science Core Collection has included 79 million records, with the total number of records across the entire platform reaching 171 million. Additionally, the system contains citation information for each article.
Web of Science excels in retrieval functionality, featuring a variety of retrieval methods and rules. It also possesses unique citation indexes, in-depth analysis tools, academic influence evaluation functions, and personalized services. These capabilities provide support to researchers in multiple research aspects, such as understanding the development context of research topics, assessing influence, and tracking the progress of research projects.
Our team used the Web of Science Core Collection as the data source, and the configured retrieval formula was “(ALL = CARD8) OR (ALL = Caspase Recruitment Domain Family Member 8)”. Through this retrieval process, a total of 603 pieces of literature were obtained, including 491 research articles, 76 reviews, and 36 pieces of literature of other types. To enhance the accuracy of the content, all literature has been downloaded, and a comprehensive full-text review has been conducted. The flow chart of the screening process is shown in Fig. 1.
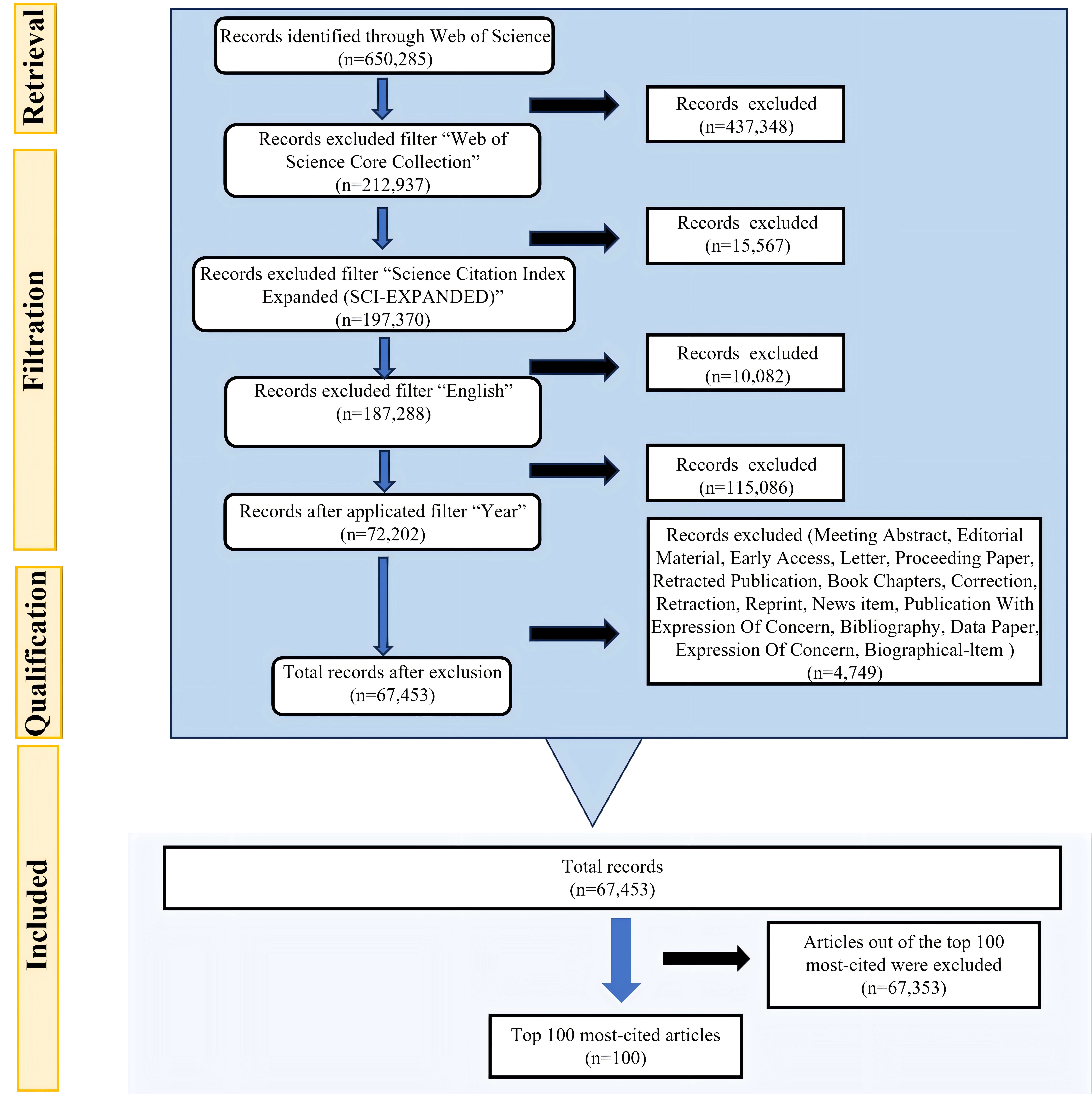 Fig. 1.
Fig. 1.
Flowchart of the search methodology and data source.
Atherosclerosis is a complex and progressive vascular disease characterized by the accumulation of lipids, inflammatory cells, and extracellular matrix components within the arterial wall [55, 56, 57, 58, 59, 60]. The development of atherosclerosis is initiated by endothelial injury, which promotes the retention and oxidation of LDL in the subendothelial space [61, 62, 63, 64]. This injury disrupts the homeostasis of the arterial wall and triggers a series of pathological events, including lipid deposition, smooth muscle cell proliferation, and immune cell infiltration [65, 66, 67, 68, 69, 70]. The retention of oxidized LDL (ox-LDL) in the intima activates endothelial cells, which secrete pro-inflammatory cytokines and adhesion molecules that recruit circulating monocytes to the site of injury [63, 71, 72]. Once monocytes are recruited, they differentiate into macrophages, which engulf ox-LDL and transform into foam cells [73, 74]. These foam cells contribute to the formation of a lipid-rich core within the plaque. As the plaque matures, smooth muscle cells migrate into the intima, forming a fibrous cap that stabilizes the plaque. However, in the presence of sustained inflammation, the plaque can become unstable, leading to rupture and thrombosis, which is a major cause of acute cardiovascular events such as myocardial infarction and stroke [75, 76].
Recent research has elucidated the crucial role of immune responses in the
pathogenesis of atherosclerosis [68, 77]. The inflammatory process is driven by
the activation of innate and adaptive immune cells, which orchestrate the
recruitment and activation of additional inflammatory mediators. Central to the
regulation of inflammation in atherosclerosis are the inflammasomes, multiprotein
complexes that mediate the activation of pro-inflammatory cytokines such as
IL-1
Immune cells play a central role in the development and progression of atherosclerosis [82]. The disease is marked by the infiltration of various immune cell types, including monocytes, macrophages, dendritic cells, T lymphocytes, and B lymphocytes, all of which contribute to the inflammatory microenvironment of the atherosclerotic plaque [83, 84, 85, 86, 87, 88].
Monocytes and macrophages are the most prominent immune cells involved in
atherosclerosis [84, 89]. In response to endothelial damage and lipid
accumulation, monocytes migrate to the plaque and differentiate into macrophages
[90, 91]. These macrophages, in turn, become foam cells by ingesting ox-LDL,
contributing to the formation of the lipid-rich core of the plaque [73, 92]. Foam
cells also release a range of pro-inflammatory cytokines, such as IL-1
The inflammatory process in atherosclerosis is tightly regulated by several
signaling pathways, with inflammasomes playing a pivotal role in modulating the
immune response [105]. Inflammasomes are large, multi-protein complexes that
respond to pathogen- or damage-associated molecular patterns and activate the
pro-inflammatory cytokines IL-1
In addition to the NLRP3 inflammasome, other inflammasomes, such as the Absent in Melanoma 2 (AIM2) and NLR Family CARD Domain-Containing Protein 4 (NLRC4) inflammasomes, are also implicated in atherosclerosis, though their roles are less well understood [109]. Importantly, these inflammasomes can be modulated by various endogenous factors, such as lipids and oxidative stress, both of which are elevated in atherosclerosis. The activation of these inflammasomes within the plaque provides a critical link between lipid metabolism, immune activation, and inflammation in the pathogenesis of atherosclerosis.
The NLRP3 inflammasome is one of the most well-characterized
inflammasomes involved in atherosclerosis, and it is tightly regulated by various
cellular components, including CARD8 [45, 46]. CARD8, a
negative regulator of NLRP3, plays a crucial role in limiting the
overactivation of the inflammasome [110]. By binding to NLRP3,
CARD8 inhibits its activation and prevents excessive production of
IL-1
The balance between CARD8 and NLRP3 activity is critical in controlling the inflammatory environment in atherosclerotic plaques. Increased expression of CARD8 in macrophages and other immune cells within the plaque may serve as a compensatory mechanism to counteract the harmful effects of excessive inflammasome activation [50]. This highlights the potential of CARD8 as a therapeutic target for modulating inflammasome activity and reducing the chronic inflammation that accelerates atherosclerotic progression (Table 1).
| Pathway/Factor | CARD8’s role | Mechanism of action | Impact on atherosclerosis |
| NLRP3 Inflammasome | CARD8 inhibits NLRP3 inflammasome activation, reducing inflammatory responses | CARD8 interacts with NLRP3 protein, regulating its activation state | Reduces immune cell infiltration into the arterial wall, alleviates plaque formation |
| IL-1 |
CARD8 may modulate IL-1 |
Regulates synthesis and secretion of IL-1 |
Elevated IL-1 |
| IL-18 | CARD8 may modulate IL-18 levels, influencing immune responses | CARD8’s action on NLRP3 inflammasome could affect IL-18 secretion | IL-18 levels correlate with worsening atherosclerosis; CARD8 modulation may reduce this effect |
| TNF- |
Indirect regulation of TNF- |
CARD8 modulates NLRP3 inflammasome activation, indirectly affecting TNF- |
Excessive TNF- |
CARD8, Caspase Recruitment Domain Family Member 8; NLRP3, NOD-like Receptor Pyrin Domain-Containing 3; IL, Interleukin; TNF, Tumor Necrosis Factor.
CARD8 is a member of the CARD family of proteins, which includes other well-known inflammasome regulators such as NLRP3, ASC, and CASP1 [113]. The CARD8 gene is located on chromosome 1, and the protein contains a CARD at its N-terminus, which facilitates interactions with other CARD-containing proteins [51]. CARD8 does not possess caspase-like protease activity but plays a crucial role in regulating the activation of inflammasomes, particularly the NLRP3 inflammasome [46].
The protein structure of CARD8 consists of a CARD domain at the N-terminal, followed by a central coiled-coil domain and a C-terminal domain [114]. The CARD domain allows CARD8 to interact with other CARD-containing proteins, such as NLRP3 and ASC, thereby modulating inflammasome activation [115]. The coiled-coil domain is thought to mediate protein-protein interactions, and the C-terminal domain is involved in the regulation of CARD8 stability and function [115]. Importantly, the structure of CARD8 allows it to function as a negative regulator of the NLRP3 inflammasome, which is crucial in controlling the inflammatory response in atherosclerosis and other inflammatory diseases.
CARD8 is primarily involved in modulating the innate immune response,
particularly through its interaction with the NLRP3 inflammasome [116].
The NLRP3 inflammasome is a multi-protein complex that is activated in
response to a variety of danger signals, including pathogen-associated molecular
patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [117, 118].
When activated, NLRP3 triggers the activation of caspase-1, which
subsequently processes pro-IL-1
CARD8 is expressed in a variety of immune cells, including macrophages,
dendritic cells, and T lymphocytes [44, 51, 121]. The expression of
CARD8 is tightly regulated in response to inflammatory stimuli, and its
levels can be upregulated in response to danger signals such as ox-LDL and other
DAMPs [122]. Studies have shown that the expression of CARD8 is
particularly high in macrophages, where it plays a critical role in controlling
the inflammatory response within atherosclerotic plaques [123]. In these cells,
CARD8 interacts with the NLRP3 inflammasome to limit the
activation of IL-1
The regulation of CARD8 expression is also influenced by various
transcription factors, including NF-
CARD8 plays a critical role in modulating the immune response in
atherosclerosis by regulating the activation of the NLRP3 inflammasome
and other inflammatory pathways. In atherosclerotic plaques, the interaction
between CARD8 and NLRP3 helps to suppress excessive
inflammasome activation, thereby reducing the release of IL-1
In macrophages, CARD8 limits the production of pro-inflammatory
cytokines, which helps to stabilize the plaque and prevent plaque rupture [43].
In addition, CARD8 modulates the activation of T lymphocytes,
particularly Tregs, which play a key role in suppressing inflammation and
promoting plaque stability [128]. By influencing both the innate and adaptive
immune responses, CARD8 contributes to the resolution of inflammation in
atherosclerosis and plays a protective role in maintaining plaque stability
[113]. Furthermore, CARD8’s regulatory function extends beyond
inflammasome inhibition [113]. While CARD8 predominantly acts as a
negative regulator of NLRP3 inflammasome activation in atherosclerosis
by binding to NLRP3 and inhibiting its oligomerization with ASC, thereby
limiting IL-1
A study by Paramel Varghese et al. [106] examined CARD8 mRNA expression in atherosclerotic vascular tissue and compared it to transplant donor arterial tissue. They found that CARD8 mRNA was highly expressed in atherosclerotic plaques compared to the expression in transplant donor vessels [51]. Another research used immunohistochemistry to examine CARD8 expression in non-atherosclerotic arteries and carotid lesions. In non-atherosclerotic vessels, CARD8 expression was primarily detected in endothelial cells and smooth muscle cells in the tunica media. In atherosclerotic carotid lesions, CARD8 was detected in the endothelial layer, smooth muscle cells, and CD68+ macrophages, suggesting that immune cells, along with vascular cells, contribute to the increased expression of CARD8 in human atherosclerotic lesions [43]. These studies indicate that CARD8 expression indeed varies between normal and atherosclerotic tissues. However, more research is needed to comprehensively understand its expression differences across diverse atherosclerosis models, such as those induced by different risk factors (e.g., high- fat diet - induced, oxidized LDL - induced).
Atherosclerosis is a chronic inflammatory disease in which immune cell activation, lipid deposition, and extracellular matrix remodeling contribute to the thickening of the arterial walls and the formation of plaques [131]. The inflammatory response plays a pivotal role in all stages of atherosclerosis, from the initiation of endothelial injury to the destabilization and rupture of advanced plaques [131]. As a negative regulator of the NLRP3 inflammasome, CARD8 has emerged as a key player in controlling the balance between inflammation and tissue repair in atherosclerosis [113].
The expression of CARD8 in atherosclerotic lesions has been shown to
influence plaque stability and progression [43]. In animal models, the
overexpression of CARD8 in macrophages leads to the suppression of
IL-1
Experimental studies have provided valuable insights into the role of
CARD8 in atherosclerosis. In ApoE-/- mice, a commonly used
animal model of atherosclerosis, CARD8 expression is upregulated in the
aortic tissues, particularly in macrophages and other immune cells within
atherosclerotic plaques [43]. Studies have shown that the deletion of
CARD8 in these mice leads to increased levels of pro-inflammatory
cytokines such as IL-1
In contrast, the overexpression of CARD8 in macrophages has been shown
to attenuate the inflammatory response in these animal models, leading to a
reduction in plaque size and improved plaque stability [43]. This protective
effect is attributed to CARD8’s ability to inhibit NLRP3
inflammasome activation, preventing the excessive release of IL-1
| Experimental manipulation | Plaque size | Cytokine levels (IL-1 |
Foam cell counts | Other outcomes | Reference |
| CARD8 deletion | Increased plaque area in aortic tissues | Elevated IL-1 |
Increased foam cell formation and macrophage infiltration | Enhanced immune cell recruitment, reduced plaque stability | [133] |
| CARD8 overexpression | Reduced plaque size | Decreased IL-1 |
Decreased foam cell numbers | Improved plaque stability, attenuated inflammatory response | [43, 113] |
| NLRP3 deletion | No significant change in plaque progression | Reduced IL-1 |
No major alteration in foam cells | Independent of NLRP3 in some ApoE-/- contexts, minimal impact on hypercholesterolemia | [134] |
| NLRP3 inhibition | Reduced atherosclerotic lesion area | Suppressed IL-1 |
Decreased foam cell accumulation | Stabilized plaques, reduced calcification | [80] |
The role of CARD8 in immune cells, particularly macrophages, has been extensively studied in the context of atherosclerosis. Macrophages are the primary immune cells involved in the formation and progression of atherosclerotic plaques. Upon recruitment to the site of injury, monocytes differentiate into macrophages, where they play a key role in phagocytosing ox-LDL and forming foam cells [136]. Foam cells contribute to plaque formation and progression by secreting pro-inflammatory cytokines that perpetuate the inflammatory cycle [137].
CARD8 has been shown to regulate the inflammatory response in
macrophages by inhibiting NLRP3 inflammasome activation [113]. In
macrophages from CARD8-deficient mice, there is a marked increase in
IL-1
CARD8 is also expressed in other immune cells, including monocytes and
T lymphocytes, which are involved in the adaptive immune response in
atherosclerosis. In monocytes, CARD8 regulates the activation of the
NLRP3 inflammasome and helps to control the production of IL-1
| Immune cell type | CARD8 expression level | Function of CARD8 in immune cells | Impact on atherosclerosis |
| Macrophages | High | Regulates NLRP3 inflammasome activation, modulates inflammatory response | Promotes progression of atherosclerosis by enhancing local inflammation |
| Monocytes | Moderate | Involved in innate immune responses, induces cytokine release | Contributes to immune cell infiltration in the arterial wall, promoting plaque formation |
| T cells | Low | Modulates T cell activation, influences Th1/Th2 balance | Indirect effect on atherosclerosis via modulation of immune microenvironment |
| Endothelial cells | Very low or none | Not directly expressed, but may mediate through immune cells | Indirect influence on atherosclerosis through immune cell activation |
Beyond its role in immune regulation, CARD8 may also influence lipid deposition within the atherosclerotic plaque. Foam cell formation is a key event in atherosclerosis, and macrophages play a central role in the uptake of ox-LDL, which contributes to lipid accumulation in the plaque [140]. Recent studies suggest that CARD8 may modulate lipid metabolism in macrophages by regulating the expression of genes involved in lipid uptake and efflux [43]. In particular, CARD8 has been shown to inhibit the expression of pro-inflammatory lipid receptors, such as scavenger receptors, which are responsible for the uptake of ox-LDL into macrophages [140]. In addition, CARD8 may influence plaque inflammation through its effects on smooth muscle cells. Smooth muscle cells contribute to the formation of the fibrous cap in advanced plaques, and their proliferation is driven by inflammatory signals [141]. By suppressing the activation of pro-inflammatory pathways in smooth muscle cells, CARD8 may help maintain plaque stability and prevent plaque rupture [141].
Clinical studies investigating the role of CARD8 in human
atherosclerosis have provided further evidence of its involvement in plaque
stability. Immunohistochemical analysis of human atherosclerotic plaques reveals
that CARD8 expression is significantly higher in stable plaques compared
to unstable plaques [43]. Moreover, higher levels of CARD8 in plaque
macrophages are associated with lower levels of IL-1
Despite promising associations with plaque stability, translating CARD8
as a biomarker into clinical practice faces several hurdles, including assay
development for detection in blood versus plaque tissue and establishing
correlations with clinical endpoints like major adverse cardiovascular events
(MACE). CARD8 is primarily an intracellular protein, complicating its
detection in circulation; current methods rely on IHC analysis of plaque tissue
or mRNA quantification via microarray in cohorts like the BiKE study, where
CARD8 expression inversely correlates with IL-1
The therapeutic potential of CARD8 as a target for atherosclerosis treatment is supported by its ability to modulate inflammation and immune cell function within plaques [43]. By enhancing CARD8 expression or activity, it may be possible to reduce the inflammatory burden in atherosclerosis, prevent plaque destabilization, and improve patient outcomes [113]. However, further studies are needed to determine the exact mechanisms by which CARD8 influences plaque biology and to evaluate its potential as a therapeutic target in clinical settings [39].
As a negative regulator of the NLRP3 inflammasome, CARD8 has emerged as a promising therapeutic target for atherosclerosis, a disease primarily driven by inflammation. The idea of modulating CARD8 expression or function to control the inflammatory response in atherosclerotic lesions offers a novel approach to treatment [113]. Given that atherosclerosis is characterized by chronic inflammation, which leads to plaque instability, targeted therapies aimed at restoring the regulatory function of CARD8 could potentially halt or even reverse disease progression [142].
Preclinical studies have highlighted the potential of CARD8 as a
therapeutic target. By inhibiting excessive activation of the NLRP3 inflammasome, CARD8 can reduce the production of IL-1
There are several strategies for targeting CARD8 in the context of atherosclerosis treatment. One promising approach is the use of small molecules that can enhance CARD8 expression or activate its function [137]. Alternatively, the development of monoclonal antibodies that mimic CARD8’s regulatory effects on the NLRP3 inflammasome could provide a more targeted and specific therapeutic intervention [111].
Although direct therapeutic strategies targeting CARD8 are still in the early stages of development, several approaches have been explored in related inflammatory diseases, which could provide insight into potential therapies for atherosclerosis [143].
One potential strategy involves the use of small molecules that modulate the activity of the inflammasome. For example, inhibitors of the NLRP3 inflammasome, such as MCC950, have shown promise in reducing systemic inflammation and preventing disease progression in models of atherosclerosis [144]. These inhibitors could work synergistically with CARD8, either by increasing its activity or by restoring its function in cases of CARD8 deficiency [145]. As the role of CARD8 in inflammasome regulation becomes clearer, drugs that specifically target CARD8’s interaction with NLRP3 could be developed to provide more precise modulation of the immune response [146].
Another potential approach involves gene therapy to enhance the expression of CARD8 in atherosclerotic plaques. Using viral vectors or mRNA-based delivery systems, the therapeutic delivery of CARD8 could help restore its normal function in immune cells, particularly macrophages, where its effect on inflammasome regulation is most significant [147]. Preclinical studies in animal models of atherosclerosis have demonstrated the potential for gene therapy to reduce plaque size and improve plaque stability, with CARD8 playing a central role in this effect [111].
Additionally, monoclonal antibodies or recombinant proteins that mimic
CARD8’s anti-inflammatory effects could offer a more targeted approach
to modulating the inflammasome in atherosclerosis [111]. These therapies would be
designed to activate CARD8’s role in inhibiting NLRP3
activation, thereby reducing IL-1
| Therapeutic strategy | Mechanism of action | Preclinical results | Challenges in preclinical research |
| Small molecule inhibitors | Inhibit CARD8 expression or function, reduce NLRP3 inflammasome activation | Small molecule inhibitors effectively reduce CARD8 expression and slow atherosclerosis progression in animal models | Selectivity, off-target effects, and potential toxicity require further validation |
| Antibody therapy | Antibodies bind to CARD8, inhibiting its function, blocking inflammasome activation | In animal models, anti-CARD8 antibodies reduced atherosclerotic plaque formation | Antibody half-life, stability, and immune tolerance remain major challenges |
| Gene editing | Knock out CARD8 gene, completely abolishing its function | CRISPR-Cas9 effectively deleted the CARD8 gene in animal models, reducing atherosclerosis symptoms | Off-target effects and long-term safety concerns need further evaluation |
| Vaccine therapy | Immunization to activate or suppress CARD8-specific functions | Immunization may activate specific immune responses, though efficacy in atherosclerosis is under further investigation | Immunogenicity and safety of the vaccine need more clinical trials |
A variety of preclinical studies have investigated the role of CARD8 in atherosclerosis and its potential as a therapeutic target [152]. In animal models, the inhibition or deletion of CARD8 exacerbates plaque formation and inflammation, leading to unstable plaques that are prone to rupture [153]. These findings suggest that restoring CARD8 function could stabilize plaques and reduce the incidence of cardiovascular events. Gene knockout studies in ApoE-/- mice have demonstrated that CARD8 deficiency accelerates the development of atherosclerotic lesions and increases the inflammatory cytokine production in the plaques [148]. Restoring CARD8 expression in these animals significantly reduces plaque size and stabilizes the lesions. Additionally, the modulation of CARD8 activity through gene therapy or small molecules has been shown to reduce the inflammatory response, improving plaque stability and reducing the risk of plaque rupture [111].
In vitro studies using macrophage and endothelial cell cultures have
further confirmed the role of CARD8 in regulating the inflammatory
response in atherosclerosis. For instance, activating CARD8 in
macrophages has been shown to inhibit NLRP3 inflammasome activation,
reduce IL-1
While the therapeutic potential of CARD8 in atherosclerosis is promising, several challenges must be addressed before it can be translated into clinical practice [154]. One major challenge is the need for specific and safe targeting of CARD8. Given the complexity of the immune system and the fact that CARD8 is involved in regulating multiple immune responses, any therapeutic intervention must be carefully designed to avoid unintended effects on other aspects of immune function [143].
Another challenge lies in the delivery of CARD8-targeting therapies to the atherosclerotic lesions. Efficient and targeted delivery of therapeutic agents to specific cells, such as macrophages in plaques, remains a significant hurdle in the field of cardiovascular disease treatment [155]. Advances in nanotechnology and targeted drug delivery systems may help overcome these obstacles, but further research is needed to optimize these approaches [156].
Finally, while preclinical studies have demonstrated the potential benefits of CARD8 modulation, clinical trials in humans are required to assess the safety and efficacy of such therapies [153]. The success of clinical trials will depend on the ability to identify suitable biomarkers for CARD8 activity and monitor the effects of treatment on plaque progression and stability [157]. Additionally, the potential for off-target effects and long-term safety must be thoroughly evaluated before CARD8-targeting therapies can be introduced into clinical practice [158].
Long-term safety challenges for CARD8-targeted therapies, which
primarily inhibit NLRP3 inflammasome activation, include unintended
immunosuppression, off-target effects on other inflammasomes such as AIM2 and
NLRC4, and compromised host defense mechanisms, potentially increasing infection
risk in chronic inflammatory conditions like atherosclerosis [159]. Unintended
immunosuppression arises from NLRP3’s role in innate immunity; chronic
inhibition may impair pathogen clearance, as evidenced by increased
susceptibility to bacterial and viral infections in preclinical models treated
with NLRP3 inhibitors like MCC950, where long-term dosing reduced
IL-1
Given the complex pathophysiology of atherosclerosis, combination therapies
targeting CARD8 along with other inflammatory pathways could offer a
more effective treatment strategy [162]. For example, combining CARD8
modulation with existing anti-inflammatory therapies, such as IL-1
Atherosclerosis remains a leading cause of cardiovascular morbidity and mortality worldwide, and despite significant advancements in therapeutic strategies, the disease continues to pose a major public health challenge. Chronic inflammation is a hallmark of atherosclerosis, and targeting key regulatory pathways involved in immune responses holds considerable promise for novel therapeutic interventions. Among the various immune modulators, CARD8 has emerged as a critical player in regulating the NLRP3 inflammasome, a central driver of inflammation in atherosclerosis.
Through its inhibitory effect on NLRP3 activation, CARD8
regulates the production of key pro-inflammatory cytokines, such as IL-1
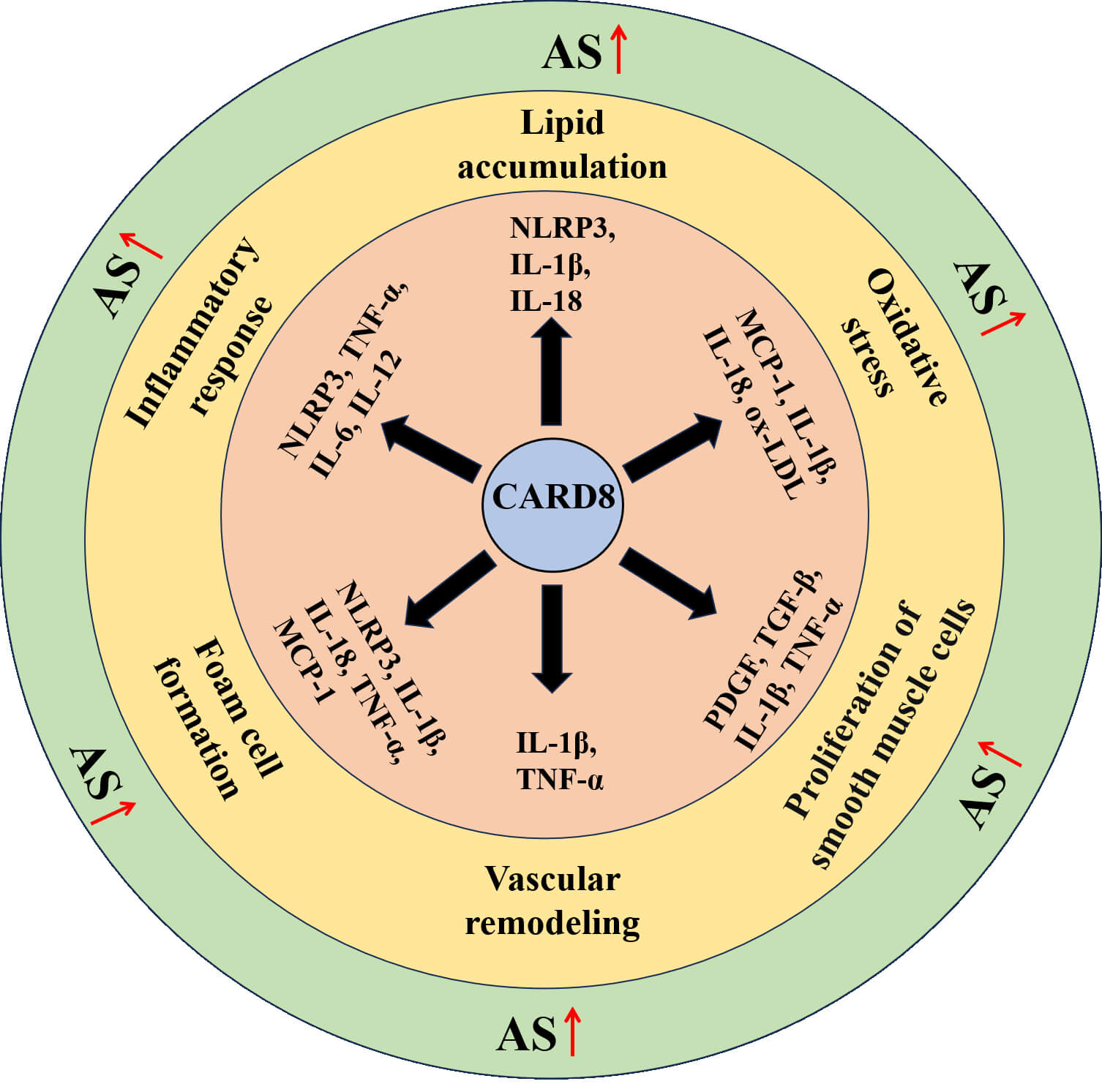 Fig. 2.
Fig. 2.
CARD8-mediated regulation and the major cardiometabolic risk factors of atherosclerosis. Arrows in red: promote; AS, atherosclerosis; MCP-1, monocyte chemoattractant protein-1; ox-LDL, oxidized low-density lipoprotein; PDGF, Platelet-Derived Growth Factor; TGF, Transforming Growth Factor.
Despite the promising preclinical data, several challenges remain in translating CARD8-targeted therapies into clinical practice. A comprehensive understanding of the molecular mechanisms by which CARD8 influences immune cell responses, along with the identification of reliable biomarkers, is essential for the development of targeted therapies. Additionally, overcoming obstacles related to the delivery of CARD8-modulating agents to atherosclerotic lesions and ensuring their safety and efficacy in long-term clinical trials are critical steps toward the successful clinical application of CARD8-targeted therapies.
Looking ahead, future research should focus on addressing these challenges by
expanding our understanding of the role of CARD8 in immune regulation
across different stages of atherosclerosis and in various immune cell
populations. Moreover, the development of combination therapies that target
CARD8 alongside other inflammatory pathways, such as IL-1
Despite the promising role of CARD8 in atherosclerosis and its potential as a therapeutic target, several important research gaps remain. First, the precise molecular mechanisms by which CARD8 regulates the NLRP3 inflammasome and how this influences atherosclerosis progression are not fully understood. While CARD8 has been established as an inhibitor of NLRP3 inflammasome activation, its detailed role in the modulation of other immune signaling pathways, such as those involved in macrophage polarization or endothelial dysfunction, requires further investigation. Understanding the broader context in which CARD8 functions could unveil additional therapeutic opportunities, especially in the context of other cardiovascular diseases that also involve chronic inflammation. Second, the role of CARD8 in different types of immune cells in the context of atherosclerosis needs to be more thoroughly explored. Although CARD8 has been predominantly studied in macrophages, its role in other immune cells, such as dendritic cells, T cells, and smooth muscle cells, is less well understood. These cells contribute to various stages of plaque development and stability, and their response to CARD8 modulation could have significant therapeutic implications. Another major challenge is the heterogeneity of atherosclerosis itself. Atherosclerotic plaques are highly variable, with different stages of progression and varying degrees of inflammation, lipid accumulation, and vascular remodeling. The development of biomarkers to identify patients at different stages of disease, and the role of CARD8 in these stages, would be crucial for determining the optimal timing and strategy for therapeutic intervention. Moreover, the effects of CARD8 modulation on plaque stability, rupture, and cardiovascular events need to be clarified through long-term clinical trials.
Future research should prioritize specific gaps to advance CARD8’s
therapeutic potential in atherosclerosis. First, elucidating CARD8’s
role in early versus advanced atherosclerotic lesions is critical, as its
expression is elevated in stable plaques with thicker fibrous caps but less clear
in early lipid-driven lesions, where NLRP3-driven inflammation may
dominate [43]. Studies in ApoE-/- mice suggest CARD8
upregulation in advanced plaques reduces IL-1
CAD, coronary artery disease; LDL, low-density lipoprotein; CARD8, Caspase Recruitment Domain Family Member 8; CARD, caspase recruitment domain; ox-LDL, oxidized LDL; Tregs, regulatory T cells; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; MCP-1, monocyte chemoattractant protein-1; DC, dendritic cell; IHC, immunohistochemical; BiKE, Biobank of Karolinska Endarterectomies; DAB, 3,3′-diaminobenzidine; MACE, major adverse cardiovascular events.
DDT and LL contributed to the conception and design of the study, data acquisition, data interpretation, and critically reviewed the article. GGZ and JRH contributed to the data analysis, data interpretation, article drafting, and revision. HQL, DDM, ZXY, XYM, YXC, and CYX contributed to study design and interpretation, and critical revision of the article. All authors have read and approved the final version submitted. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
Youth Research Foundation Project of General Hospital of Hunan University of Medicine (project number: QNJJ202501), Health Commission of Hunan Province 2023 National Clinical Key Specialty Major Scientific Research Project (Z2023005), Hunan Provincial People’s Hospital Medical Alliance Special Research Fund Project (LY-2024-27).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.