

 , Maomao Zhao 1,2,3,4,†, Lu Bai 1,2,3,4,†, Jing Zhao 1,2,3,4, Hanxiang Gao 1,2,3,4,*
, Maomao Zhao 1,2,3,4,†, Lu Bai 1,2,3,4,†, Jing Zhao 1,2,3,4, Hanxiang Gao 1,2,3,4,* , Ming Bai 1,2,3,4,*
, Ming Bai 1,2,3,4,*1 Department of Cardiology, First Hospital of Lanzhou University, 730000 Lanzhou, Gansu, China
2 Gansu Key Laboratory of Cardiovascular Diseases, The First Hospital of Lanzhou University, 730000 Lanzhou, Gansu, China
3 Gansu Clinical Medical Research Center for Cardiovascular Diseases, The First Hospital of Lanzhou University, 730000 Lanzhou, Gansu, China
4 The First School of Clinical Medicine, Lanzhou University, 730000 Lanzhou, Gansu, China
†These authors contributed equally.
Abstract
The relationship between inflammation and atrial fibrillation (AF) has recently attracted significant research interest. Epicardial adipose tissue (EAT) contributes to the pathogenesis of AF through its inflammatory, metabolic, and electrophysiological effects and may also influence AF outcomes. Inflammatory cells within EAT release key proinflammatory cytokines, including interleukin (IL)-1β and tumor necrosis factor-α (TNF-α), which promote cardiomyocyte apoptosis and fibrosis. These changes compromise cardiac electrophysiological stability and elevate the risk of arrhythmias. Moreover, increased EAT thickness and volume have been identified as critical biomarkers for AF risk, providing new insights into AF diagnosis and treatment. However, despite compelling evidence of a strong association between EAT and AF, further studies are needed to fully elucidate the mechanisms underlying the role of EAT and assess its potential as a therapeutic target. This review aimed to explore the specific mechanisms of inflammation-related EAT in AF and evaluate the clinical potential of EAT as a biomarker and therapeutic target.
Keywords
- epicardial adipose tissue (EAT)
- atrial fibrillation (AF)
- inflammation
- biomarker
Atrial fibrillation (AF) represents a widespread and complex cardiac rhythm disorder, showing a strong correlation with advancing age [1]. Among individuals aged 45 years and above, the probability of developing AF during their lifetime is approximately one in four. Global epidemiological data indicate a 33% surge in AF cases over the past twenty years, with current estimates suggesting nearly 37.57 million affected individuals worldwide, representing about half a percent of the global population [2]. This upward trend is particularly evident in nations with higher economic development, though the rate of increase is more pronounced in regions with intermediate economic status [1, 3, 4, 5]. Emerging research has established significant connections between inflammation-related epicardial adipose tissue (EAT) and various systemic conditions, such as arterial plaque formation [6], cardiac rhythm disturbances, immune system irregularities [7], age-related changes [8], metabolic disorders, and excessive body weight [8, 9]. The development of AF involves a complex interplay of electrical conduction abnormalities, structural changes in atrial tissue, and impaired atrial muscle cell function, with inflammatory processes playing a pivotal role in these mechanisms. This analysis seeks to explore the specific pathways through which inflammatory EAT contributes to AF development and progression, potentially providing novel approaches for cardiovascular disease management and prevention.
EAT is a metabolically active fat compartment primarily composed of adipocytes, vascular elements, and immune cells [10]. Pathological changes in EAT have been strongly associated with several cardiovascular diseases, including AF [11], coronary artery disease (CAD) [12] and heart failure [13]. Table 1 (Ref. [11, 12, 13, 14]) lists important research results related to EAT. The thickening of EAT has been recognized as an independent risk factor for CAD, with evidence showing that EAT thickness correlates with the degree of coronary stenosis, particularly in individuals with obesity and metabolic syndrome [15, 16]. Additionally, in chronic heart failure, both inflammatory responses and dysregulated fat metabolism in EAT contribute to an increased cardiac burden. The pathological alterations in EAT, characterized by the dynamic increase in both adipocytes and inflammatory cells, can impair cardiac function, further exacerbating the symptoms of heart failure [17]. Elevated EAT levels are frequently linked to the onset of cardiovascular conditions such as CAD, heart failure, and disturbances in lipid metabolism [16, 17].
| Author (year) | Research topic | Study design | Sample size | Image/measurement method | Main finding | Evidence grade | Refs. |
| Mazurek et al. (2003) | “Human epicardial adipose tissue is a source of inflammatory mediators” | Laboratory investigation | 45 | Surgical sampling + biochemical analysis | EAT secretes pro-inflammatory cytokines (such as IL-6, TNF- |
2B | [14] |
| Thanassoulis et al. (2010) | “Pericardial fat is associated with prevalent atrial fibrillation” | Cross-sectional study | 3201 | CT | Each 1 standard deviation (SD) increase in pericardial fat volume was associated with a 28% higher risk of atrial fibrillation (OR = 1.28, p = 0.01), independent of BMI. | 2A | [11] |
| Mahabadi et al. (2013) | “Association of epicardial fat with cardiovascular risk factors and incident myocardial infarction” | Prospective cohort study | 4093 | CT was used to measure pericardial fat thickness | For every 1 SD increase in EFV, the risk of coronary artery calcification score (CACS) increased by 18% (p |
1B | [12] |
| Rabkin SW (2017) | “Is reduction in coronary blood flow the mechanism by which epicardial fat produces left ventricular diastolic dysfunction?” | Clinical cross-sectional study | 100–150 | Cardiac CT/MRI (volume) or ultrasound (thickness) | Through thickening, compression and secretion of vasoactive substances, EAT leads to decreased coronary microvascular blood flow reserve, which leads to limited left ventricular diastolic function. | 2A | [13] |
Abbreviations: EAT, epicardial adipose tissue; MRI,
magnetic resonance imaging; EFV, epicardial fat volume; Refs, references;
IL, interleukin; TNF-
In recent decades, there have been numerous studies on atrial fibrillation and EAT, and Table 2 (Ref. [18, 19, 20, 21, 22, 23, 24]) lists the key studies in this regard. The volume and thickness of EAT are strongly linked to the onset and severity of AF. Increased EAT thickness is a major risk factor for AF, as its excessive accumulation leads to atrial structural remodeling, which promotes AF. EAT contributes to AF development by secreting inflammatory cytokines, which can induce myocardial cell inflammation and apoptosis [25, 26]. Specifically, inflammatory mediators in EAT can increase the duration of myocardial action potential and resting membrane potential, destabilizing the heart’s electrical activity and enhancing AF susceptibility. Electrophysiological remodeling is a key mechanism in AF involving structural and functional changes in atrial cells. Research has shown that the distribution of high dominant frequency (DF) regions in AF patients correlates with EAT volume, suggesting that EAT contributes to AF through inflammation and electrical remodeling [27, 28]. Changes in DF reflect this remodeling process, particularly in persistent AF. High DF regions are often hotspots for electrophysiological remodeling, and their electrical activity characteristics can be identified using DF mapping techniques. The stability of DF is closely linked to AF persistence, with stable DF typically indicating stable atrial electrical activity sources [29]. Notably, the volume of EAT and levels of inflammatory biomarkers are higher in patients with persistent AF compared to those with paroxysmal AF, indicating that EAT may play different roles at various stages of AF [30].
| Author (year) | Research topic | Study design | Sample size | Image/measurement method | Main finding | Refs. |
| Wong et al. (2011) | Pericardial fat is associated with atrial fibrillation severity and ablation outcome | Prospective cohort study (case-control) | 102 AF patients and 20 controls | Cardiac MRI quantified EAT volume | EAT volume was independently correlated with the presence, severity, ablation and recurrence of AF. | [18] |
| Friedman et al. (2014) | Pericardial fat is associated with atrial conduction: the Framingham Heart Study | Cross-sectional study | 1946 cases | CT was used to measure pericardial fat thickness | EAT thickness was positively correlated with P-wave duration (p |
[19] |
| Nalliah et al. (2020) | Epicardial adipose tissue accumulation confers atrial conduction abnormality | Cross-sectional studies combined with in vitro cell experiments | 19 cases | CT+ electrophysiological mapping + histological analysis + cell experiment + proteomics | The volume of EpAT in the anterior wall of the right atrium was significantly correlated with slower conduction and increased electrical signal complexity, but the EAT in both atria had no such correlation. | [20] |
| Lin et al. (2012) | Adipocytes modulate the electrophysiology of atrial myocytes | Laboratory investigation | Not specified | In vitro electrophysiological recording + fluorescence microarray technique | The low levels of inflammatory cytokines in the supernatant of epicardial adipocytes cultured alone suggest that the interaction between adipocytes and cardiomyocytes may be realized through paracrine mechanism. | [21] |
| Shaihov-Teper et al. (2021) | Extracellular vesicles from epicardial fat facilitate atrial fibrillation | Prospective observational studies combined with in vitro experiments and animal model validation | Epicardial fat (eFat) samples were collected from 62 patients | Extracellular vesicles (EVs) isolation + electrophysiological experiment | The eFat in AF patients secreted more EVs, and was rich in pro-inflammatory (IL-1 |
[22] |
| Haemers et al. (2017) | Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria | Observational study combined with animal model experiments | 92 cases (human) + unspecified number (animal) | Histological analysis + imaging examination + inflammation analysis | The degree of subepicardial fat infiltration was higher in patients with permanent atrial fibrillation. AF sheep showed dense fiber-fat infiltration in the left atrium, consistent with human samples. | [23] |
| De Coster et al. (2018) | Arrhythmogenicity of fibro-fatty infiltrations | Computer simulation research | Not specified | Mathematical model | Adipose infiltration is more likely to induce arrhythmia than fibrosis alone (30% non-conductive tissue is required). | [24] |
TGF-
Recent studies have highlighted the significant association between EAT and both the onset and prognosis of AF. EAT enlargement may contribute to AF through multiple mechanisms. Specifically, adipocytes within EAT secrete pro-inflammatory cytokines, which lead to chronic inflammation in cardiac tissue, resulting in myocardial fibrosis and electrophysiological changes that promote both the initiation and persistence of AF [25, 30]. Moreover, free fatty acids released from EAT may adversely affect cardiac electrophysiology by increasing myocardial cell excitability, thereby triggering AF [26]. EAT’s metabolic activity is tightly connected to heart function, and metabolic abnormalities may predispose the heart to AF [31]. Several studies have shown that EAT volume is significantly associated with AF recurrence. For example, patients with EAT volumes greater than 92 cm3 who undergo catheter ablation are nearly twice as likely to experience AF recurrence compared to those with smaller EAT volumes [32]. Furthermore, the EAT volume surrounding the atria is especially linked to AF recurrence, with larger volumes of EAT decreasing the likelihood of maintaining sinus rhythm [33, 34]. These findings suggest that EAT plays a pivotal role in AF recurrence. EAT volume and its metabolic activity may serve as valuable prognostic markers for AF patients. Increased EAT volume has also been shown to correlate with a higher risk of cardiovascular events in AF patients [35, 36]. Additionally, EAT changes may predict treatment success; for example, a significant reduction in EAT volume following catheter ablation could indicate a favorable response to treatment and a lower risk of recurrence. Tracking EAT changes provides an opportunity for clinicians to adjust treatment plans based on individual patient needs, potentially improving treatment outcomes.
Inflammatory cytokines are pivotal in initiating atrial electrophysiological and
structural changes predisposing individuals to AF [37, 38]. Serum levels of
inflammatory markers, including C-reactive protein (CRP), tumor necrosis
factor-
Atrial remodeling is a fundamental mechanism underlying AF, involving
electrophysiological disturbances and structural alterations in the atria.
Inflammatory mediators activate atrial fibroblasts, promoting fibrosis. Atrial
fibrosis further impairs atrial electrical conduction, creating a vicious cycle
that increases the risk of AF [51, 52]. Myocardial fibrosis, characterized by
increased collagen deposition in the myocardial interstitium, leads to
significant structural and functional changes. This fibrotic process is closely
regulated by pro-fibrotic factors such as transforming growth factor-beta
(TGF-
EAT exhibits complex interactions between its neuroendocrine, immune, and inflammatory responses, which influence arrhythmia development through multiple mechanisms. These interactions influence arrhythmogenesis through multiple pathways, including changes in cardiac electrophysiological properties, chronic myocardial damage mediated by inflammation, and neuroregulation. Bioactive substances secreted by EAT, such as leptin, directly affect myocardial cell membrane potentials and action potential duration, which increases arrhythmia risk [30, 59]. Chronic inflammation in EAT raises cytokine levels, inducing myocardial apoptosis and fibrosis, destabilizing cardiac electrical activity, and promoting arrhythmias. Furthermore, the neuroendocrine function of EAT affects autonomic nervous system balance, with sympathetic activation and parasympathetic inhibition leading to irregular heart rate and increased arrhythmia risk. Metabolic dysfunction, often seen in obesity, disrupts EAT function, exacerbating inflammation and perpetuating a cycle that increases arrhythmia risk. The interplay between the neuroendocrine, immune, and inflammatory functions of EAT forms a complex network that collectively impacts heart health. Further investigation into these mechanisms will provide essential insights for developing novel therapies for arrhythmias and offer new perspectives in managing cardiovascular diseases.
Inflammatory cytokines play a central role in driving atrial
electrophysiological and structural remodeling, which predisposes individuals to
AF [37, 38]. Elevated serum levels of inflammatory markers, including CRP,
TNF-
Current research emphasizes the assessment of inflammatory markers in EAT, a metabolically active fat depot surrounding the heart. Techniques such as tissue biopsy, gene expression profiling, and biochemical assays enable the evaluation of EAT’s cellular composition and inflammatory mediator expression (e.g., RNA analysis of pro-inflammatory genes) [50]. Targeting these inflammatory pathways may offer novel therapeutic strategies to mitigate AF progression and improve cardiovascular outcomes.
Atrial remodeling—encompassing electrophysiological dysfunction and structural
fibrosis—is a hallmark of AF pathophysiology. Inflammatory mediators activate
atrial fibroblasts, triggering collagen deposition and interstitial fibrosis.
This fibrotic process disrupts electrical signal propagation, creating a
self-perpetuating cycle that sustains AF [51, 52]. Key pro-fibrotic factors such
as TGF-
EAT functions as a neuroendocrine-immune organ, with its inflammatory activity intricately linked to arrhythmogenesis. Bioactive molecules secreted by EAT, including leptin, directly influence cardiomyocyte membrane potentials and action potential duration, increasing susceptibility to arrhythmias [30, 59]. Chronic inflammation within EAT elevates systemic cytokine levels, promoting myocardial apoptosis, fibrosis, and electrical instability.
EAT also interacts with the autonomic nervous system (ANS): sympathetic hyperactivity and parasympathetic suppression—mediated by EAT-derived neuroendocrine factors—predispose to heart rate variability and arrhythmia. In obesity-related metabolic dysfunction, EAT expansion exacerbates inflammation, creating a vicious cycle that amplifies arrhythmia risk. This triad of neuroendocrine, immune, and inflammatory mechanisms in EAT underscores its role as a critical modulator of cardiac electrophysiology and structural integrity.
The synergistic effects of inflammation, atrial remodeling, and EAT-mediated pathways form a complex network driving AF pathogenesis. Elucidating these interactions—particularly the cross-talk between inflammatory markers, fibrotic pathways, and neuroendocrine-immune regulation—holds significant translational potential. Future therapies targeting EAT inflammation or its downstream mediators may improve AF management and reduce cardiovascular morbidity.
Inflammatory signaling pathways are key mechanisms by which EAT contributes to
the development of AF (Fig. 1). EAT secretes several pro-inflammatory cytokines,
including TNF-
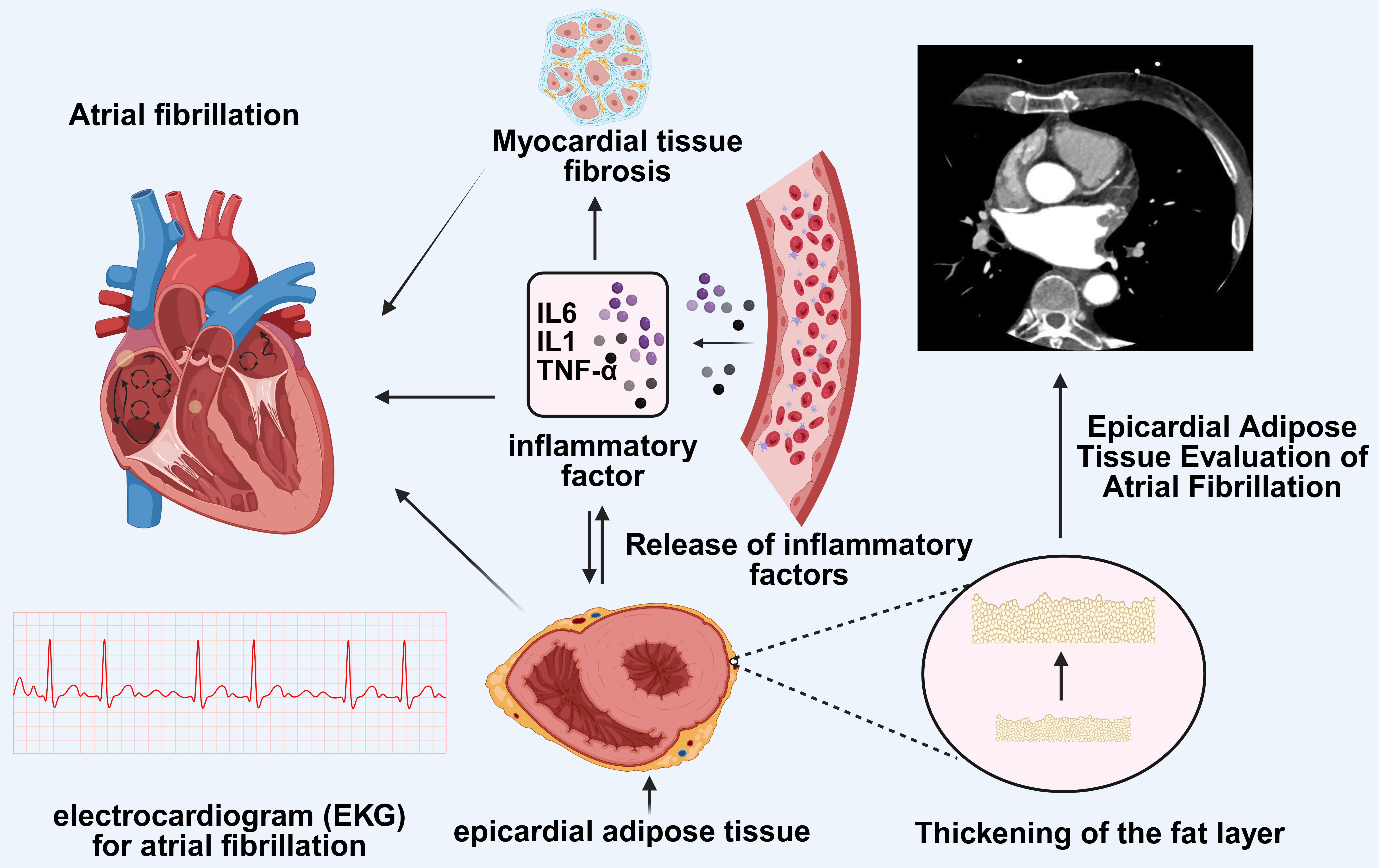 Fig. 1.
Fig. 1.
Imaging features of EAT and the impact of inflammation in EAT on
atrial fibrillation. AF is a common arrhythmia associated with multiple factors.
The figure depicts the role of EAT in AF. The imaging features of EAT can be
evaluated using computed tomography or cardiac magnetic resonance imaging, which
reveals the thickening of the adipose layer. EAT releases inflammatory cytokines
such as IL-6, IL-1, and TNF-
The Janus tyrosine kinase (JAK)-signal transducer and activator of the
transcription 3 (STAT3) signaling pathway plays a pivotal role in the heart’s
inflammatory response, particularly through the action of IL-6, which activates
JAK-STAT3 and triggers an inflammatory cascade that downregulates gap junction
proteins in cardiac tissue. This process leads to electrical remodeling of the
atrium, contributing to the onset of AF [83, 84]. Moreover, the activation of the
Smad signaling pathway is closely linked to both inflammation and fibrosis in
cardiac pathology. Upregulation of inflammatory processes commonly accompanies
fibrosis in the heart, and excessive activation of the TGF-
Inflammatory signaling pathways play a central role in the contribution of EAT
to the development of AF [60] (Fig. 1) (Table 3, Ref. [30, 59, 60, 64, 65, 66, 67, 69, 70, 71, 72, 73, 74, 75, 76, 80, 81, 82, 83, 84, 85, 86, 87]). Among these, the JAK-STAT3, TGF-
| Signaling pathways/routes | Involved factors/molecules | Mechanisms of action | Effects | Refs. |
| Inflammatory response pathway | TNF- |
Endocrine and paracrine actions trigger myocardial inflammation | Affect the electrical activity of atrial myocytes, resulting in irregular heart rhythm; accelerate arteriosclerosis and increase the risk of atrial fibrillation. | [60] |
| Atrial fibrosis pathway | TGF- |
Promote atrial collagen deposition and fibrosis | Myocardial extracellular matrix remodeling; collagen deposition and atrial structural changes. | [64, 65, 66, 67] |
| Oxidative stress pathway | ROS and CaMKII | Enhance kinase activity, oxidize ion channels | The formation of a higher oxidative stress state increases the production of ROS and reduces the level of antioxidants to aggravate the oxidative damage of the atrium. | [69, 70, 71] |
| Lipid infiltration pathway | Free fatty acids | Cause electrophysiological changes, myocardial structural disorder | The conduction of cardiomyocytes is slowed down, the lateral cell connection is lost and the myocardial structure is disturbed, causing conduction delay and reentry. | [30, 72, 73] |
| Autonomic nervous pathway | Adrenergic and cholinergic | Change cell membrane potential, cause electrophysiological change | The activity of the sympathetic nerve affects the electrical conductivity of the heart and the metabolism of the outer adipose tissue, which aggravates atrial fibrillation, while the activation of the parasympathetic nerve can help maintain the stability of the heart and reduce the occurrence of atrial fibrillation. | [74, 75, 76] |
| RAAS pathway | Ang II, TGF- |
Cause myocardial fibrosis, upregulate gene expression | By activating RAAS to produce high levels of Ang II, EAT stimulates fibroblast proliferation and collagen synthesis, leading to remodeling and fibrosis of myocardial tissue. | [80, 81] |
| NF-κB signaling pathway | NF-κB (p65, p50), TNF- |
Mediate NF-κB translocation, upregulate inflammatory factor expression | Overactivation of NF-κB in EAT may exacerbate the electrophysiological instability of the heart, thereby increasing the risk of AF. | [59, 82, 85, 86, 87] |
| JAK-STAT3 pathway | JAK, STAT3, IL-6, TGF- |
Trigger inflammatory cascade, downregulate gap junction proteins | IL-6 produced by EAT triggers an inflammatory cascade, down-regulates cardiac gap connexin, leads to atrial electrical remodeling, and induces AF. | [83, 84, 86, 87] |
| Smad signaling pathway | TGF- |
Smad signaling pathway | If the TGF- |
[85, 86, 87] |
RAAS, renin-angiotensin-aldosterone system; MMP, matrix metalloproteinase; ROS,
reactive oxygen species; CaMKII, Ca2+/calmodulin-dependent protein
kinase-II; Ang II, angiotensin II; NF-
In addition to these pathways, individual cytokines and mediators secreted by
EAT play distinct roles in AF pathogenesis. TNF-
The interplay between these signaling pathways and cytokines creates a vicious
cycle of inflammation and fibrosis in EAT, which drives atrial remodeling and
increases the risk of AF [86, 87]. Targeting these pathways, such as inhibiting
NF-
The metabolic alterations in EAT during inflammatory states are complex, involving changes in adipocyte function, immune cell infiltration, and the activation of various metabolic signaling pathways. Chronic low-grade inflammation disrupts the metabolic function of EAT, which in turn increases the risk of conditions such as insulin resistance and metabolic syndrome. In the context of AF-associated inflammation, EAT is a specialized form of fat storage that influences cardiac health and disease. Particularly, inflammatory changes in EAT are critical in modulating cardiac function. Under physiological conditions, EAT helps regulate the toxic concentration of fatty acids between the myocardium and the surrounding vasculature while secreting anti-inflammatory and anti-fibrotic cytokines to exert protective effects [68]. However, during pathological inflammation, the quantity and diversity of inflammatory cells in EAT increase, which reflects localized inflammatory responses [69]. Inflammation within the heart is a well-established contributor to arrhythmogenesis, and the thickening of EAT can alter the electrophysiological properties of the heart and promote structural remodeling, thereby increasing the risk of arrhythmias such as AF [70, 71].
EAT normally contains a controlled number of inflammatory cells that contribute
to immune homeostasis and tissue repair. In inflammatory conditions, however,
there is a significant increase in the infiltration of inflammatory cells,
particularly macrophages and T lymphocytes. This accumulation exacerbates local
inflammatory responses and releases pro-inflammatory cytokines such as
TNF-
Adipocytes in EAT undergo a series of morphological alterations under
inflammatory conditions, which not only affect their size and shape. Still, they
may also have far-reaching consequences on their function, thereby influencing
metabolic health. Adipocyte hypertrophy, characterized by increased cell size due
to lipid accumulation, is a common feature of EAT during chronic inflammation,
especially in obese individuals. In obesity, hypertrophy leads to the
accumulation of lipid droplets within adipocytes, which alters their shape and
structure [90]. Moreover, inflammation may lead to changes in adipocyte numbers,
often through hyperplasia, the formation of new adipocytes from mesenchymal stem
cells (MSCs). Certain signaling pathways are activated during inflammation,
promoting this transformation and forming more mature adipocytes [90].
Interestingly, adipocytes in inflammatory environments may exhibit irregular
shapes, departing from the typical round or oval morphology. These morphological
alterations are closely associated with uneven lipid distribution, cell membrane
changes, and cytoskeleton reorganisation, indicating adipocyte adaptive responses
to inflammatory signals [91]. Multiple signaling pathways within adipocytes are
reactivated during inflammation, leading to these morphological changes. For
instance, pro-inflammatory cytokines such as TNF-
EAT undergoes profound changes in its cytokine secretion profile during
inflammatory states, a phenomenon that plays a key role in the pathogenesis of
cardiovascular diseases. In particular, anti-inflammatory cytokines such as
adiponectin are downregulated, while pro-inflammatory cytokines, including CRP,
are upregulated. This disruption in the balance of cytokine secretion is
considered a significant factor in the development and progression of
cardiovascular pathologies [59, 94]. Compared to subcutaneous adipose tissue, EAT
exhibits a markedly higher expression of pro-inflammatory cytokines like IL-6 and
TNF-
The inflammatory biomarkers in EAT suggest it could be a valuable biomarker for AF. By measuring the levels of inflammatory cytokines and other markers in EAT, it is possible to assess both the risk and prognosis of AF. Additionally, EAT thickness and its inflammatory condition may offer new perspectives for personalized treatment strategies. According to research by Korantzopoulos et al. [52], elevated inflammatory markers in EAT are closely linked to the development and recurrence of AF, with more pronounced localized inflammation observed within the atrium. By monitoring changes in these inflammatory markers, clinicians could better predict patient outcomes in AF. Targeting EAT inflammation through interventions, such as anti-inflammatory drugs, may help lower AF incidence [98]. Despite existing studies providing initial evidence, additional clinical and basic research is needed to validate these findings further. Continued investigation may lead to the development of novel therapeutic approaches that not only reduce the burden of AF but also enhance patients’ quality of life.
EAT demonstrates unique clinical value in the early identification and prognostic management of AF. Its core strength lies in providing dynamic information on cardiac metabolic status through non-invasive methods, offering objective evidence for clinical decision-making. Imaging techniques such as cardiac computed tomography (CT) enable precise quantification of EAT volume and spatial distribution. By establishing EAT volume thresholds, clinicians can identify high-risk populations, particularly in obese or metabolic syndrome patients, where this structural biomarker complements traditional risk assessment tools. Dynamic changes in EAT serve as a biological indicator of therapeutic response. For example, reduced EAT volume post-catheter ablation is associated with lower AF recurrence rates [30], while decreased EAT-derived inflammatory markers (e.g., IL-6) following lifestyle interventions may reflect improved cardiometabolic health [56, 99]. Furthermore, persistent EAT proliferation is closely linked to long-term AF recurrence risk [96, 99], providing a basis for personalized follow-up strategies. EAT exerts both local and systemic effects: its secreted inflammatory mediators directly promote atrial electrical remodeling and indirectly exacerbate AF risk by aggravating systemic pathologies such as hypertension and insulin resistance [100]. This dual role positions EAT as a critical nexus between arrhythmias and metabolic cardiovascular diseases, enhancing its utility in comprehensive risk stratification models.
Applying inflammation-targeted therapies in cardiovascular diseases has made
remarkable strides in recent years. Biologic agents, particularly those
inhibiting IL-1
Current randomized controlled trials (RCTs) are actively investigating the efficacy of anti-inflammatory drugs in AF patients. For example, colchicine, while early studies indicated potential protective effects against postoperative AF, recent clinical trials have yielded conflicting results. The COP-AF trial demonstrated that colchicine failed to significantly reduce AF incidence after non-cardiac thoracic surgery [111], and the IMPROVE-PVI trial did not confirm its efficacy in decreasing AF recurrence post-pulmonary vein isolation [110]. Collectively, these outcomes highlight that monotherapy with non-specific anti-inflammatory agents may be insufficient to markedly improve AF outcomes, whereas targeted interventions against specific inflammatory pathways (e.g., IL-1, IL-6, or IL-17) may hold greater therapeutic promise. Building on these insights, future research may explore combined anti-inflammatory strategies to optimize therapeutic efficacy. For example, combining cytokine-targeted therapies (e.g., IL-1 inhibitors) with non-specific anti-inflammatory agents (e.g., statins) could synergistically inhibit inflammatory cascades through multi-pathway mechanisms, thereby reducing AF recurrence risk.
EAT disrupts cardiac electrophysiology by promoting arrhythmia through electrotonic coupling between resident stem cells and cardiomyocytes [112]. Conversely, therapeutic stem cells can mitigate this risk by normalizing electrical coupling between cells, thereby reducing EAT-related conduction delays. In addition to electrical regulation, stem cells may play a paracrine role—secreting bioactive factors that improve the pathological microenvironment of EAT and indirectly inhibit its arrhythmic activity [113]. Globally, multiple clinical trials are evaluating the safety and therapeutic efficacy of human pluripotent stem cells (hPSCs) in arrhythmia management, with a primary focus on their cardiomyogenic differentiation potential and functional cardiac repair capabilities [114]. Parallel investigative efforts are exploring combined gene-editing and stem cell therapies, where genetic modifications (e.g., enhanced gap junction protein expression) are employed to augment the antiarrhythmic properties of transplanted cells [115]. While preliminary outcomes appear promising, widespread clinical adoption of these approaches remains limited by scalability and long-term safety considerations. Notably, emerging trials target the inflammatory microenvironment of EAT, investigating stem cell-mediated immunomodulation of CD8+ T cells and other immune populations to attenuate arrhythmogenic triggers [116]. Stem cell therapy has shown great potential in inhibiting the arrhythmic effects of EAT and may provide a new strategy for the treatment of arrhythmia. However, research is still in its early stages, and more clinical trials and mechanistic studies are needed to verify its safety and efficacy.
The relationship between EAT and AF is intricate and multifaceted. EAT contributes to the onset of AF and influences its prognosis. Thus, a detailed assessment of EAT can offer valuable information for managing AF patients. This review highlights the role of EAT in AF through inflammatory, metabolic, and electrophysiological mechanisms. We propose that routine EAT evaluation be integrated into clinical practice for AF patients to support personalized treatment plans and assess the impact of interventions aimed at reducing EAT volume on AF outcomes. While existing research has elucidated the connection between EAT and AF, further prospective studies are required to validate EAT as a biomarker and to assess its clinical applicability in AF management. These studies will contribute to a more robust mechanistic model to guide clinical decisions.
HG and MB designed the research study. JL, MZ, and LB all contributed to writing the article. JL was responsible for creating the tables; MZ handled the overall content revision and polishing of the article; LB was in charge of creating Fig. 1. JL, MZ, LB, JZ, HG, and MB participated in reviewing and editing of the article. All authors contributed to the conception and editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This review was conducted following the Declaration of Helsinki and approved by the Ethics Committee of LZU No. 1 Hospital, China (study number LDYYLL-2024-784).
Not applicable.
This work was supported by grants from the Science and Technology Planning Project of Lanzhou City (NO.2021RCCX0009) and the Special Fund for Civil-Military Integration Development of Gansu Province (NO.2060303).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.