


 , Jiao Yang 3,†, Zeyuan Yang 4,5, Ting Liu 1, Bingqian Zeng 1, Mingxi Ma 1, Ying Liu 1, Shuanglan Xu 1,*
, Jiao Yang 3,†, Zeyuan Yang 4,5, Ting Liu 1, Bingqian Zeng 1, Mingxi Ma 1, Ying Liu 1, Shuanglan Xu 1,* , Xiqian Xing 1,6,*
, Xiqian Xing 1,6,*1 Department of Pulmonary and Critical Care Medicine, The Affiliated Hospital of Yunnan University, 650021 Kunming, Yunnan, China
2 College of Clinical Medicine, Dali University, 671003 Dali, Yunnan, China
3 Department of Pulmonary and Critical Care Medicine, First Affiliated Hospital of Kunming Medical University, 650032 Kunming, Yunnan, China
4 State Key Laboratory of Primate Biomedical Research, 650500 Kunming, Yunnan, China
5 Institute of Primate Translational Medicine, Kunming University of Sciences and Technology, 650500 Kunming, Yunnan, China
6 Key Laboratory of Respiratory Disease Research of Department of Education of Yunnan Province, 650021 Kunming, Yunnan, China
†These authors contributed equally.
Abstract
Pulmonary hypertension (PH) is characterized by an abnormally high pressure within the pulmonary arteries, which can be attributed to various factors. Severe diseases affecting pulmonary vessels may result in heart failure and potentially lead to death; these conditions are linked to significant mortality and unfavorable outcomes. Approximately 1% of adults worldwide have PH, and this condition may affect up to 10% of people older than 65 years. Currently, the mechanisms involved in the development of PH are not fully known and are thought to result from multiple coordinated factors. This lack of understanding remains a bottleneck in clinical practice. Numerous studies have confirmed that pulmonary artery endothelial cell (PAEC) dysfunction plays an important role in occlusive pulmonary vascular remodeling and the pathogenesis of PH. Src homology region 2 domain-containing phosphatase-1 (SHP-1) is a regulatory molecule that negatively modulates various cellular mediators and growth factors, primarily playing a negative regulatory role in signal transduction pathways. This review mainly presents an in-depth exploration of the key signaling pathways through which SHP-1 regulates the expression of endothelial cells (ECs), thereby influencing various physiological functions, including proliferation, migration, oxidative stress, angiogenesis, apoptosis, autophagy, the inflammatory response, and vascular permeability. Furthermore, the potential mechanisms through which endothelial SHP-1 plays a role in pulmonary vascular remodeling in PH are discussed. These findings underscore SHP-1 as an encouraging therapeutic target for preventing and managing PH.
Keywords
- pulmonary hypertension
- SHP-1
- endothelial cells
- vascular remodelling
Pulmonary hypertension refers to a disease that involves increased pulmonary
vascular resistance. The pathological process of PH is characterized by pulmonary
vascular remodelling, which involves excessive proliferation of pulmonary artery
smooth muscle cells (PASMCs) and pulmonary artery endothelial cells (PAECs),
distal pulmonary artery muscularization, vascular occlusion, plexiform lesions,
and abnormal accumulation of inflammatory cells [1, 2, 3, 4]. Alterations in pulmonary
vascular tone and remodelling contribute to a progressive increase in pulmonary
vascular resistance, ultimately culminating in a spectrum of clinical syndromes
associated with right heart failure and, in severe cases, death [5]. The
diagnostic criteria for haemodynamics are outlined as follows: A mean pulmonary
artery pressure (mPAP) of
Recent research has shown that among individuals undergoing standard treatment for PH, the use of sotatercept results in decreased pulmonary vascular resistance and increased haemodynamic metrics and exercise ability, as evaluated using the 6-minute walk test [9, 10]. These findings provide a new potential treatment strategy for PH [11]. Although current treatment methods can improve the quality of life of patients, none are curative, which presents a significant challenge in clinical practice [12]. In the early stages of PH development, EC injury and apoptosis are predominant [13, 14, 15]. Conversely, in the later stages of PH, EC overproliferation and antiapoptotic mechanisms prevail, resulting in significant vascular remodelling [16]. Recent studies have shown that endothelial cell dysfunction, injury, and immune‒inflammatory responses, along with metabolic abnormalities, epigenetic changes, endothelial‒mesenchymal transition (EndMT), and the release of growth factors and chemokines from endothelial cells (ECs), induce the proliferation of SMCs and play a role in the structural changes associated with pulmonary vascular remodelling. This process increases pulmonary artery pressure and pulmonary vascular resistance [17].
Src homology region 2 protein tyrosine phosphatase-1 (SHP-1), which is
encoded by the gene protein tyrosine phosphatase non-receptor type 6 (PTPN6), is an essential component of the protein tyrosine
phosphatase (PTP) family [18, 19]. SHP-1 functions as
a negative regulator in various cellular signalling pathways [19, 20], primarily
by dephosphorylating tyrosine residues on various signalling proteins (such as
signal transducer and activator of transcription (STAT), protein kinase B (Akt) and
Src). This dephosphorylation modulates tumour, inflammatory, and
metabolic pathways [21], inhibiting signal transduction by targeting tyrosines in
proteins. These factors significantly influence various critical biological
activities of related cells and are essential for maintaining normal cellular
functions and the integrity of the immune system [18]. Although SHP-1 is
predominantly expressed in haematopoietic cells, it has also been identified in
nonhaematopoietic cells, such as ECs [22]. Research indicates that bovine aortic
ECs contain endogenous SHP-1 [23]. Additionally, this molecule functions
as a negative modulator, blocking superoxide generation in these cells [24]. In
ECs under hypoxia, SHP-1 inhibits reactive oxygen species (ROS)
generation, reduces the stability of hypoxia-inducible factor 1-alpha
(HIF-1
We analysed the cell-type specific expression of SHP-1 based on single-cell RNA-seq data derived from the lung tissue samples of PH mice and retrieved from the GSE154959 dataset in the Gene Expression Omnibus (GEO) database. Specifically, SHP-1 is highly expressed in several immune cell populations, including macrophages, B cells, monocytes, granulocytes and dendritic cells. In contrast, SHP-1 expression is significantly inhibited in ECs, whereas SHP-1 expression is almost absent in nonimmune cells, such as adipocytes, epithelial cells, and hepatocytes (Fig. 1, Ref. [26]). This unique expression profile suggests that SHP-1 is involved primarily in the regulation of immune cell functions. However, its low-level expression in ECs may indicate a specific role in vascular homeostasis or pulmonary vascular remodelling.
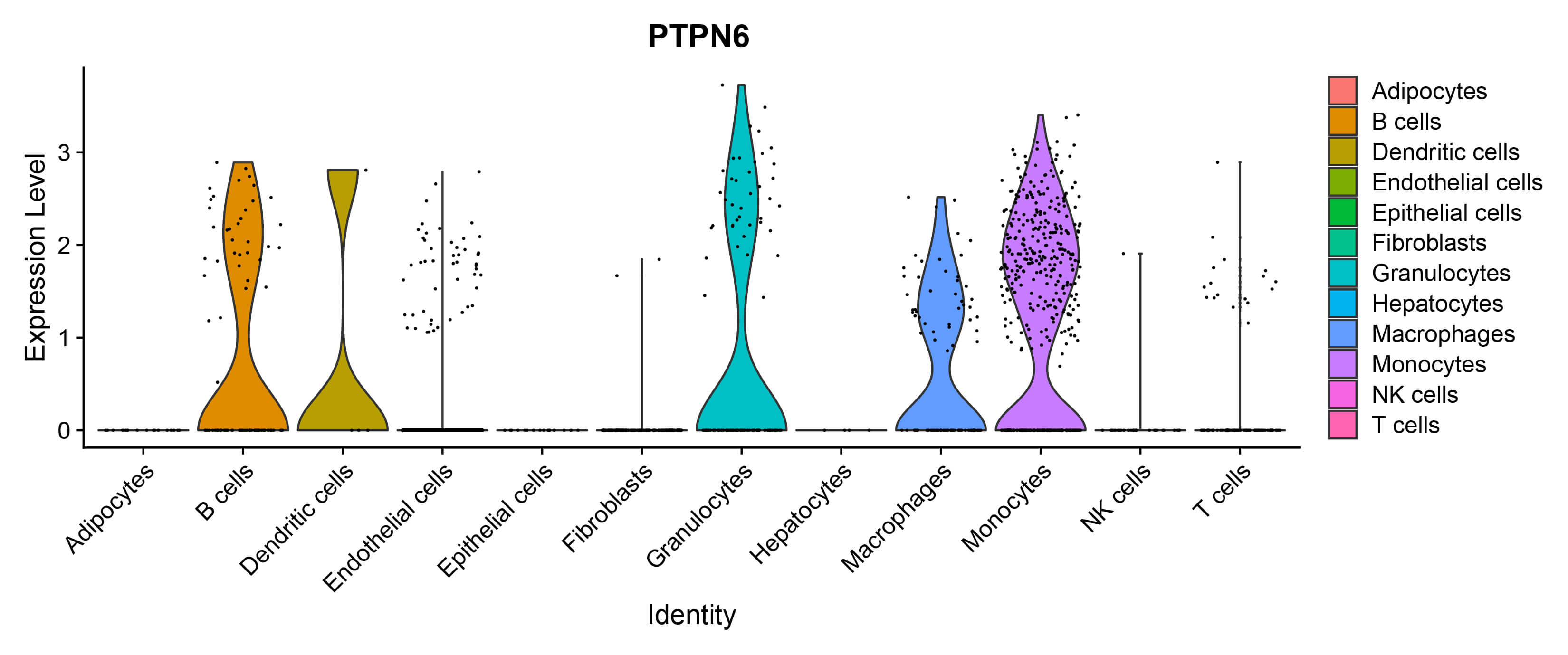 Fig. 1.
Fig. 1.
SHP-1 expression in different cell subpopulations was analysed using the GEO database GSE154959 [26]. SHP-1, Src homology region 2 domain-containing phosphatase-1; GEO, Gene Expression Omnibus; NIK, natural killer T cells, NKT cells; PTPN6, protein tyrosine phosphatase non-receptor type 6.
Therefore, the aim of this review is to explore the role and mechanisms of PAECs in the pathogenesis of PH. The possible mechanisms of endothelial SHP-1 in PH-related pulmonary vascular remodelling are discussed in detail, which may provide novel therapeutic targets and insights for the prevention and treatment of PH.
The genetic roles and mechanisms of PAECs in PH encompass multiple aspects, including abnormalities in transcription factors [27, 28, 29], mitochondrial dysfunction [30, 31], cell death pathways [30, 32], EndMT [33, 34], genetic variations, and metabolic abnormalities [34, 35]. Together, these mechanisms lead to PAEC dysfunction, which results in changes in the pulmonary vasculature and advances the progression of PH. Importantly, the most frequently identified genetic factor associated with familial PH is bone morphogenetic protein receptor type II (BMPR2) [35]. Some individuals with PH have a genetic predisposition, such as patients carrying a heterozygous abnormality in the gene encoding BMPR2 and a mutation in the activin-like kinase (Alk)-1 receptor [5, 36, 37]. Mutant mice have increased susceptibility to hypoxia-induced PH, along with impaired endothelium-dependent vasodilation within the pulmonary vasculature [38]. Heterozygous Bmpr2 knockout leads to EC injury and persistent PH in mice.
Epigenetic inheritance describes how gene expression can be altered without any modification to the DNA sequence itself. This mechanism is influenced by factors such as DNA methylation, histone modifications, and noncoding RNA [39]. A recent epigenome-wide association study (EWAS) revealed a total of 865,848 differentially methylated cytosine-phosphate-guanine (CpG) sites in the peripheral blood samples of patients suffering from PH [40], underscoring the widespread occurrence of epigenetic dysregulation. Within vascular ECs, modifications to histones are crucial for disease progression. The targeted inhibition of important elements within the histone H3 lysine 4 (H3K4) methyltransferase complex, specifically absent, small, or homeotic 2 (ASH2) and WD repeat-containing protein 5 (WDR5), significantly ameliorates hypoxia-induced PH in mice, confirming the critical role of histone methylation regulation [41].
Furthermore, studies utilizing the pulmonary thromboembolism (PTE) rat model have demonstrated that the miR-124/polypyrimidine tract binding protein 1 (PTBP1)/pyruvate kinase M (PKM) signalling axis facilitates pulmonary artery intimal remodelling through the mediation of metabolic reprogramming [42]. Collectively, these findings indicate that (1) epigenetic mechanisms, including DNA methylation and histone modifications, drive abnormal endothelial cell proliferation and vascular remodelling by regulating the expression of key genes and that (2) energy metabolism disorders resulting from metabolic reprogramming further exacerbate the pathological thickening of the pulmonary artery intima. These two mechanisms may be interrelated and jointly contribute to the progression of pulmonary hypertension.
The primary trigger for PH is the dysfunction of PAECs, which is predominantly characterized by the generation of associated active factors and alterations in coagulation within the pulmonary endothelium. This dysfunction leads to abnormal contractions of the pulmonary vasculature, in situ thrombosis, and the remodelling of vascular structures, ultimately contributing to the onset and progression of PH. This condition represents an endothelial pathological state resulting from an imbalance between substances that induce contraction and those that promote vasodilation [43].
Studies have confirmed that PAEC dysfunction disrupts the pathological proliferation and migration of adjacent PASMCs, ultimately leading to thickening of the vascular wall’s medial layer and a progressive increase in pulmonary vascular resistance [44, 45].
Mice with defects in ECs and haematopoietic cells that encode prolyl-4
hydroxylase 2 (PHD2) exhibit severe occlusive vascular
remodelling and right heart failure. In particular, the pulmonary vascular
lesions of these mice significantly increased EC proliferation. Reactivation of
hypoxia-inducible factor 2
The above studies elucidate the central role of EC dysfunction in the
pathogenesis of PH. This dysfunction leads to pulmonary vasoconstriction,
thrombus formation, and medial thickening through an imbalance of active factors,
coagulation abnormalities, and HIF-2
In PH, inflammation is characterized by (1) elevated levels of cytokines, chemokines, and adipokines and (2) varying degrees of inflammatory and immune cell infiltration surrounding and within the walls of small pulmonary arteries [51]. Furthermore, one study revealed the presence of tertiary lymphoid tissues (tLTs) in the lungs of patients with idiopathic pulmonary arterial hypertension (IPAH), which may be associated with aberrant immune system activation and autoantibody production [52]. Another study demonstrated that anti-endothelial cell antibodies (AECAs) are detectable in patients with systemic sclerosis (SSc) and are correlated with a greater incidence of vascular lesions and related symptoms. AECAs can activate ECs and lead to apoptosis in patients. Additionally, PH can also occur in patients, contributing to increased mortality [53]. A study conducted by Sasaki N’s team [54] suggested that the induction of PAEC apoptosis by a combination of anti-endothelial cell antibodies and activated natural killer cells may play a crucial role in the vascular damage associated with PH in patients with mixed connective tissue disease. These findings underscore the significant role of the immune system in PH.
Oxidative stress is recognized as a critical factor leading to EC injury and functional impairment [55]. Increases in ROS production lead to an imbalance in the signalling between reactive nitrogen species (RNS) and nitric oxide (NO) [56], as well as to DNA damage [57]. This imbalance results in abnormal proliferation, injury, and apoptosis of ECs. Oxidative stress can also promote the development of PH by disrupting the NO signalling pathway. As a crucial vasodilatory mediator, the synthesis of NO is regulated primarily by endothelial nitric oxide synthase (eNOS). Under pathological conditions associated with PH, dysfunction of eNOS leads to its uncoupling, subsequently disrupting NO signalling and significantly impairing the capacity for vasodilation. This pathological alteration not only exacerbates vasoconstrictive responses but also further accelerates the progression of pulmonary arterial hypertension [56].
Research has demonstrated that oestradiol directly inhibits the proliferation of ECs and improves haemodynamics. By enhancing mitochondrial autophagy, oestradiol also inhibits PH [58]. Conversely, it enhances EC angiogenesis in foetal lambs with persistent PH, which reduces the expression of the autophagy protein beclin-1, leading to autophagy defects [59]. Moreover, autophagy accelerates the transition from an apoptotic phenotype to a hyperproliferative phenotype in pulmonary vascular ECs associated with HIV-related PH [60].
Singh et al. [61] reported increased expression and activity of fatty acid synthase (FAS) in hypoxic human pulmonary artery smooth muscle cells (HPASMCs). The inhibition of FAS promotes HPASMC apoptosis and reduces autophagy, which reduces pulmonary vascular remodelling and endothelial dysfunction [61].
EndMT induced by dysfunctional PAECs is considered the initial step and a key pathological factor in the occurrence of PH [62]. In PH, EndMT directly promotes structural changes in the vascular wall by causing PAECs to lose their endothelial characteristics and acquire the migratory and proliferative abilities of mesenchymal cells [33, 62, 63]. In PH associated with congenital heart disease, high shear stress (HSS) can directly induce EndMT, thereby initiating vascular remodelling [64]. Numerous studies have demonstrated that apoptosis, inflammation, and metabolic abnormalities, such as oxidative stress in PAECs, can induce EndMT. These findings suggest that EndMT may serve as a compensatory response following endothelial injury [65]. This dual regulatory role positions SHP-1 as a pivotal therapeutic target for modulating vascular remodelling processes [66].
Studies have demonstrated that SHP-1 is expressed in various epithelial tissues, including haematopoietic cells and ECs [24, 67]; however, its expression level in ECs remains relatively low. In human microvascular endothelial cells (HMECs), SHP-1 is predominantly localized in the nucleus, with only moderate expression observed in the cytoplasm [25].
SHP-1 plays a critical role in ECs by protecting them from the upregulation of adhesion molecules and the harmful effects of thrombosis under inflammatory conditions. Under hypoxic or ischaemic conditions, SHP-1 promotes the development of blood vessels by suppressing oxidative stress. In ischaemic illnesses, SHP-1 suppresses the production of ROS, which in turn inhibits the proliferation and survival of ECs [25].
For example, in a diabetic mouse model, hyperglycaemia impairs the vascular
regenerative capacity of ischaemic muscles by upregulating SHP-1
expression in ECs, which inhibits the activity of angiogenic factors [68]. In an
in vitro model of chronic obstructive pulmonary disease (COPD), the
expression level of SHP-1 was significantly decreased. SHP-1
overexpression reversed the effects of cigarette smoke extract (CSE) on
endothelial cell migration, epithelial‒mesenchymal transition (EMT), and the production of proinflammatory factors. Moreover, it
mitigated the inflammatory response by inhibiting the P65 and PI3K/AKT
signalling pathways [69]. In the diabetic state, SHP-1 promotes
endothelial cell senescence and contributes to abnormal collateral vessel
formation by diminishing the proangiogenic effects of nuclear factor erythroid
2-related factor 2 (Nrf2) and VEGF, ultimately impeding blood
flow reperfusion. However, the overexpression of dominant-negative SHP-1
(dnSHP-1) effectively reverses these pathological effects [62]. In
diabetic peripheral arterial disease, SHP-1 reduces endothelial cell
migration and capillary formation by negatively regulating the vascular
endothelial growth factor receptor 2 (VEGFR2) and platelet derived growth factor
receptor beta (PDGFR-
Comprehensive evidence indicates that maintaining moderately high SHP-1 expression is crucial for controlling inflammation and ensuring endothelial homeostasis. The lack of expression of this molecule has emerged as a common pathological feature in various vascular diseases. These findings underscore the importance of SHP-1 as a vital target for research in the context of vascular diseases and inflammatory responses.
SHP-1 can indirectly affect the phosphorylation of VEGFR2 by dephosphorylating Src family kinases (such as Lyn and Fyn) [71]. This dephosphorylation depends on the interaction between SHP-1 and the SH2 domain of Src family receptors [72]. Upon VEGF stimulation, the phosphatase activity of SHP-1 is activated, leading to the dephosphorylation of specific tyrosine residues (such as Y996, Y1059, and Y1175) on VEGFR2, thereby inhibiting VEGFR2 signalling. This dephosphorylation attenuates VEGFR2-mediated downstream signalling pathways, such as the activation of extracellular signal-regulated kinase (ERK) and Akt, consequently suppressing the proliferation and DNA synthesis of vascular ECs [71]. Cellular communication network factor 1 (CCN1), also known as cysteine-rich protein sixty-one, is a stromal cell protein that interacts with integrins and is secreted by the cell. Cardiovascular system development is highly important in human life. CCN1 enhances SHP-1 activity by binding to VEGFR2, leading to VEGFR2 dephosphorylation and the inhibition of endothelial cell proliferation [73].
Furthermore, this study revealed that acetyl-11-keto-boswellic acid (AKBA) can upregulate the expression and activity of SHP-1. The upregulation of SHP-1 by AKBA leads to reduced VEGF expression and downregulated phosphorylation of VEGFR2 and STAT3, thus inhibiting angiogenesis. Overall, these findings underscore the critical role of SHP-1 in regulating endothelial cell angiogenesis [74].
Clearly distinguishing the mechanisms underlying changes in vascular permeability between PH and acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) is crucial. Both ALI and ARDS are marked by a breakdown of the alveolar-capillary barrier, which is evident through a strong inflammatory reaction that causes damage to both endothelial and alveolar epithelial cells injury, ultimately culminating in the accumulation of protein-rich pulmonary edema [75]. ARDS signifies the critical phase of ALI, marked by significant formation of hyaline membranes, collapse of alveoli, and persistent hypoxemia [76, 77]. Conversely, the changes in vascular permeability seen in PH mainly stem from endothelial dysfunction that occurs throughout the chronic remodeling of the pulmonary vasculature. This condition presents as perivascular edema rather than as exudation into the alveolar spaces, and the underlying mechanisms differ fundamentally from the acute inflammatory damage seen in ALI/ARDS [78].
Cytokine TNF superfamily member 15 (TNFSF15) is produced primarily by vascular ECs, and receptor activation leads to trimerization with VEGFR2 and death receptor 3 (DR3). This process affects the activity of SHP-1 phosphatase, which further inhibits the phosphorylation of VEGFR2 [79]. In addition, Chu et al. [80] reported that thrombospondin-1 (TSP-1) binds to VEGFR2 via its interaction with STAT3 while recruiting SHP-1 to inhibit the phosphorylation of VEGFR2; thus, TSP-1 reduces the phosphorylation level of VEGFR2 and VEGF-induced endothelial cell migration. The thrombospondin type 1 repeat (TSR) domain inhibits tube formation [80].
Additionally, SHP-1 phosphatase activity is enhanced by a novel aliphatic isohydroxamic acid ester derivative (WMJ-S-001), resulting in the inhibition of VEGFR2 phosphorylation within the VEGF-A-VEGFR2 signalling pathway, which ultimately decreases the cytogenic activity of vascular ECs [81].
Studies have shown that hyperglycaemic and hypoxic environments upregulate the
phosphatase activity of SHP-1, which inhibits VEGF signalling.
This process impairs the functional ability of ECs and inhibits angiogenesis [25, 82, 83]. In addition, N(
In an in vitro study, SHP-1 inhibition promoted the ability of
TNF-
As a tyrosine phosphatase with an SH2 domain, SHP-1 can directly bind to JAK2 and dephosphorylate its substrate STAT3. As a result, it negatively regulates the activation of the JAK/STAT3 signalling pathway, which helps maintain ECs under normal conditions [86, 87].
The levels and activity of SHP-1 control critical functions of ECs, such as proliferation, migration, and angiogenesis. For example, in a high-glucose environment, the inhibition of SHP-1 activity can result in hyperactivation of the JAK/STAT3 signalling pathway, leading to abnormal endothelial cell injury and angiogenesis [88]. Additionally, the expression and activity of SHP-1 are modulated by various factors. Under certain pathological conditions, SHP-1 expression may be downregulated, or its activity may be suppressed, resulting in the aberrant activation of the JAK/STAT3 signalling pathway [89, 90, 91].
This study revealed that naringenin can inhibit JAK2/STAT3 signalling pathway activation while increasing the expression of SHP-1, which improved hypertension during pregnancy. Sufficient evidence indicates that SHP-1 must be expressed and activated to suppress oxidative stress, inflammatory responses, and JAK2/STAT3 signalling pathway activity. This alleviation of damage to vascular endothelial cell damage and vasoconstriction further regulates the development and differentiation of ECs [92]. Angiopoietin 1 (Ang1) inhibits cell proliferation; in this context, the induction of SHP-1 dampens Ang1-mediated interleukin 6 (IL-6)-induced stimulation of the JAK/STAT3 signalling pathway, thus reducing IL-6-induced endothelial cell permeability and suppressing the vascular immune‒inflammatory response [93]. Moreover, some studies have shown that inhibiting Phloretin activates SHP-1 to phosphorylate STAT3. This process can ultimately induce apoptosis and autophagy in vascular ECs [94, 95].
SHP-1 can bind to epidermal growth factor receptor (EGFR) and dephosphorylate its downstream substrates, thereby suppressing EGFR-mediated ERK activation [96]. SHP-1 suppresses angiogenesis and inflammatory responses through the dephosphorylation of key signalling molecules, such as ERK and c-Jun N-terminal kinase (JNK) [97, 98].
Stimulation of bovine aortic ECs with VEGF and epidermal growth
factor (EGF) significantly increased the
phosphorylation of ERK. However, treatment with TNF-
Knockout of the connexin 37 (Cx37) gene in mice might increase SHP-1 activity, which in turn could lead to the dephosphorylation of proteins such as myosin light chain 2 (MLC2), ERK, and protein kinase B through various mechanisms. The angiotensin II (Ang II) signalling cascade involves the phosphorylation of proteins generated after the activation of Ang II at the AT1 receptor (AT1R) in ECs. Protein dephosphorylation may interfere with important cellular physiological processes, such as contraction, proliferation, and survival [99].
HIF-1 consists of an oxygen-regulated
Under hypoxic conditions, SHP-1 knockdown increases ROS in ECs
and further induces ROS to upregulate the expression of the
HIF-1
Under hyperglycaemic conditions, SHP-1 is activated and binds to DR3, the receptor for tumour necrosis factor ligand-related molecule 1A (TL1A). This activation inhibits the dephosphorylation of Src by SHP-1. Consequently, when glucose levels are elevated, SHP-1 binds to the receptor of TL1A, also called death receptor 3 (DR3). This action of SHP-1 prevents the dephosphorylation of Src, which then activates vascular endothelial-cadherin (VE-cadherin). As a result, EC integrity is impaired, leading to vascular leakage [107].
SHP-1 plays a crucial role in maintaining vascular haemostasis within
the body. During the inflammation of ECs caused by TNF-
Many findings present strong evidence that SHP-1 negatively regulates endothelial cell function via tyrosine phosphatase activity. The activation of SHP-1 inhibits the coagulant activity of ECs; however, loss of function or expression is able to attenuate this effect (Table 1, Ref. [23, 24, 25, 70, 73, 74, 79, 80, 81, 82, 83, 84, 85, 88, 92, 93, 94, 95, 97, 98, 99, 107, 108, 109, 110]).
| Inhibits phosphorylation levels of VEGFR2 | ||||
| SHP-1 activation | Nature of study | Regulation | EC dysfunction | Reference |
| ↑ | In vitro, in vivo | – | Proliferation | [73] |
| ↑ | In vitro, in vivo | – | Vascular permeability | [79] |
| ↑ | In vitro, in vivo | – | Angiogenesis | [74] |
| ↑ | In vitro, in vivo | – | Angiogenesis | [81] |
| ↑ | In vitro, in vivo | – | Migrate | [80] |
| ↑ | In vitro, in vivo | – | Angiogenesis | [25, 82, 83] |
| ↑ | In vitro, in vivo | – | Oxidative stress | [84] |
| ↓ | In vitro, in vivo | + | Angiogenesis | [85] |
| ↓ | In vitro | + | Proliferation | [110] |
| Inhibits the JAK2/STAT3 signalling pathways | ||||
| SHP-1 activation | Nature of study | Regulation | EC dysfunction | Reference |
| ↓ | In vitro | + | Angiogenesis | [88] |
| Endothelial cell injury | ||||
| ↑ | In vivo | – | Growth | [92] |
| ↑ | In vitro | – | Immune inflammation | [93] |
| ↑ | In vitro, in vivo | – | Apoptosis, autophagy | [94, 95] |
| Inhibits ERK phosphorylation | ||||
| SHP-1 activation | Nature of study | Regulation | EC dysfunction | Reference |
| ↑ | In vitro, in vivo | – | Angiogenesis | [97] |
| inflammation | [98] | |||
| ↑ | In vitro, in vivo | – | Proliferation | [23, 70] |
| ↑ | In vivo | – | Proliferation | [99] |
| SHP-1 is involved in regulating the levels of ROS and HIF-1 | ||||
| SHP-1 activation | Nature of study | Regulation | EC dysfunction | Reference |
| ↑ | In vivo | – | Proliferation | [25] |
| Other avenues | ||||
| SHP-1 activation | Nature of study | Regulation | EC dysfunction | Reference |
| ↑ | In vitro, in vivo | – | Immune inflammation | [107] |
| ↑ | In vivo | – | inflammation | [24] |
| ↑ | In vitro | – | Oxidative stress | [108] |
| ↑ | In vitro | – | Proliferation | [23] |
| ↓ | In vitro | + | Proliferation | [109] |
Signals may initiate (
SHP-1, Src homology region 2 domain-containing phosphatase-1; VEGFR2, vascular endothelial growth factor receptor 2; EC, endothelial cell; JAK2, janus kinase 2; STAT3, signal transducer and activator of transcription 3; ERK, extracellular signal-regulated kinase; ROS, reactive oxygen species; HIF-1, hypoxia-inducible factor 1.
SHP-1 plays a crucial role in regulating EC function and angiogenesis
by modulating the phosphorylation of VEGFR2 and its downstream
signalling pathways. Molecules such as CCN1, limonin, AKBA, and
WMJ-S-001 activate SHP-1 and inhibit VEGFR2
phosphorylation, thereby suppressing endothelial cell proliferation, migration,
and angiogenesis [73, 74, 81]. Under conditions such as hyperglycaemia and
hypoxia, the activity of SHP-1 is increased. By suppressing the
overactivation of the VEGF and JAK/STAT3 signalling pathways,
SHP-1 contributes to endothelial cell dysfunction and impaired
angiogenesis [25, 82, 83]. Furthermore, the inhibition of SHP-1 in
HUVECs promotes VEGFR2 expression and drives endothelial cell
proliferation [109]. The activation of SHP-1 induces dephosphorylation
of the ERK protein, thereby modulating endothelial cell proliferation
[23, 70, 99]. Under hypoxic conditions, SHP-1 regulates endothelial cell
proliferation by controlling HIF-1
Phloretin inhibits the phosphorylation of STAT3 by activating SHP-1, which induces apoptosis and autophagy in ECs [94, 95]. Under hypoxic conditions, the inhibition of TNFR-1 or SHP-1 in HUVECs significantly increases the expression of the antiapoptotic factor Bcl-xL while decreasing the expression of the proapoptotic factor Bax, thereby effectively suppressing apoptosis in these cells [109].
SHP-1 suppresses angiogenesis and inflammatory responses by
dephosphorylating key signalling molecules, including ERK and
JNK [97, 98]. Studies have shown that Ang1 activates SHP-1 to
inhibit IL-6-induced endothelial cell permeability and inflammatory responses
[108]. Furthermore, the inhibition of SHP-1 exacerbates
TNF-
Under hyperglycaemic conditions, SHP-1 is activated and binds to DR3, impairing its ability to dephosphorylate Src. This process induces the activation of VE-cadherin, which destabilizes endothelial cell integrity and contributes to vascular leakage [107].
In HUVECs, SHP-1 negatively regulates Rac1 activation by suppressing PI3K activity, thereby modulating NAD(P)H oxidase-dependent superoxide production and significantly reducing oxidative stress levels in ECs [24].
In summary, endothelial SHP-1 may suppress the development and progression of PH through multiple mechanisms. Specifically, SHP-1 inhibits EC migration and proliferation, reducing key drivers of vascular remodelling; SHP-1 modulates immune-inflammatory responses and oxidative stress to mitigate endothelial cell damage; SHP-1 suppresses pathological angiogenesis by inhibiting signalling pathways such as VEGF; and SHP-1 regulates vascular permeability while inhibiting apoptosis and autophagy to maintain endothelial cell function and stability. These combined effects inhibit pulmonary vascular remodelling and prevent the progression of PH, highlighting SHP-1 as a potential therapeutic target for PH treatment (Fig. 2).
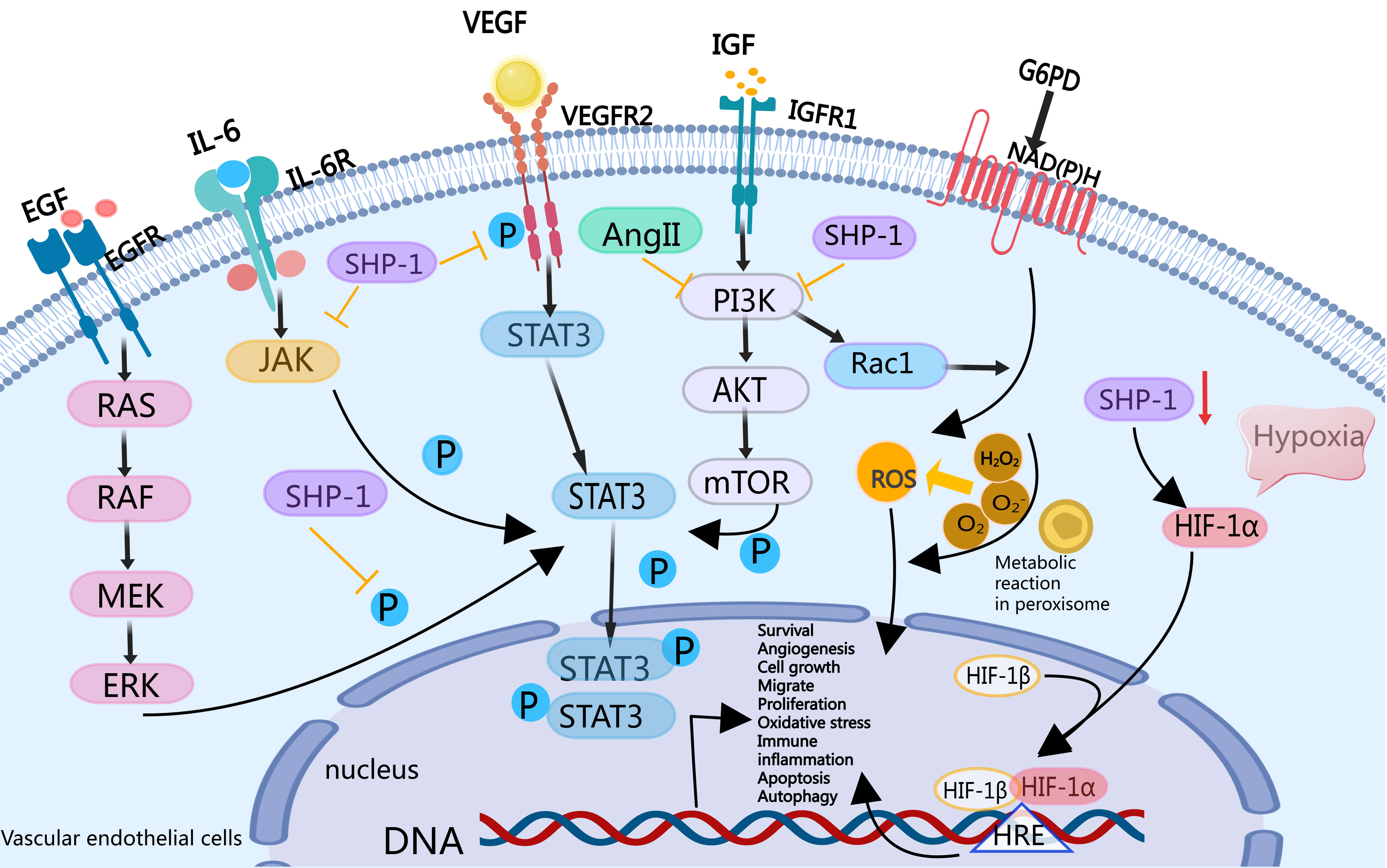 Fig. 2.
Fig. 2.
SHP-1 may regulate PAEC function to inhibit
signalling pathways that result in PH vascular remodelling. SHP-1
inhibits VEGFR2/IGFR1, the EGFR/ERK-mediated STAT3
signalling pathway, and the IL-6-induced JAK/STAT3 signalling
pathway; and the PI3K/Rac1 signalling pathway negatively regulates NADPH
oxidase (NOX)/vascular peroxidase 1 (VPO1) pathway-derived ROS. SHP-1 inhibits EC migration and
proliferation, immune inflammation, activation, oxidative stress,
vasoconstriction, generation, vascular permeability, apoptosis, autophagy, and
other important pathophysiological processes in the development of PH.
Additionally, the silencing of SHP-1 leads to an increase in ROS and hypoxia-inducible factor 1 alpha (HIF-1
The SHP-1 activators exert antiproliferative and pro-apoptotic effects by
inhibiting B cell receptor (BCR) signalling pathway, as evidenced by the
downregulation of p-Lyn. This inhibition may also indirectly influence tumour
angiogenesis [111, 112]. The overexpression of SHP-1 counteracts the migration of
endothelial cells and the release of inflammatory factors triggered by CSE,
indicating that SHP-1 has a protective function in chronic inflammatory vascular
conditions, including vascular lesions associated with COPD [69]. Angiogenesis is
influenced by SHP-1 through the regulation of TGF-
In certain types of cancer, the activation of SHP-1 may foster a microenvironment that is favourable for tumours, indicating that caution is warranted when using SHP-1 inhibitors to treat vascular-related tumours [87]. SHP-1 suppresses overly active immune responses within Treg cells; nonetheless, a lack of this protein might result in T-cell impairment, which can influence the development of vascular autoimmune disorders [114].
Modulators of SHP-1 have the potential to play dual roles in vascular disease treatment: they may enhance the healing of atherosclerotic or diabetic vascular lesions by providing anti-inflammatory benefits and protecting endothelial cells as an activator, whereas they may facilitate the regeneration of blood vessels in ischaemic tissues under certain circumstances as an inhibitor. Additional research is essential to clarify the mechanisms specific to different tissues and explore avenues for clinical application.
On the basis of current knowledge, SHP-1 is suspected to play a significant role in the development and maintenance of pulmonary endothelial dysfunction associated with PH, potentially offering new therapeutic innovations for this condition. Several experiments could be conducted to test this hypothesis, including in situ studies on tissues from patients with and without PH to verify the expression and localization of the SHP-1 protein in various cell types within remodelled pulmonary artery walls. Because PH is typically diagnosed at advanced stages and patients are often treated with multiple therapeutic agents, tracking SHP-1 expression and its targets in preclinical models of PH at different stages of its development is crucial. Such analyses include using the chronic Sugen-hypoxia model, models severe PH induced by monocrotaline, and the combination model in rats. Furthermore, in vitro experiments could be designed to investigate the molecular mechanisms regulated by SHP-1 in dysfunctional PAECs from PH patients by manipulating SHP-1 expression levels in PAECs from patients without PH. Using haemodynamic data from adult knockout or conditionally overexpressing SHP-1 mice or rats and evaluating the efficacy of SHP-1 agonist treatments in preclinical models are both essential to strengthening these observations. In these in vivo studies, it will be important to thoroughly assess cardiac function to ensure that these approaches do not negatively impact ventricular performance, including adaptive hypertrophy of the right ventricle. Taken together, these data could be used to determine whether restoring SHP-1 expression is a promising novel intervention in the treatment of PH.
PH, pulmonary hypertension; PAECs, pulmonary artery endothelial cells; SHP-1, Src homology region 2 domain-containing phosphatase-1; mPAP, mean pulmonary artery pressure; EMT, endothelial mesenchymal transition; PTP, protein tyrosine phosphatase; GEO, Gene Expression Omnibus; ECs, endothelial cells; BMPR2, type II bone morphogenetic protein receptor; Alk, activin-like kinase; ASH2, absent, small, or homeotic 2; WDR5, WD repeat-containing protein 5; VEGF, vascular endothelial growth factor; PHD2, prolyl-4 hydroxylase 2; Cas, Crk-associated substrate; VWF, von Willebrand factor; IL-6, interleukin 6; JNK, c-Jun N-terminal kinase; NOX, NADPH oxidase; VPO1, vascular peroxidase 1; FAS, fatty acid synthase; HPASMCs, human pulmonary artery smooth muscle cells; EndMT, endothelial-mesenchymal transition; siRNA, small-interfering RNA; HIF-2
The datasets [ANALYZED] for this study can be found in the [Gene Expression Omnibus] [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154959].
XZ wrote the article, SX drafted the manuscript, and both XZ and SX participated in the revision of the article and made significant contributions to the main concept and design of the article. XX and JY proposed the topic of the article, provided help and advice in writing the article, critically reviewed important intellectual content. ZY analysis and interpretation Fig. 1. TL and BZ helped perform the analysis the article with a constructive conclusion. MM and YL co-founded Table 1. All authors contributed to the conception and editorial changes in the manuscript. All authors read and approved of the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the National Natural Science Foundation of China [grant numbers 82160016, 82560078]; Famous Doctors of High-level Talent Training Support Program of Yunnan Province [grant numbers YNWR-MY-2020-013]; The Special and Joint Program of the Yunnan Provincial Science and Technology Department and Kunming Medical University [grant numbers 202201AY070001-265, 202301AY070001-189]; The Science Research Foundation of Yunnan Provincial Education Department [grant numbers 2024J0016, 2024Y918]; Yunnan Provincial Innovation Team for Respiratory and Pulmonary Circulation Diseases [grant numbers 202405AS350018]; Yunnan Fundamental Research Projects [grant numbers 202501CF070056]; The Yunnan University Medical Research Foundation [grant numbers YDYXJJ2024-0026].
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.