



 , De-Gang Mo 3, Jing-Xian Bai 1,2, Qian-Feng Han 2, Heng-Chen Yao 1,2,*
, De-Gang Mo 3, Jing-Xian Bai 1,2, Qian-Feng Han 2, Heng-Chen Yao 1,2,*
1 Shandong First Medical University, 250117 Jinan, Shandong, China
2 Department of Cardiology, Liaocheng People's Hospital, 252000 Liaocheng, Shandong, China
3 School of Medicine, Qingdao University, 266000 Qingdao, Shandong, China
Abstract
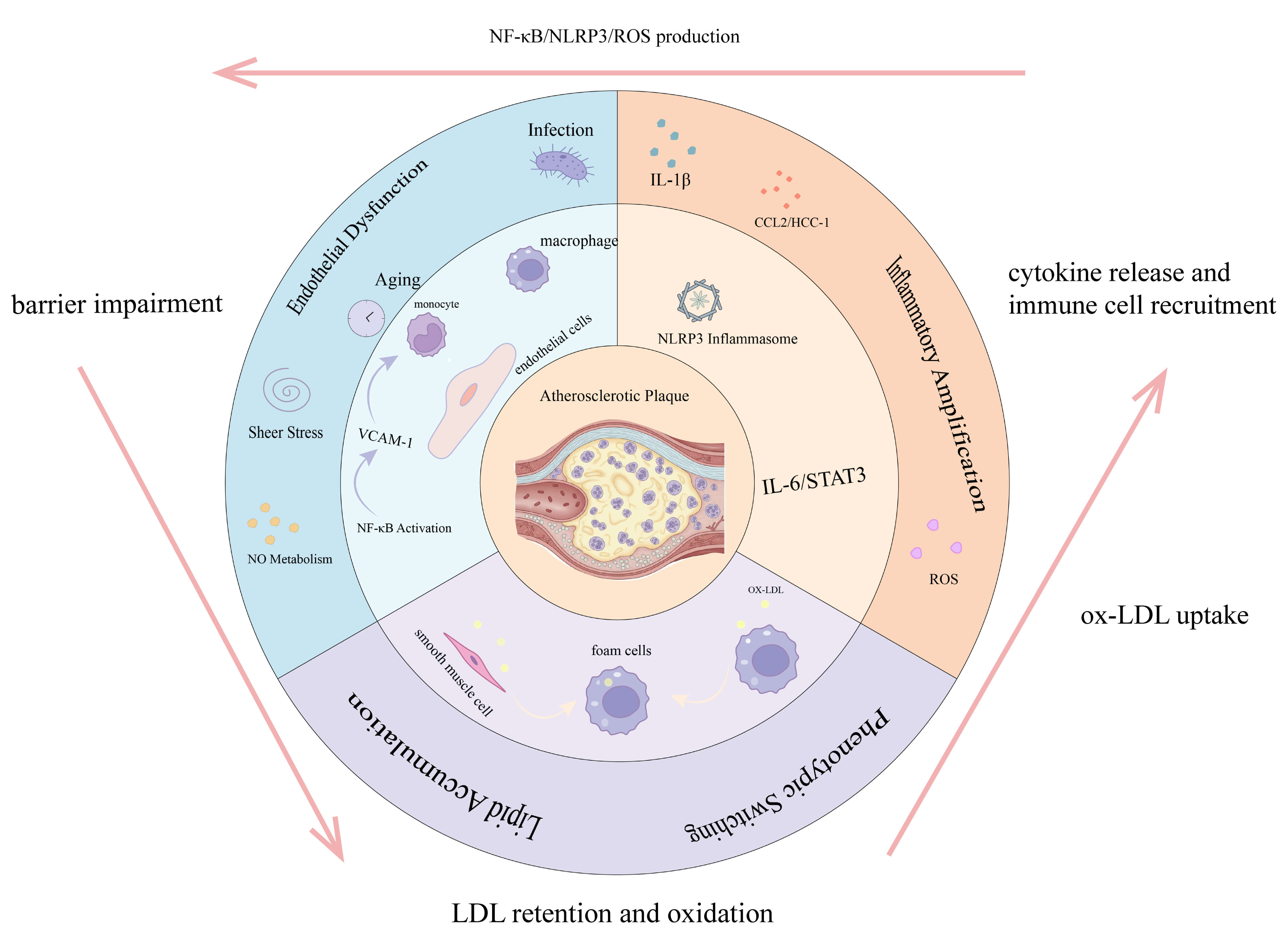
Atherosclerosis, a leading cause of global mortality, is a chronic inflammatory disease driven by a vicious cycle of endothelial dysfunction, dysregulated lipid metabolism, and persistent inflammation. This review examines the mechanisms through which diverse triggers initiate the cycle. We discuss key cellular and molecular events, such as the detrimental phenotypic switching of vascular smooth muscle cells. We also describe the processes through which various upstream signals converge on core inflammatory hubs, such as the Toll-like receptor 4 (TLR4)/nuclear factor-κB (NF-κB) pathway and the nucleotide-binding oligomerization domain, leucine-rich repeat-containing family, pyrin domain-containing-3 (NLRP3) inflammasome. By integrating these established mechanisms with recent findings on novel regulators, including the chemokine hemofiltrate CC chemokine 1 (HCC-1) and cell surface glycoRNA, this review identifies several potential new biomarkers. Overall, this review aimed to provide a comprehensive understanding of the pathogenesis of atherosclerosis, informing future research and the development of targeted interventions.
Graphical Abstract
Keywords
- atherosclerosis
- endothelial dysfunction
- dysregulated lipid metabolism
- inflammation
- biomarker
Cardiovascular diseases (CVDs) are the leading cause of global mortality, with coronary artery disease (CAD) being the most common form. CAD is caused by atherosclerosis and was responsible for 9.44 million deaths and 185 million disability-adjusted life years (DALYs) in 2021, according to the Global Burden of Disease study [1]. DALYs is a composite measure of disease burden that combines years of life lost due to premature mortality (YLL) and years lived with disability (YLD).
Atherosclerosis is fundamentally a chronic inflammatory disease characterized by lipid deposition and plaque formation in the artery wall [2]. Its progression is driven by a vicious cycle involving three core processes. The cycle begins with endothelial dysfunction, which allows lipids such as low-density lipoprotein (LDL) to be retained in the arterial wall. This lipid accumulation then triggers a persistent inflammatory response that, in turn, worsens endothelial function and lipid handling, driving plaque growth and eventual rupture [3].
Understanding the synergy between endothelial dysfunction, lipid metabolism, and inflammation is critical for the development of more effective therapies. This review systematically explores the interplay of these three core mechanisms in the inflammatory regulation of atherosclerosis, integrating emerging research with established pathways. The goal is to provide new perspectives on coronary atherosclerosis and inform the development of novel biomarkers and targeted treatments.
Endothelial cells (ECs) form a continuous monolayer lining the vascular lumen, serving as a biological barrier that precisely regulates the transmembrane transport of nutrients and signaling mediators. Endothelial dysfunction is a core pathological event in atherosclerosis [4] and is characterized by reduced biosynthesis of endothelium-derived nitric oxide (NO) and accumulation of reactive oxygen species (ROS). This leads to impaired vasodilation, activation of a pro-inflammatory phenotype, and an imbalance between procoagulant and anticoagulant activities, significantly increasing the risk of major adverse cardiovascular events (MACE). EC dysfunction can be triggered by various factors that compromise the endothelial barrier through specific signaling pathways, leading to monocyte infiltration [5]. The relevant factors and mechanisms are shown in Fig. 1.
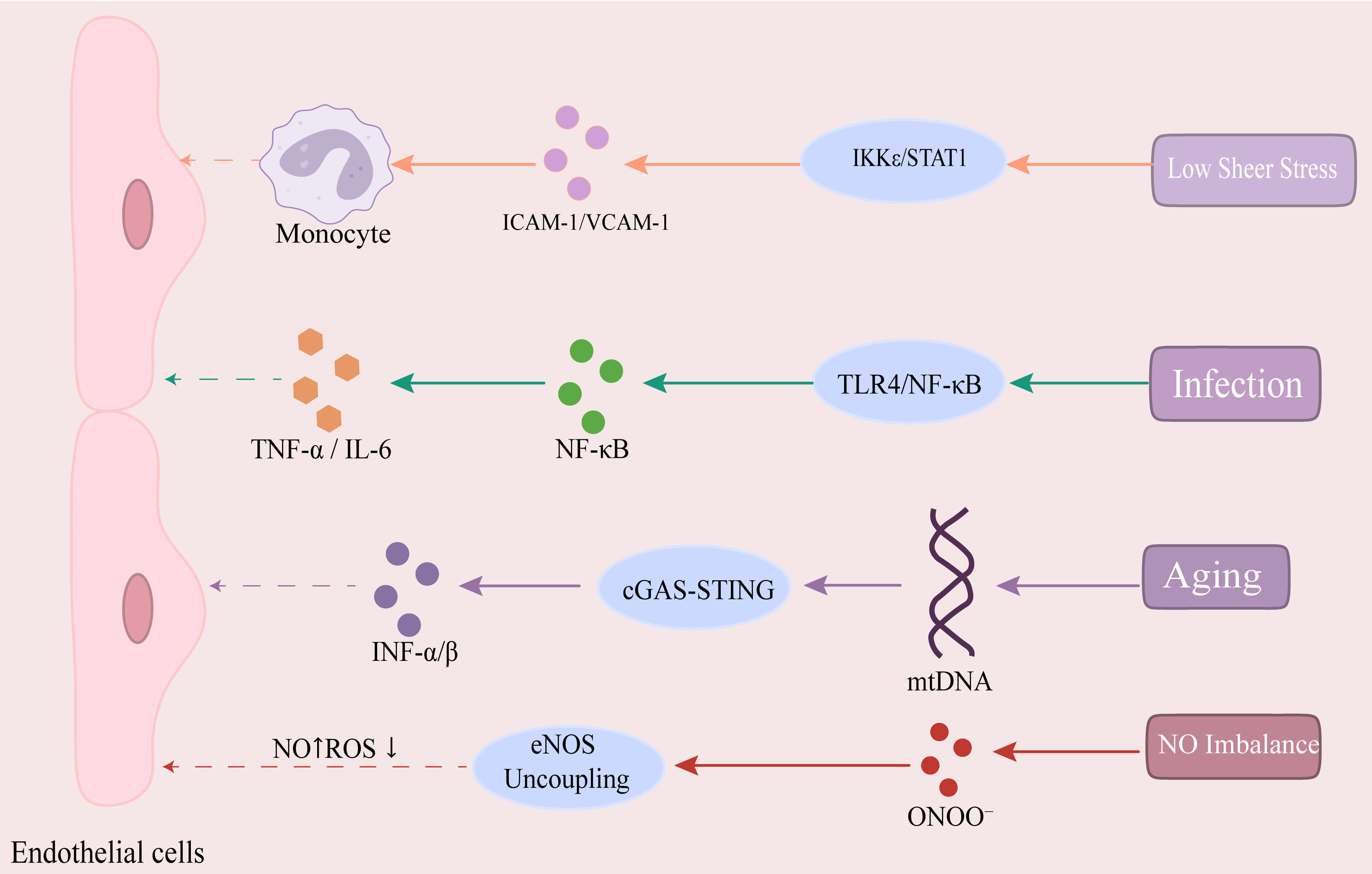 Fig. 1.
Fig. 1.
Key pathways in endothelial cell dysfunction. Low shear stress
(LSS) activates the I
Endothelial activation is a key hallmark of dysfunction, primarily driven by
pro-inflammatory cytokines such as TNF-
Low shear stress (LSS) is recognized as a critical initiator of endothelial
dysfunction. LSS is a pathological state in which the frictional force of blood
flow on the vessel wall is significantly reduced. This condition occurs most
commonly at vascular bends and bifurcations and is an important indicator of
hemodynamic disturbance [7]. LSS directly alters endothelial cell structure and
function and stimulates the migration and proliferation of vascular smooth muscle
cells and monocytes [8]. LSS activates the I
Emerging evidence shows that shear stress can affect endothelial autophagy, with defective autophagy subsequently promoting atherosclerosis. Studies using cellular and animal models have suggested the mechanosensitive channel Piezo1 is activated under LSS and oxidized low-density lipoprotein (ox-LDL) stimulation. This leads to the nuclear translocation of Yes-associated protein (YAP), which in turn inhibits autophagy [13]. Another preclinical investigation has highlighted the potential role of SRY-related High Mobility Group box transcription factor 4 (SOX4), which was observed to be highly expressed in vascular areas with LSS in both human tissues and in mouse models. Supporting this observation, the experimental overexpression of SOX4 in isolated mouse ECs and aortic roots resulted in the loss of endothelial markers. In a separate in vitro model, treatment with the antidiabetic drug metformin was shown to reverse cytokine-induced SOX4 expression in human umbilical vein ECs (HUVECs). Collectively, these preclinical data suggest that SOX4 is a potential regulator, but its definitive role in human disease requires further validation [14].
Beyond its effects on autophagy, disturbed blood flow also promotes
atherosclerosis by inducing the protein CCN1 (also known as cysteine-rich angiogenic inducer 61). In vitro studies have
demonstrated that oscillatory shear stress significantly upregulates CCN1
expression in HUVECs and mouse aortic ECs. This upregulation helps create a
positive feedback loop whereby CCN1 and integrin
Beyond physical stresses like disturbed blood flow, infectious agents are
another major cause of endothelial damage. General viral infections can activate
macrophages to release inflammatory cytokines (e.g., TNF, IL-6), which disrupt
endothelial tight junctions and degrade the vascular basement membrane by
upregulating trypsin and matrix metalloproteinase-9 (MMP-9), leading to increased
vascular permeability and inflammation [17]. Following Influenza A virus (IAV)
infection, elevated levels of IAV mRNA and viral antigens were observed in the
arterial wall and perivascular adipose tissue (PVAT) of pregnant mice. IAV
induced expression of the antiviral mediator IFN-
In vitro cellular experiments have also revealed that Gram-negative bacterial endotoxin (LPS) induces ROS accumulation, which activates the ERK1/2/STAT1 pathway and upregulates inflammatory molecules such as VCAM-1 and various cytokines. At the same time, LPS increases the expression of HMGB1 and the receptor for advanced glycation end-products (RAGE). The subsequent secretion of HMGB1 and its binding to RAGE disrupts EC-cell junctions, thereby promoting atherosclerosis [21]. During the immune response against pathogens, the phenomenon of molecular mimicry may induce cross-reactivity, causing the immune system to mistakenly target structurally similar self-proteins within the vascular wall, thereby triggering chronic autoimmune-mediated vascular injury [22].
These preclinical findings, which link pathogens to vascular damage, are echoed in clinical observations. Data from a multicenter registry indicate that patients with ST-segment elevation myocardial infarction (STEMI) during the first wave of the COVID-19 pandemic experienced longer ischemic times and higher rates of adverse events [23]. However, this clinical data does not establish a direct causal relationship between the worse outcomes and accelerated atherosclerosis in these patients.
By disrupting NO metabolic homeostasis, oxidative stress becomes a core driver
of endothelial dysfunction. Under physiological conditions, ECs catalyze the
conversion of L-arginine to L-citrulline via endothelial nitric oxide synthase
(eNOS), synthesizing NO molecules with vasodilatory and anti-inflammatory
properties. The activity of eNOS is dually regulated by calcium-dependent
phosphorylation (e.g., Akt-mediated modification at Ser1177) and
calcium-independent mechanisms (e.g., binding to heat shock protein 90) [24].
However, under conditions of oxidative stress, eNOS becomes uncoupled, reducing
NO production and increasing ROS [25]. This “NO-ROS imbalance” impairs the
vasodilation capacity and promotes the expression of inflammatory factors via the
NF-
Additionally, oxidative stress inhibits the bioavailability of tetrahydrobiopterin (BH4). BH4 is an essential cofactor for electron transfer in the eNOS catalytic cycle. Under oxidative stress, excess O2- oxidizes BH4 to BH2, which can competitively replace BH4 and weaken its role in eNOS catalysis. This ultimately results in reduced NO synthesis and dysfunction of eNOS. However, in patients with CAD, direct supplementation with BH4 analogs failed to improve endothelial function and was associated with an increase in BH2 levels [26].
Recent studies have identified vascular endothelial protein tyrosine phosphatase (VE-PTP) as a key regulator of endothelial homeostasis. This enzyme negatively modulates eNOS activity by dephosphorylating members of signaling complexes such as the Tie-2 receptor, CD31, VE-cadherin, and vascular endothelial growth factor receptor 2 (VEGFR2, a key receptor tyrosine kinase) [27]. Oxidative stress can upregulate VE-PTP expression. The resulting dephosphorylation inhibits the Tie-2/Akt/eNOS signaling axis, which reduces NO synthesis and causes abnormal vascular tone and endothelial barrier disruption. Targeted inhibition of VE-PTP has been shown to restore eNOS function, improve endothelium-dependent vasodilation, and reduce vascular resistance in animal models of hypertension [28]. These observations suggest that VE-PTP is a potential therapeutic target, although it remains to be determined whether such benefits are directly transferable to the distinct pathological context of atherosclerosis.
The intrinsic process of aging is a primary driver of endothelial dysfunction, as demonstrated in aged mice and in vitro models using HAECs. With advancing age, a study has shown that protective eNOS expression decreases, whereas aging markers (e.g., p53, p21) and components of the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway increase. Notably, inhibiting the cGAS-STING pathway reversed these changes in both animal and cellular models. Mechanistically, this process is driven by the cGAS-STING pathway, which acts as a sensor for age-related damage. During senescence, DNA leaks within the cell are sensed by cGAS, which in turn activates STING. Activated STING subsequently induces type I interferon production, leading to a state of sterile inflammation that further accelerates endothelial senescence [29].
In addition to the cGAS-STING pathway, abnormal activation of RhoA/Rho kinase
(ROCK, a key downstream effector of the small GTP-binding protein RhoA) signaling
is another central mechanism in aging-related endothelial damage. ROCK activation
in the vasculature can disrupt the VE-cadherin/
Further research into the mechanisms of vascular aging has identified the RNA-binding protein Grb10-interacting GYF protein 2 (GIGYF2) as a key regulator. GIGYF2 is overexpressed in senescent human ECs and in the aortas of aged mice. Overexpression of GIGYF2 in young ECs induces senescence, while silencing or knocking out GIGYF2 in aged cells and mice reduces senescence markers and improves vascular function by enhancing NO production. Mechanistically, GIGYF2 stabilizes Staufen double-stranded RNA binding protein 1 (STAU1) mRNA, increasing its protein translation and thereby activating the mechanistic target of rapamycin complex 1 (mTORC1)/ribosomal protein S6 kinase 1 (S6K1) signaling axis. Activated mTORC1 then inhibits autophagy, causing abnormal protein aggregation, and impairs Sirtuin1 (SIRT1)-dependent eNOS function [33]. This cascade accelerates endothelial aging and dysfunction, suggesting that targeting the GIGYF2-STAU1-mTORC1 pathway may be a novel therapeutic strategy for age-related cardiovascular diseases.
In addition to the above triggers, major atherosclerotic risk factors such as
hypertension and hyperglycemia also contribute to endothelial dysfunction.
Hyperglycemia exerts its effects primarily through advanced glycation
end-products (AGEs). The binding of AGEs to their receptor (RAGE) on human
coronary artery ECs (HCAECs) activates the p38 and ERK1/2 signaling pathways,
reduces eNOS expression and induces oxidative stress, ultimately leading to
endothelial dysfunction [34]. Similarly, in mouse models, hyperglycemia has been
shown to promote endothelial dysfunction by inducing the expression of functional
adhesion molecules in the endothelium. Treatment with empagliflozin lowers blood
glucose levels and reduces the expression of P-selectin, E-selectin, and VCAM-1
[35]. As a hemodynamic factor, hypertension shows a positive correlation between
its severity and the extent of endothelial dysfunction. Under hypertensive
conditions, sustained mechanical stress and oxidative stress lead to increased
production of ROS, which exacerbates endothelial injury. Hypertension also
activates the angiotensin II (Ang II) signaling pathway, inducing NF-
The interaction between ECs and immune cells comprises the core network that regulates vascular inflammation. A key regulator within this network is the transcription factor Gata6, which is highly expressed in healthy ECs. This was demonstrated in mouse models, where EC-specific deletion of Gata6 resulted in significantly reduced monocyte infiltration and smaller atherosclerotic lesions. Mechanistically, Gata6 directly controls the target gene Cytidine monophosphate kinase 2 (Cmpk2) that mediates immune cell recruitment. Gata6 deletion lowers Cmpk2 expression, which in turn reduces monocyte adhesion and inflammatory foam cell formation via the Cmpk2-Nlrp3 pathway. Gata6 also directly regulates another target, the chemokine C-C motif chemokine ligand 5 (CCL5), which is similarly involved in monocyte adhesion and migration [37]. These findings suggest that targeting Cmpk2 or CCL5 could be a novel therapeutic avenue for atherosclerosis, though further studies are needed to validate the clinical efficacy and safety of this approach.
Another protein involved in mediating the immune response in atherosclerosis is epithelial-stromal interaction 1 (EPSTI1). The expression of EPSTI1 is significantly upregulated in human atherosclerotic plaques compared to healthy arteries. In vitro experiments have further clarified its role by demonstrating that overexpression of EPSTI1 in HUVECs enhances THP-1 (an immortalized human monocytic cell line derived from an acute monocytic leukemia patient) monocyte adhesion through upregulation of the adhesion molecules VCAM-1 and ICAM-1 [38]. These findings suggest that EPSTI1 contributes to atherosclerosis by promoting monocyte recruitment to the endothelium, and therefore its targeting may represent a novel therapeutic strategy.
As a counterbalance to pro-inflammatory molecules, the endothelium also
expresses protective proteins like endothelial-specific thrombomodulin (TM) that
suppress excessive immune activation. The TM-thrombin complex catalyzes the
conversion of protein C into its activated form (APC), which inhibits
IKK
GlycoRNA is primarily found on the cell surface and plays a critical role in neutrophil recruitment in vivo. Recent studies have identified the presence of cell surface RNA (glycoRNA) on neutrophils, where it plays a critical role in inflammatory responses. The elimination of glycoRNA markedly diminishes the recruitment of neutrophils to inflammatory sites, as well as reducing their adhesion to and transmigration across ECs [40]. Another study showed that glycoRNA-coated neutrophil membrane-coated siMT1-loaded nanoparticles (GlycoRNA-NP-siMT1) can specifically deliver siMT1 to abdominal aortic aneurysm (AAA) lesions. GlycoRNA-NP-siMT1 mitigates pathological remodeling of the abdominal aorta by reducing neutrophil infiltration and inhibiting neutrophil extracellular trap (NET) formation [41], thus offering new possibilities for glycoRNA-targeted therapy. However, research on glycoRNA is still in its early stages, and its role in atherosclerosis has not been fully validated. Additional large-scale studies are needed to confirm its mechanism of action.
The preceding sections have detailed how diverse triggers, including abnormal
hemodynamics, infection, and aging, contribute to endothelial dysfunction. These
factors do not act in isolation but instead form a synergistic network, often
converging on a few core pathological hubs such as the NF-
A defining feature of atherosclerosis is lipid accumulation, which begins when apolipoprotein B (apoB)-containing lipoproteins are retained within the arterial wall. Small lipoprotein particles, such as LDL and VLDL remnants, cross the endothelial barrier and enter the intima. Here, they are trapped by the extracellular matrix, leading to a high concentration of lipids within the vessel wall [42]. Lipid accumulation triggers a local inflammatory response, creating a self-perpetuating vicious cycle that drives the progression of atherosclerosis, as illustrated in Fig. 2 [43].
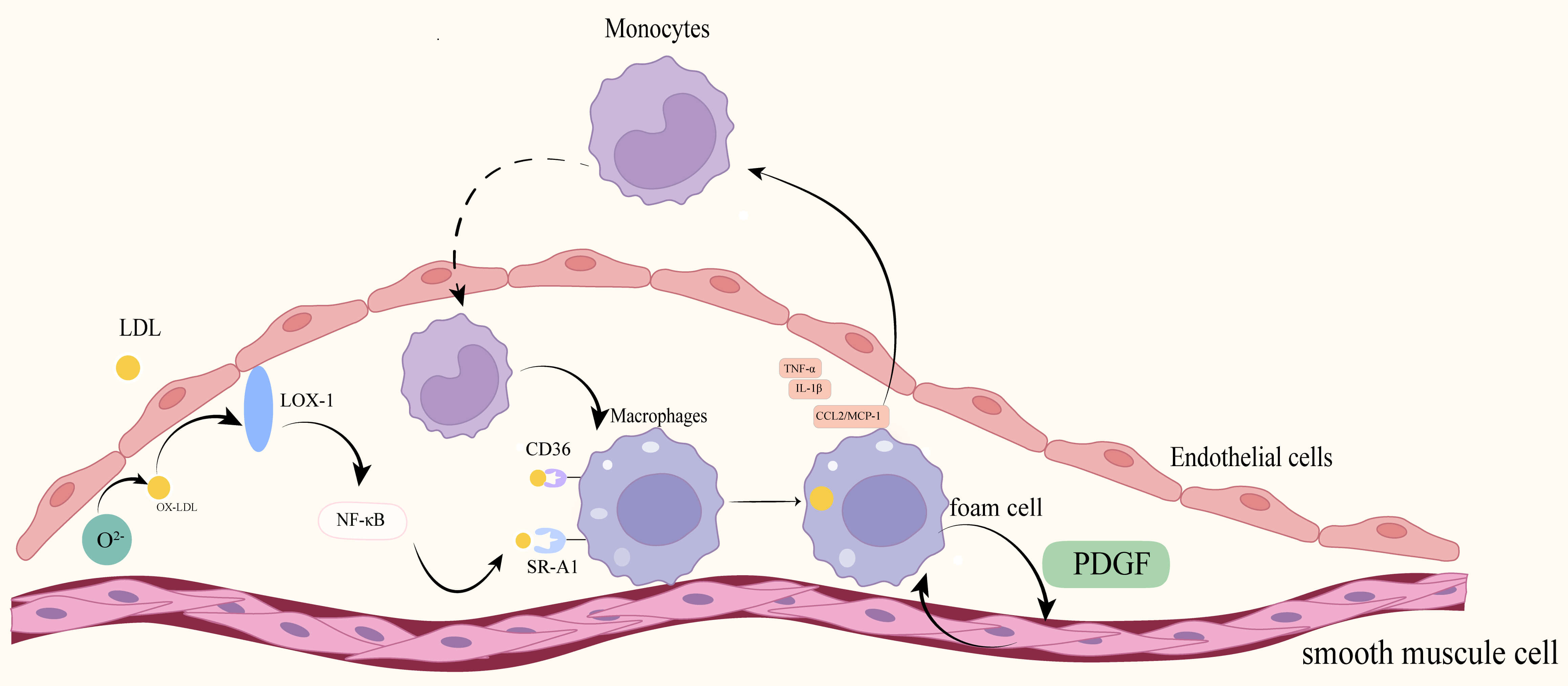 Fig. 2.
Fig. 2.
Lipid accumulation and inflammation. LDL particles are retained
beneath the endothelium and undergo oxidative modification under conditions of
oxidative stress and myeloperoxidase (MPO) catalysis, leading to the formation of
oxidized low-density lipoprotein (ox-LDL). Ox-LDL binds to the lectin-like
oxidized low-density lipoprotein receptor-1 (LOX-1) on endothelial cells (ECs),
activating the NF-
The process of lipid accumulation and inflammation profoundly affects VSMCs, the principal component of the artery’s middle layer (tunica media). While they normally confer structural stability, VSMCs can also undergo a detrimental phenotypic switch in atherosclerosis, transforming them into a pro-inflammatory state and participating in lipid uptake [42]. The significance of this is highlighted by lineage-tracing studies, which show that up to 50% of foam cells in atherosclerotic plaques originate from transdifferentiated VSMCs [44]. This transformation is actively driven by lipids. One key mechanism, identified by in vitro studies using rat VSMCs and confirmed in vivo with mice, is the mtROS/c-Fos/lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) signaling axis. Ox-LDL stimulates mitochondrial ROS (mtROS), which activates the transcription factor c-Fos to upregulate the scavenger receptor LOX-1. This further increases lipid uptake, accelerating the conversion of VSMCs into foam cells [45]. Other pathways, such as cholesterol-induced reprogramming of the miR-143/145-myocardin axis, also contribute to this macrophage-like transformation [46].
The behavior of VSMCs within the plaque is governed by a complex network of counteracting signals. While some pathways promote their harmful transformation, others offer crucial protection. For instance, the serine-threonine kinase Akt1 acts as a key survival signal by suppressing the pro-apoptotic factor forkhead box protein O3a (FoxO3a), thereby protecting VSMCs from cell death and mitigating adverse arterial remodeling [47]. Another key protective factor is the nuclear deacetylase SIRT6, whose expression in VSMCs is markedly reduced in both human and murine atherosclerotic plaques. Mechanistically, SIRT6 prevents VSMC senescence by maintaining telomere integrity, a function that is dependent on its deacetylase activity. The importance of this mechanism was confirmed in a preclinical study on apolipoprotein E (ApoE)-/- mice, wherein VSMC-specific overexpression of functional SIRT6 reduced atherosclerosis, but overexpression of a deacetylase-deficient version worsened features of plaque instability [48].
Similarly, the extracellular matrix protein CCN2 secreted by VSMCs has been identified as another protective regulator. In mouse models, SMC-specific deletion of CCN2 resulted in larger atherosclerotic lesions that showed elevated endoplasmic reticulum stress (ERS) and increased lipid uptake. Single-cell analyses suggest that CCN2 helps to maintain a healthy VSMC phenotype by suppressing an ERS-endocytosis axis that would otherwise promote a harmful macrophage-like transformation [49]. Conversely, defects in protective mechanisms like autophagy can amplify VSMC death and accelerate disease progression.
These findings provide supportive evidence for the role played by VSMCs in atherosclerosis. Understanding the balance between the various detrimental pathways and the impaired protective pathways is crucial for developing therapies aimed at maintaining a healthy VSMC phenotype.
A key cellular process for managing the lipid accumulation that drives VSMC transformation is lipophagy, a specialized form of autophagy that degrades lipid droplets. This process involves enclosing lipid droplets in autophagosomes, which then fuse with lysosomes where enzymes break down the lipids. The resulting free cholesterol can then be used by the cell, or removed via the ATP-binding cassette transporter G1 (ABCG1) transporter [50, 51]. Lysosomal pH imbalance or diminished cathepsin activity can obstruct autophagic flux, leading to abnormal lipid droplet accumulation. Properly functioning lipophagy is critical for lipid homeostasis, and its impairment is a shared pathogenic mechanism in metabolic diseases like atherosclerosis [52]. Recent preclinical research has identified a specific regulator of this process in VSMCs, the P2RY12 receptor, which acts as a significant suppressor of autophagy. Mechanistic studies have shown that P2RY12 inhibits key steps in the autophagic machinery, including maturation of MAP1LC3/LC3 (Microtubule-Associated Protein 1 Light Chain 3), a key protein marker of autophagy. The importance of this mechanism was confirmed in ApoE-/- mouse models, where pharmacological blocking of the P2RY12 receptor enhanced VSMC autophagy and consequently reduced the progression of atherosclerosis [53].
Acid-sensing ion channel 1 (ASIC1) influences lipophagy by impeding cholesterol efflux in macrophages [54]. In vitro experiments have shown that ASIC1 activation promotes phosphorylation of the signaling protein receptor-interacting protein 1 (RIP1, also known as RIPK1) and the master autophagy regulator, transcription factor EB (TFEB). In cellular models, phosphorylation of TFEB hindered its nuclear translocation and suppressed the expression of essential lysosomal genes, ultimately disrupting the lipophagy process and causing lipid accumulation [55]. Based on these findings, the targeting of regulatory pathways such as the P2RY12 axis in VSMCs and the ASIC1-TFEB axis in macrophages has been proposed as a novel therapeutic strategy. However, significant further research is required to validate these specific mechanisms in the human context and to assess the long-term safety and efficacy of such targeted interventions.
Building on the concept of cellular lipid clearance, reverse cholesterol transport (RCT) is a critical systemic process in which macrophages remove cholesterol from the arterial wall using a pathway that is heavily dependent on the transporter ABCA1 [56]. The regulation of RCT is complex and involves various factors, including non-coding RNAs. In mouse models, the long non-coding RNA (lncRNA) AI662270 was found to inhibit cholesterol efflux by directly attenuating the expression and activity of ABCA1 [57]. Conversely, lncRNA PCA3 was found to be downregulated in cellular studies of ox-LDL-induced foam cells, whereas miR-140-5p was highly expressed. A possible mechanism is that lncRNA PCA3 increases expression of the transcription factor regulatory factor X7 (RFX7, a transcription factor) and ABCA1 by competitively binding miR-140-5p, thus promoting cholesterol efflux [58].
Several other molecules have also been shown to regulate the RCT pathway. The gut microbiota metabolite indole-3-propionic acid (IPA) promotes RCT in preclinical models, reportedly through the miR-142-5p/ABCA1 pathway [59]. Similarly, the adipokine Asprosin was shown to increase cholesterol efflux in cellular and mouse models by activating the p38/Elk-1 signaling cascade to boost the transcription of ABCA1 and ABCG1 [60].
Conversely, other factors can impair RCT. One such molecule is Tumor necrosis
factor-alpha-induced protein 1 (TNFAIP1), initially identified as being induced
by TNF-
Adding to the complexity of molecular regulation in atherosclerosis, the lipid-sensing receptor triggering receptor expressed on myeloid cells 2 (TREM2) found on macrophages appears to have multifaceted and sometimes contradictory roles [65]. On the one hand, some research has indicated a detrimental function. For example, Guo et al. [66] reported that overexpression of TREM2 in macrophages upregulated the scavenger receptor cluster of differentiation 36 (CD36), which then increased lipid uptake and the formation of foam cells. On the other hand, TREM2-deficient macrophages have lower survival and impaired phagocytosis under lipid-loading conditions, thereby exacerbating necrotic core formation. Conversely, TREM2 activation protects against atherosclerosis by limiting necrotic core formation [66]. This apparent discrepancy demonstrates the highly context-dependent function of TREM2, which likely varies with the disease stage or the plaque micro-environment. Therefore, future therapeutic strategies targeting TREM2 must be highly nuanced and involve stage- or cell-specific modulation rather than uniform activation or inhibition.
Sirtuin 6 (Sirt6) is a histone deacetylase that enhances plaque stability by
promoting macrophage autophagy and lipophagy [67]. It achieves this partly by
inhibiting Wnt1/
CD36 is a scavenger receptor regulated by palmitoylation. It plays a key role in atherosclerosis by acting as a receptor for pro-atherosclerotic, oxidized high-density lipoprotein (ox-HDL) [71, 72, 73]. HDL is a cholesterol carrier that mediates RCT [74]. Preclinical studies, primarily in vitro, have shown that ox-HDL can catalyze the palmitoylation of CD36, causing it to cluster in lipid raft microdomains. This single modification is thought to initiate a vicious cycle in which raft-localized palmitoylated CD36 not only increases the uptake of ox-HDL, but also simultaneously triggers pro-inflammatory signaling via the c-Jun N-terminal kinase (JNK) cascade and impairs lipid droplet clearance by inhibiting autophagy [75]. Furthermore, co-immunoprecipitation experiments suggest that palmitoylated CD36 forms a complex with the innate immune receptor TLR4. Inhibition of this CD36-TLR4 interaction was observed to reduce lipid accumulation [76].
In essence, these findings indicate that CD36 palmitoylation is a critical molecular switch that can transform the CD36 receptor into a potent driver of both lipid accumulation and inflammation.
The intracellular breakdown of lipids is carried out by key enzymes such as
adipose triglyceride lipase (ATGL). This enzyme binds to the surface of lipid
droplets (LDs) and catalyzes the breakdown of triglyceride (TG) to release free
fatty acids (FAs) [77]. In atherosclerosis models, endothelial deficiency of ATGL
has been linked to vascular lipid accumulation and dysfunction. The proposed
mechanism involves the induction of ERS by TG accumulation, which in turn
promotes inflammation via the NF-
The atherosclerotic environment also disrupts the function of the adhesome, a
protein complex that regulates cell adhesion, with Kindlin3 (K3) being a vital
component for macrophage function. In vitro studies with macrophage
cultures show that ox-LDL reduces K3 levels. This weakens the K3-integrin
Beyond lipid accumulation, specific molecular axes that tightly couple lipid metabolism with inflammatory signaling are now understood to be critical drivers of atherosclerosis. These regulatory hubs operate at multiple cellular levels.
Proprotein convertase subtilisin/kexin type 9 (PCSK9) negatively modulates the
low-density lipoprotein receptor (LDLR) via lysosomal degradation, while also
directly promoting macrophage inflammatory responses [82]. Moreover, PCSK9 was
found to increase TLR4/NF-
Another key node that acts within the cytoplasm is the ADP-ribosylation factor-like protein 11 (ARL11)/Janus kinase 2 (JAK2)/STAT1 axis. ARL11 is an ADP-ribosylation factor-like GTPase that activates JAK2 to promote STAT1 phosphorylation. It is highly expressed in atherosclerotic plaques [85], and its silencing was observed to reduce both lipid deposition and plaque area in atherosclerosis models. The ARL11 signaling cascade has also been shown to drive pro-inflammatory M1 macrophage polarization [86].
The protein perilipin 1 (PLIN1) is a crucial protective gatekeeper at the lipid droplet surface. Dysfunction of this lipid droplet-coating protein can lead to ectopic lipid deposition and metabolic inflammation. Evidence from clinical genomics studies has shown that deficiency of PLIN1 promotes inflammatory cytokine secretion, while its overexpression in cellular models inhibits ox-LDL uptake and increases cholesterol efflux [87]. Underscoring its clinical relevance, human genome-wide association studies (GWAS) have linked loss-of-function PLIN1 mutations to increased coronary artery calcification scores [88, 89].
In summary, we have described how lipid accumulation in atherosclerosis is far from a passive process. It triggers a cascade of cellular dysfunctions, most notably the pro-inflammatory phenotypic switching of VSMCs. The maintenance of cellular lipid homeostasis depends on a delicate balance between lipid uptake, lipophagy, and cholesterol efflux through RCT. These processes are tightly controlled by a complex network of molecular regulators that include non-coding RNAs, signaling kinases, cell surface receptors, and lipid droplet-associated proteins. Disruption of these regulatory axes provides a direct mechanistic link between disordered lipid metabolism and the amplification of inflammatory responses.
The inflammatory response is now recognized as the central driving force throughout the entire progression of atherosclerosis [90]. Infiltrating plasma lipoproteins undergo modifications that activate resident inflammatory cells, which then release inflammatory signals that recruit more circulating leukocytes to the site, further amplifying the response and establishing a “lipid infiltration-inflammation activation-cell recruitment” positive-feedback loop [91]. This chronic inflammation involves intricate cross-talk between the innate and adaptive immune systems, ultimately driving plaque progression and leading to clinical cardiovascular events.
The inflammatory response in atherosclerosis is orchestrated by several core
signaling hubs. This review will focus on key nodes in the inflammatory signaling
pathways that function as master regulators of the downstream inflammatory
cascade, including the NLRP3 inflammasome and major cytokines such as
IL-1
NLRP3-IL-1
IL-6 Signaling Network: In atherosclerosis, the IL-6/signal transducer and activator of transcription 3 (STAT3) axis promotes the expression of endothelial adhesion molecules, increases monocyte adhesion and foam cell formation, and induces the proliferation of smooth muscle cells and MMP expression, thereby destabilizing plaques. Proteomic analyses have identified CXCL10 as a downstream mediator of IL-6 that recruits CD8+T cells and pro-inflammatory CD68+CD163– macrophages to the plaque shoulder, leading to increased plaque vulnerability [94].
Mechanisms involving the NLRP3 inflammasome and IL-6/STAT3 pathway in atherosclerosis (Fig. 3).
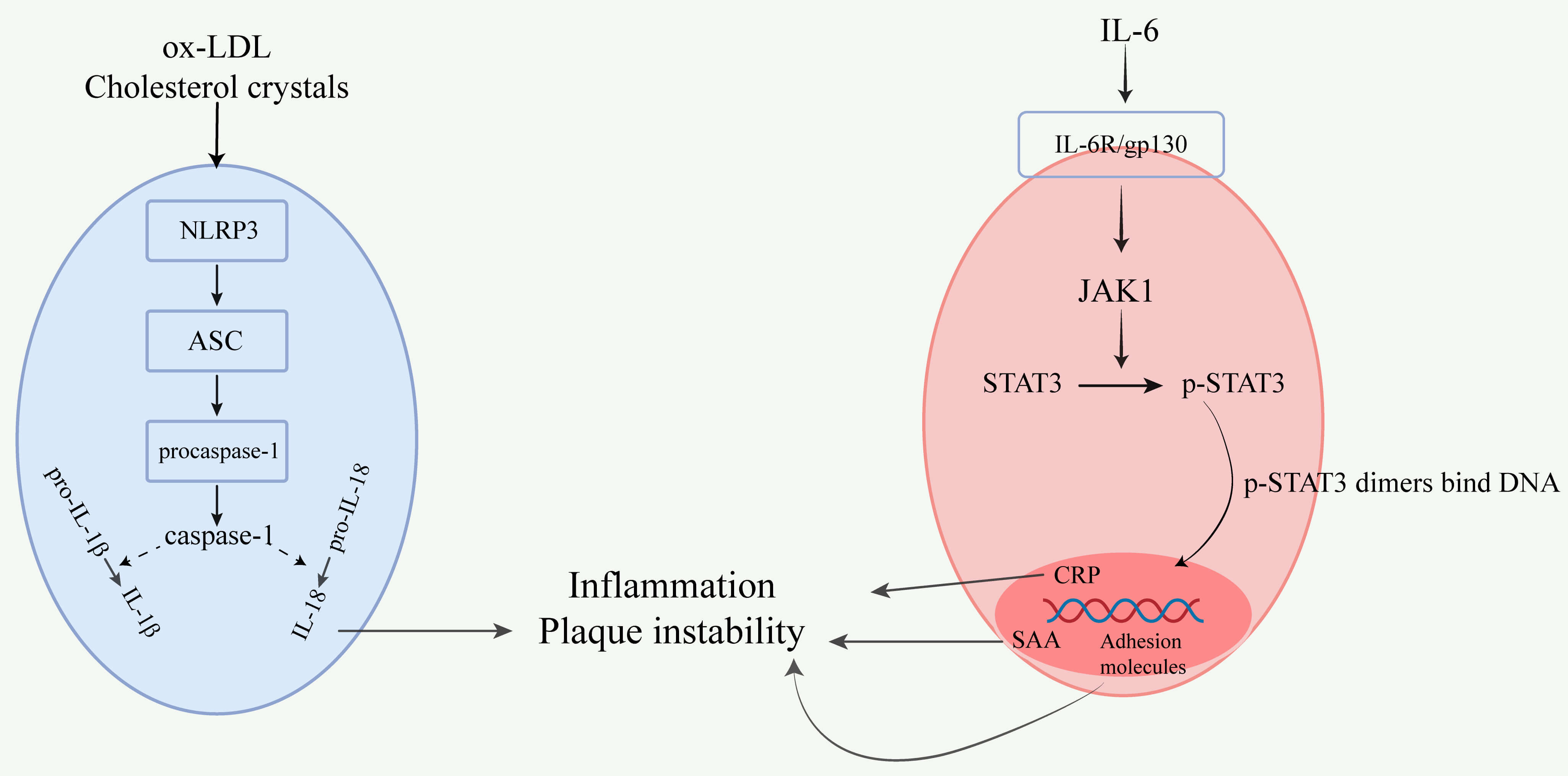 Fig. 3.
Fig. 3.
Mechanisms involving the NLRP3 inflammasome and IL-6/STAT3
pathway in atherosclerosis. In atherosclerosis, the NLRP3 inflammasome is
activated by ox-LDL and cholesterol crystals, leading to the assembly of NLRP3
with ASC (apoptosis-associated speck-like protein) and procaspase-1 into an
inflammasome complex. This process activates caspase-1, which cleaves
pro-IL-1
IL-1
This section will focus on the spatial dynamics of inflammation by exploring how chemokines and their receptors orchestrate the precise recruitment and positioning of immune cells within the local microenvironment of the atherosclerotic plaque.
CCL2-CCR2 Axis: Endothelial denudation and pro-thrombotic components of
atherosclerotic plaques can trigger chemokine release [97]. Preclinical studies
have shown that C-C chemokine receptor type 2 (CCR2) antagonists reduce plaque
IL-1
CCL5/RANTES: CCL5/RANTES is a chemokine that mediates the chemotaxis and
activation of T cells, monocytes, mast cells, and dendritic cells. It interacts
with chemokine receptors (CCR1, CCR3, CCR4, and CCR5) and plays a key role in the
inflammatory process [99]. In an accelerated atherosclerosis model, CCR5
antagonism promoted the formation of stable plaques with fewer macrophages [100].
Treatment with HT-C6, a synthetic derivative of the natural olive antioxidant
hydroxytyrosol, has been shown in cellular models to inhibit NF-
Pathological Effects of HCC-1 (CCL14): Hemofiltrate chemokine 1 (HCC-1), also
known as CCL14. Clinical and bioinformatic analyses have shown that
HCC-1 expression is elevated in the serum and atherosclerotic plaques of
patients. Moreover, it is positively correlated with disease occurrence and the
switch from stable to unstable plaques. In ApoE-/- mice, the
overexpression of HCC-1 increased inflammatory factors, macrophage accumulation,
and pyroptosis within plaques, thereby decreasing their stability. Complementary
cellular studies demonstrated that HCC-1 directly promotes pro-atherogenic
processes such as monocyte adhesion and pro-inflammatory M1 macrophage
polarization. These effects are reportedly mediated through the
NF-
The growing understanding of the central role played by inflammation in
atherosclerosis has spurred the development of targeted, anti-inflammatory
therapies. The CANTOS trial, for instance, provided pivotal proof-of-concept by
demonstrating that targeting of the IL-1
Table 1 (Ref. [97, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119]) provides a comprehensive summary of these and other key inflammatory mediators, detailing their specific mechanisms, therapeutic agents that target them, and the current status of clinical or preclinical evidence for intervention.
| Mediator | Core mechanism | Clinical/Preclinical intervention status |
| NLRP3 | Ox-LDL and cholesterol crystals activate the NLRP3 inflammasome, leading to caspase-1 activation and the release of IL-1 |
MCC950 (a small-molecule NLRP3 inhibitor) significantly reduces plaque burden in atherosclerotic mouse models [107]. |
| IL-1 |
Activation of the NLRP3 inflammasome triggers IL-1 |
CANTOS trial: Canakinumab (anti-IL-1 |
| IL-6 | IL-6R/gp130 activates the JAK1/STAT3 pathway, inducing acute-phase proteins (CRP, SAA) and adhesion molecules, thereby exacerbating endothelial inflammation and macrophage polarization [105]. | Tocilizumab (anti-IL-6R) has shown improvement of vascular function in a small-scale trial of acute myocardial infarction [109]. |
| TNF- |
TNF- |
Anti-TNF- |
| IL-17A | Th17 cells secrete IL-17A, inducing ECs and macrophages to produce IL-6, TNF- |
Secukinumab (anti-IL-17A) shows neutral effects on vascular inflammation and cardiometabolic biomarkers in psoriasis patients [111]. ASCVD studies are ongoing. |
| IL-23 | IL-23 drives Th17 cells to secrete IL-17A and promotes inflammatory activation of macrophages and expression of matrix-degrading enzyme MMP-9 via the STAT3/retinoic acid-related orphan receptor gamma t (ROR |
Anti-IL-23 monoclonal antibodies (e.g., tildrakizumab) have demonstrated safety in autoimmune disease patients [112]. Investigation of their potential for cardiovascular protection is being evaluated. |
| CCL2-CCR2 | CCL2 binds to CCR2 to mobilize Ly6Chigh monocytes from the bone marrow and guide their infiltration into plaques, increasing pro-inflammatory gene expression in macrophages [97]. | Inhibiting the CCL2/CCR2 axis may stabilize atherosclerotic plaques and reduce complications such as acute coronary syndromes [113]. |
| CCL5-CCR5 | CCL5 recruits CCR5+ monocytes and T cells into plaques, promotes M1 polarization of macrophages, and induces MMP-2/9 expression, leading to matrix degradation. | Maraviroc (CCR5 antagonist) reduces plaque size and inflammation in the ritonavir-induced atherosclerosis model [114]. It also inhibits NADPH oxidase 1 (Nox1) expression to reduce vascular inflammation [115]. |
| CXCL8 (IL-8) | IL-8 is secreted by ECs and macrophages and promotes the chemotaxis of neutrophils and monocytes via C-X-C chemokine receptor (CXCR) 1/2, enhancing ROS production and expression of adhesion molecules. | Reparixin (CXCL8 receptor antagonist) reduces endothelial damage in an in vitro model of ischemia-reperfusion [116]. |
| CXCL10-CXCR3 | CXCL10 recruits CXCR3+ Th1 cells and natural killer cells (NK) to the lesion site, where they secrete IFN- |
CXCR3 antagonists block the direct migration of CXCR3+ effector cells into plaques, modulate inflammatory responses, and reduce arterial inflammation [117]. |
| CXCL12-CXCR4 | CXCL12/CXCR4 activates glycogen synthase kinase 3 beta (GSK3 |
CXCR4 inhibitor AMD3100 blocks abdominal aortic aneurysm expansion and rupture in mice, and reduces inflammation and immune cell accumulation [118]. |
| CX3CL1-CX3CR1 | CX3CL1 (fractalkine) is expressed by activated ECs and macrophages, mediating adhesion and chemotaxis to the lesion site via CX3CR1 on CD14+CD16– monocytes. Overactivation of this axis is associated with increased susceptibility to coronary atherosclerosis. | CX3CR1 antagonist KAND567 (an antagonist of the human CX3CR1 receptor) blocks CX3CL1 signaling, reducing post-myocardial infarction, immune cell recruitment and inflammation, and decreasing infarct size [119]. |
| CCL20-CCR6 | CCL20 is highly expressed in atherosclerotic plaques, binding to CCR6 to promote the migration of Th17 cells and CCR6+ monocytes to the lesion site, and facilitating matrix degradation and the spread of inflammation through MMP-2/9. | CCR6 antagonists remain in the preclinical research stage. |
ABCA1, ATP-binding cassette transporter A1.
Collectively, the various cytokines and chemokines discussed above form a
dynamic and complex inflammatory interactome that plays a central role in the
microenvironment of the atherosclerotic plaque. A clear regulatory hierarchy
exists within this network. For instance, upstream signaling hubs centered on the
NLRP3 inflammasome can, upon activation, drive maturation and release of the key
downstream effectors IL-1
Operating at the post-transcriptional level, microRNAs (miRNAs) function as
systemic integrators in the complex regulatory network of atherosclerosis, acting
as molecular bridges that connect core pathological processes. For instance, the
lipid-centric miR-33, co-transcribed with its host gene SREBP, links
cholesterol synthesis with the inhibition of cholesterol efflux by targeting
ABCA1, while also promoting pro-inflammatory macrophage polarization [120]. The
classic “inflamma-miR”, miR-155, creates a vicious cycle by translating
inflammatory signals into endothelial injury via eNOS suppression, and further
inflammation via NF-
The incidence and complications of atherosclerosis exhibit sex dimorphism. Due to the protective effects of estrogen against atherosclerosis, the onset of CAD in women is delayed by 10–15 years compared to men. After menopause, traditional risk factors such as hypertension, dyslipidemia, and diabetes have a greater impact on the development of CVD in women [123]. Sex differences are also observed in plaque size, composition, and rupture risk. Premenopausal women tend to develop stable, diffuse lesions, whereas men are more prone to acute plaque rupture. In postmenopausal women, the triggering mechanisms for ACS may be more closely associated with systemic endothelial dysfunction and a hypercoagulable state [124]. These variations extend to the genetic level. A Study using reproductive models has found that, compared with the XY genotype, the XX genotype upregulates key enzymes involved in both free radical scavenging during injury and processes common to celiac disease, thereby enhancing the bioavailability of dietary fats [125]. This, in turn, provides the material basis for elevated blood lipids and plaque formation. Such findings suggest that the biological basis of women may not be inherently “protected”, but they may instead have a higher genetic predisposition to dyslipidemia. This underscores the need to adopt a sex-specific perspective in future research and therapeutic strategies.
Atherosclerosis is a chronic inflammatory disease driven by endothelial dysfunction, lipid metabolism disorders, and persistent inflammation. It remains the leading cause of cardiovascular disease incidence and mortality worldwide. Here, we summarize the complex interactions among the core mechanisms, highlighting how key molecular pathways collaboratively regulate plaque initiation, progression, and rupture. Furthermore, we explore emerging regulatory factors such as glycoRNA and HCC-1, which provide new insights into the modulation of inflammation and also demonstrate potential as novel diagnostic biomarkers.
Future research will extend beyond single molecules or pathways, and focus instead on revealing the full spectrum of the disease through advanced research tools. Multi-omics integration techniques that combine genomics, transcriptomics, proteomics, and metabolomics data offer an unprecedented multidimensional molecular profile of the disease, enabling in-depth phenotyping of patients. This approach not only uncovers the complete flow of molecular information from static genetic risks (genotype) to dynamic disease manifestations (phenotype), but also facilitates the discovery of novel biomarker combinations associated with plaque instability [126]. At the same time, artificial intelligence (AI) and machine learning (ML) have revolutionized our ability to analyze these vast and complex datasets. AI/ML algorithms excel in developing cardiovascular risk prediction models that outperform traditional scoring systems, such as QRISK3 and ASCVD/PCE [127, 128]. They have also achieved groundbreaking advances in cardiovascular imaging. In particular, deep learning-based radiomics can extract subtle texture features from standard anatomical images, such as coronary computed tomography angiography. Such features are imperceptible to the human eye and are related to inflammation in perivascular adipose tissue (PCAT), enabling non-invasive quantification of local vascular inflammation [129].
On the therapeutic front, future strategies are likely to become more targeted and intelligent. Nanomedicine offers innovative solutions to the delivery challenges of nucleic acid drugs, such as antisense oligonucleotides and siRNA. Nanocarriers constructed from materials like chitosan and gold nanoparticles can precisely deliver drugs to atherosclerotic plaques [130]. Furthermore, “smart” delivery systems, such as pH-low insertion peptides (pHLIP), can utilize the acidic microenvironment of plaques to trigger targeted drug release to specific cells such as macrophages within the lesion (e.g., the delivery of anti-miR-33) [131]. This strategy integrates diagnosis with treatment, thereby maximizing efficacy while minimizing systemic side effects, and heralding the era of “theranostics”.
In summary, our improved understanding and management of atherosclerosis is leading to a new era of multidimensional, systematic, and personalized approaches, thanks to the clarification of core pathological mechanisms, the application of cutting-edge research tools, and the development of precision treatment strategies. Collectively, these advances hold great promise for ameliorating the management of atherosclerosis, achieving true individualized treatment, and ultimately reducing its global health burden.
The article has been drafted by ML and DM. JB prepared all figures. HY and QH edited and finalized the manuscript. All authors contributed to the conception and editorial changes. All authors have read and approved the final version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the Natural Science Foundation of Shandong Province (No.ZR2016HM49), Horizontal research project of Shandong University (No.3450012001901 and 23460012711702), and Traditional Chinese Medicine Science and Technology Development Plan Project of Shandong Province (No.2019-0891).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.