


 , Mohammad Hamza 2,†, Aslam Malik 3
, Mohammad Hamza 2,†, Aslam Malik 31 Division of Cardiology, Department of Medicine, Johns Hopkins University, Baltimore, MD 21218, USA
2 Department of Internal Medicine, TidalHealth, MD 21801, USA
3 Department of Internal Medicine, St. Luke's Healthcare, Sellersville, PA 18960, USA
†These authors contributed equally.
Abstract
Transthyretin cardiac amyloidosis (ATTR-CA) is an increasingly recognized and underdiagnosed cause of heart failure (HF), encompassing both preserved (HFpEF) and reduced (HFrEF) ejection fraction phenotypes. Once identifiable only following a biopsy, the advent of bone scintigraphy has dramatically improved noninvasive detection and detected a higher community prevalence, particularly among older patients with unexplained left ventricular hypertrophy. ATTR-CA arises from misfolding of transthyretin (TTR), leading to amyloid fibril deposition within the myocardium, which impairs cardiac compliance, conduction, and output. This review explores the evolving epidemiology of ATTR-CA in HF, mechanisms of disease progression, and key features for screening, emphasizing clinical red flags, biomarkers, and imaging features. This review also addresses the nuanced role of guideline-directed medical therapy in this population, where neurohormonal agents may offer limited benefit or be poorly tolerated due to restrictive physiology and autonomic dysfunction. Crucially, the emergence of amyloid-specific therapies, including TTR silencers, stabilizers, and degraders, has transformed the therapeutic landscape, offering mortality and morbidity benefits that were previously unavailable. Early diagnosis and individualized management, integrating conventional and amyloid-targeted approaches, are essential to improving outcomes in this complex and increasingly treatable cardiomyopathy.
Keywords
- transthyretin cardiac amyloidosis
- heart failure
- heart failure with reduced ejection fraction
- guideline-directed medical therapies
- amyloid-specific therapies
Transthyretin cardiac amyloidosis (ATTR-CA) has increasingly been recognized as an important etiology of heart failure (HF), driven in part by the advent of bone-avid, technetium-labeled pyrophosphate tracers specific to ATTR-CA [1]. These imaging agents have revealed an unexpectedly high community prevalence of a disease that was previously diagnosed only through endomyocardial biopsy [2].
Once thought to be associated almost exclusively with HF with preserved ejection fraction (HFpEF), recent data demonstrate that a substantial proportion of patients with ATTR-CA present with HF with reduced ejection fraction (HFrEF) at the time of diagnosis [3]. As a clinical mimic of HFpEF, timely diagnosis of ATTR-CA is critical—particularly given the emergence of amyloid-specific therapeutics that offer morbidity and mortality benefits in this unique patient population.
This review will explore the evolving epidemiologic paradigm of ATTR-CA in HF, the pathophysiologic mechanisms leading to both HFpEF and HFrEF phenotypes, and the current evidence on the use of guideline-directed medical therapies (GDMT) in patients with ATTR-CA, including mechanistic explanations for their limited tolerability in this setting. We will also examine the data for amyloid-specific therapies, focusing on strategies to reduce transthyretin production, stabilize the native protein, and explore the ongoing efforts to promote resorption of existing amyloid fibrils.
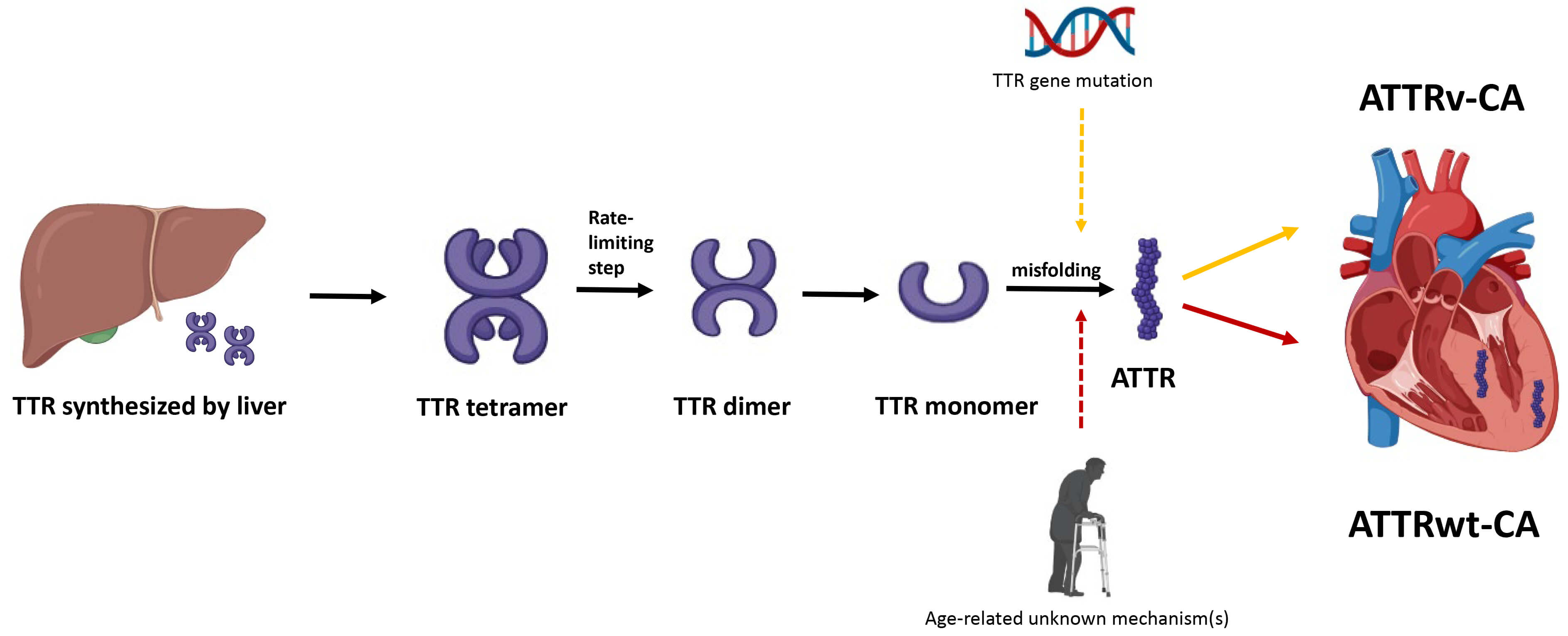
The pathogenesis of ATTR-CA centers on the destabilization and misfolding of the transthyretin (TTR) protein, a tetramer primarily synthesized in the liver that transports thyroxine and retinol (vitamin A) in the plasma (Fig. 1). ATTR-CA is divided into 2 subtypes. Hereditary or variant ATTR-CA (ATTRv-CA) arises from mutations in the TTR gene, with over 130 mutations identified to date, which weaken the tetramer’s structural integrity. In wild-type ATTR-CA (ATTRwt-CA), age-related biochemical changes—though not fully understood—similarly reduce tetramer stability. The tetramer dissociates into monomer, which subsequently misfolds into amyloid fibrils. These fibrils deposit in tissues, including the heart, leading to progressive organ dysfunction. The degree of cardiac involvement at the time of diagnosis is the most important prognostic indicator.
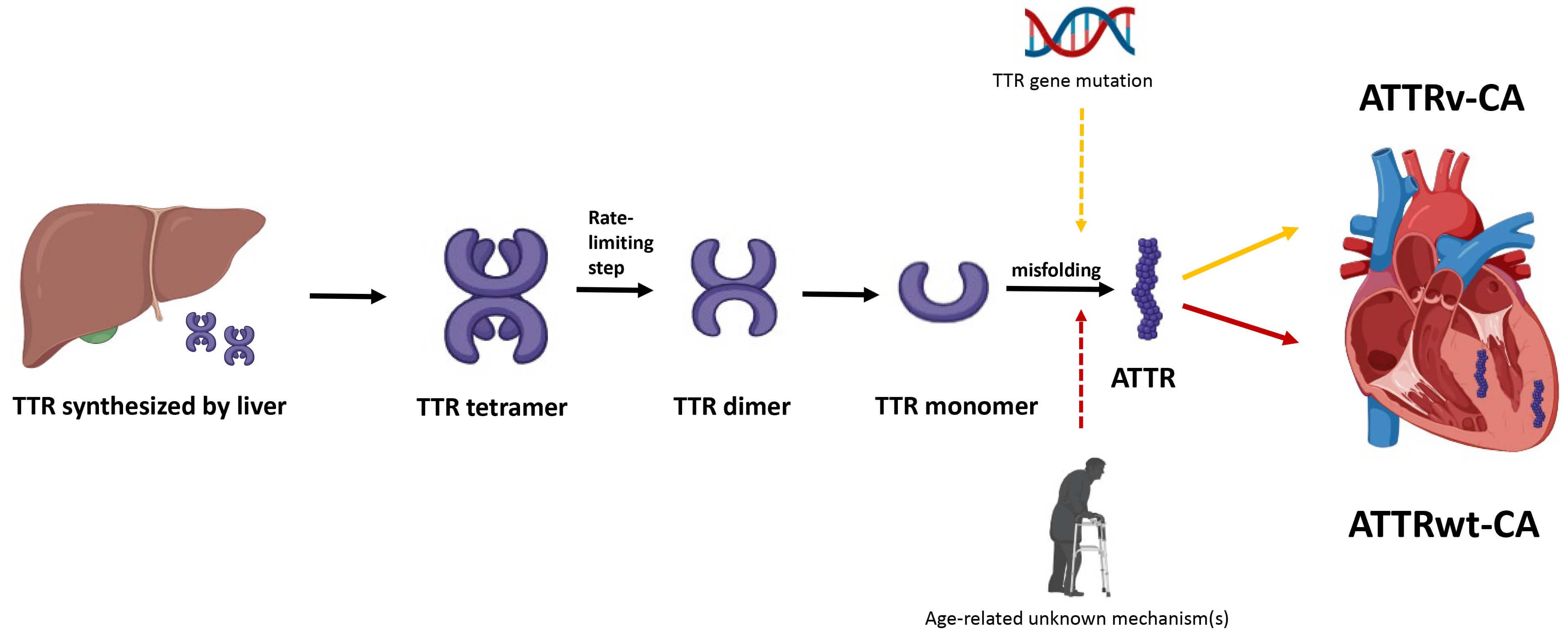 Fig. 1.
Fig. 1.
Pathogenesis of transthyretin cardiac amyloidosis. Hepatically-derived TTR undergoes misfolding, either due to mutation in TTR gene or secondary to age-related unknown mechanisms, and the resulting ATTR is subsequently deposited in the myocardial interstitium. TTR, transthyretin; ATTR, transthyretin amyloid fibril; ATTRv-CA, variant or hereditary transthyretin cardiac amyloidosis; ATTRwt-CA, wild-type transthyretin cardiac amyloidosis.
ATTR-CA is now increasingly recognized as a significant and underdiagnosed contributor to HFpEF, though its true prevalence remains elusive. Estimates range widely—from as low as 3% to as high as 20% —largely due to methodological variability across studies [4, 5, 6, 7, 8]. These differences include patient selection criteria, age cutoffs, left ventricular (LV) wall thickness thresholds, and the degree of diagnostic scrutiny.
A recent Spanish study comprising 387 older individuals with HFpEF and LV
hypertrophy (LVH, defined as wall thickness
The prevalence of ATTRwt-CA and ATTRv-CA is also markedly different. ATTRwt-CA is a predominant subtype in elderly patients with HF. In a prospective study evaluating 120 patients with HFpEF, the prevalence of ATTRwt-CA, defined as positive nuclear scintigraphy and absence of TTR gene mutations, was found to be 13% [9]. On the other hand, ATTRv-CA is seen among certain mutations that have predilection to cardiac involvement. ATTRv-CA is a progressive disease affecting multiple systems, with clinical manifestations that vary from mainly polyneuropathy to predominant cardiomyopathy [10]. One of the important variants is V122I ATTRv-CA, which is present almost exclusively in Afro-Caribbean population in the United States, and predominantly involves cardiomyopathy [11]. Another mutation is T60A ATTRv-CA, which is predominantly seen in individuals of Irish descent [12]. According to the Irish Amyloidosis Network, the clinical presentation of T60A ATTRv-CA in Ireland usually involves individuals developing symptoms in their seventies, with neuropathy emerging as the initial and predominant feature, preceding the development of HF–driven cardiac involvement [13].
To summarize, determining the true prevalence of ATTR-CA in HFpEF remains challenging. Most studies focus on patients with clear phenotypic signs, often missing early disease in those without left ventricular thickening or typical red flags. As a result, the overall burden is likely underestimated, and opportunities for early, disease-modifying treatment are missed. Broader, systematic screening using sensitive tools—such as bone scintigraphy, advanced imaging, and circulating biomarkers—is essential to detect subclinical cases, better understand disease progression, and enable timely intervention.
ATTR-CA, once considered a condition confined to HFpEF, is now increasingly recognized in patients with HFrEF. Approximately one-third of patients present with HFrEF at the time of ATTR-CA diagnosis, reflecting ‘burnt-out’ or advanced disease and portending a worse prognosis compared with those who have HFpEF [14]. However, the prevalence of ATTR-CA exclusively among patients with HFrEF remains underexplored. In a meta-analysis of 11 studies including 3303 patients with HF, the pooled prevalence of ATTR-CA in HFrEF was estimated at 11.3%, derived from only two studies, compared with pooled prevalence of 15% in HFpEF [15]. In a separate study of 75 patients with unexplained HF and systolic LV dysfunction, the prevalence of wild-type ATTR-CA was approximately 9% [16]. These findings underscore the need for larger, dedicated studies to more accurately define the burden of ATTR-CA in HFrEF, which in turn will help refine community-based prevalence estimates of ATTR-CA.
ATTR-CA is characterized by the extracellular deposition of ATTR as insoluble amyloid fibrils within the myocardium, leading to progressive myocardial stiffening [17]. The loss of myocardial compliance increases LV filling pressures and impairs ventricular relaxation, a hallmark feature of early-stage disease. As the disease progresses, amyloid accumulation also affects the conduction system, coronary microvasculature, and valves, exacerbating cardiac dysfunction. Amyloid deposition in the atria and conduction pathways can lead to arrhythmias such as atrial fibrillation [18] and various degrees of heart block [19]. Microvascular dysfunction due to amyloid infiltration impairs coronary perfusion, contributing to subendocardial ischemia and myocyte death despite unobstructed epicardial coronary arteries. The progressive myocardial infiltration increases wall thickness without true hypertrophy, leading to a pseudo-hypertrophic appearance and further compromising cardiac output. In later stages, systolic function may deteriorate, resulting in overt HFrEF.
Furthermore, the pathophysiological cascade in ATTR-CA involves neurohormonal activation secondary to decreased cardiac output. This compensatory mechanism—driven by activation of the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system—exacerbates fluid retention, vasoconstriction, and ventricular remodeling. Combined with the fixed stroke volume imposed by the stiffened ventricle, these processes culminate in the classic clinical syndrome of HFpEF, progressing in many cases to advanced, treatment-refractory HF. The infiltrative process is particularly insidious in ATTRwt-CA, which typically presents in older individuals with subtle symptoms and is often misdiagnosed as hypertensive heart disease or hypertrophic cardiomyopathy (HCM).
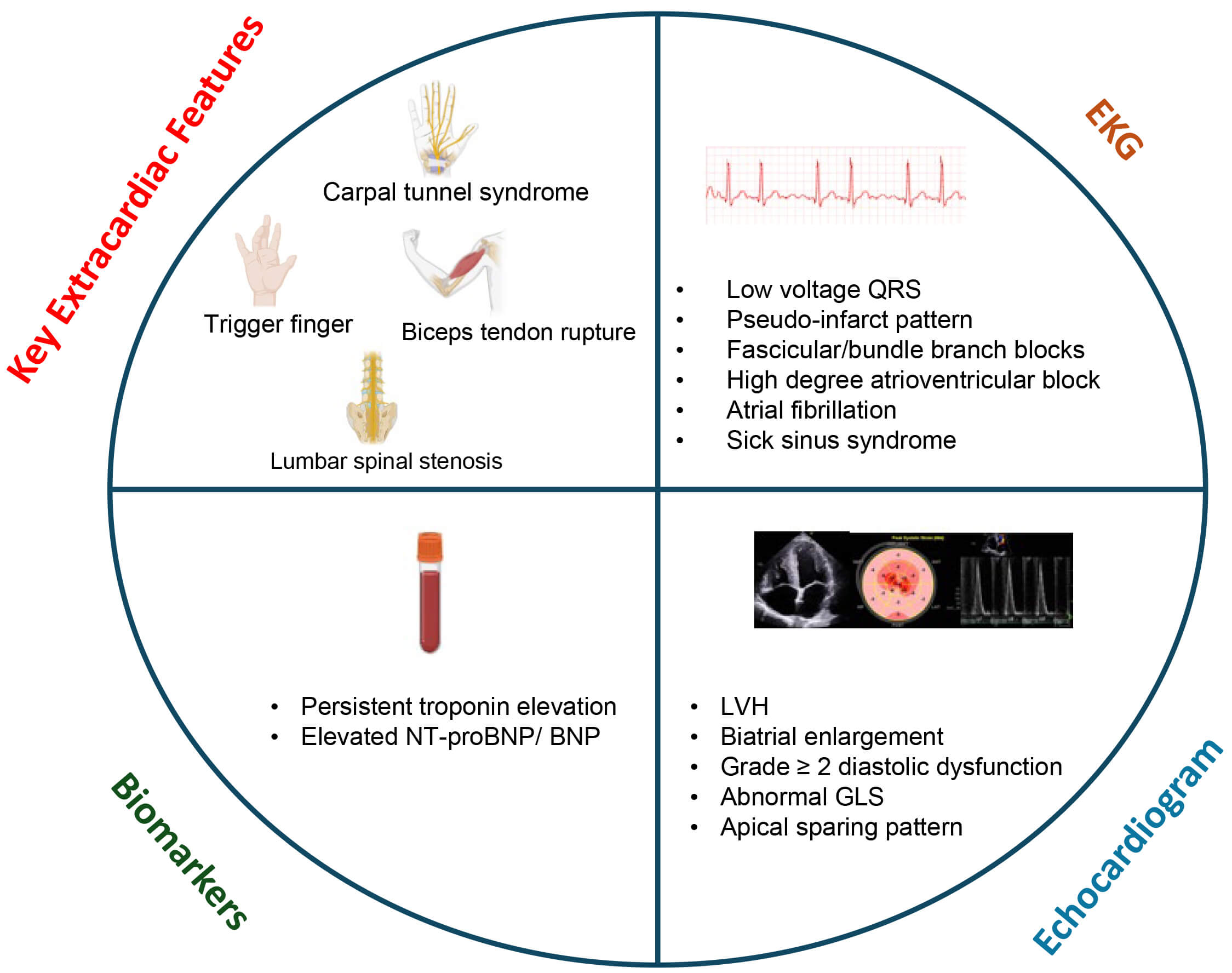
While ATTR-CA and HF, particularly HFpEF, have some demographic, clinical and
echocardiographic commonalities, there are certain clues on clinical history,
laboratory workup, electrocardiogram (EKG) and echocardiography that can raise
initial suspicion for ATTR-CA [20] (Fig. 2). Patients with ATTR-CA, more
specifically ATTRwt-CA, often have extracardiac features that are predominantly
musculoskeletal, resulting from ATTR deposition in the ligaments and tendons.
Carpal tunnel syndrome is probably the most common musculoskeletal manifestation
that has a prevalence of up to 50% in patients with ATTR-CA and is known to
precede the onset of cardiac dysfunction by 5–10 years in patients with ATTR-CA
[21]. Roughly 10% of patients undergoing idiopathic carpal tunnel release
surgery have biopsy-confirmed amyloid deposits in the tenosynovial sheath [22].
Trigger finger, which involves the same tendon sheath as carpal tunnel syndrome,
is also commonly reported in patients with ATTR-CA, and biopsy during trigger
finger release surgery has demonstrated a 2% yield for ATTR-CA [23]. Spontaneous
biceps tendon rupture is another musculoskeletal manifestation of ATTR-CA, and a
single center reported prevalence of 33% in patients with ATTRwt-CA [24].
Finally, about one-third of patients aged
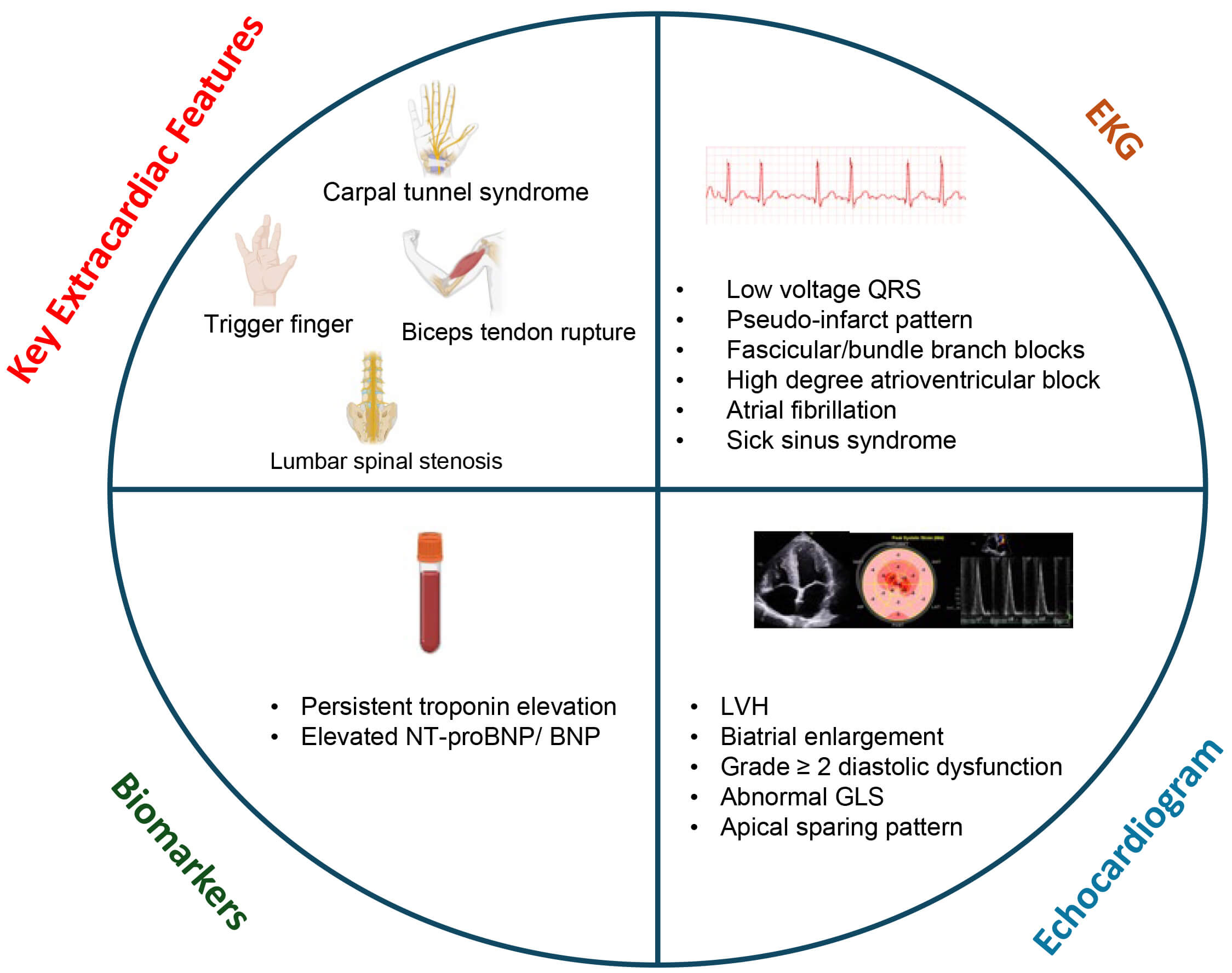 Fig. 2.
Fig. 2.
Screening transthyretin cardiac amyloidosis in heart failure. These red flag features on clinical exam, laboratory evaluation, EKG and echocardiography may raise suspicion for cardiac amyloidosis. GLS, global longitudinal strain; LVH, left ventricular hypertrophy; EKG, electrocardiogram; NT-proBNP, N-terminal pro-brain natriuretic peptide.
Cardiac troponin is the preferred biomarker for identifying myocardial injury
and is commonly elevated in patients with ATTR-CA. Numerous mechanisms have been
suggested to explain myocardial injury in ATTR-CA, including direct cytotoxicity
of amyloid precursors, interstitial infiltration of amyloid fibrils, coronary
microvascular dysfunction, concomitant coronary artery disease, diastolic
Persistent elevation of cardiac troponin levels may have prognostic value for ATTR-CA. Troponin is used to risk-stratify patients and predict mortality in both ATTRwt-CA and ATTRv-CA. Higher baseline troponin levels are associated with worse outcomes. Along with NT-proBNP, troponin forms the basis for Mayo staging system that predicts survival [28]. However, there are certain limitations with the use of troponin as a prognostic tool in HF. The lack of standardization in measuring troponin levels remains a challenge, as various generations of assays from different manufacturers have introduced variability, leading individual centers to favor specific assays [29]. Moreover, the relationship between variations in absolute troponin levels and corresponding changes in disease progression or status has not been established. Expert consensus suggests a 30% relative increase, using a high-sensitivity assay, as a better indication of ATTR-CA progression rather than a pre-specified absolute level, but this needs to be examined using robust data [30]. Finally, troponin levels are also influenced by kidney function due to impaired excretion [31], and since patients with ATTR-CA have varying degrees of kidney dysfunction, a cautious interpretation of variation in troponin levels is warranted.
NT-proBNP is the gold standard biomarker in HF used in routine clinical practice
[32]. While NT-proBNP does not provide diagnostic utility specific to ATTR-CA, it
is an important biomarker for prognostication. NT-proBNP levels are important
markers for tracking disease progression, and have been incorporated in various
amyloidosis staging systems [28]. In a study of 869 UK patients with ATTR-CA
(553 ATTRwt-CA, 316 ATTRv-CA), a three-stage system based on NT-proBNP and
estimated glomerular filtration rate (eGFR) thresholds (Stage I: NT-proBNP
Low voltage on EKG is a key finding in ATTR-CA and, while not specific, its presence in the setting of marked LVH should raise suspicion for the disease. Unlike true hypertrophy, ATTR-CA involves pseudo-LVH due to non-conducting amyloid fibril deposition and extracellular expansion, leading to disproportionately low QRS voltages on EKG despite increased myocardial wall thickness [35, 36]. In addition, ATTR-CA can be characterized by a ‘pseudo-infarct’ pattern, which refers to the presence of Q waves or QS complexes on the EKG that mimic previous myocardial infarction, typically in the anterior or inferior leads, but without corresponding evidence of coronary artery disease [37]. This is reflective of delayed conduction in amyloid-infiltrated myocardial tissue rather than ischemic injury, and can have diagnostic utility when seen together with low-voltage QRS and LVH.
EKG changes may also help in the assessment of disease progression. The development of advanced atrioventricular (AV) block and PR interval prolongation could signify disease progression, and could appear in isolation or together with bundle branch block patterns. Furthermore, the need for pacemaker implantation for bradyarrythmias (atrial fibrillation with slowed ventricular response, sick sinus syndrome and high-degree AV block) is also suggestive of progressive amyloid burden.
ATTR-CA and garden-variety HFpEF have some overlapping features on
echocardiogram. While LVH can be seen in non-amyloid HFpEF, LV thickening in
ATTR-CA is significantly greater [4, 38], and is often symmetric [39]. However,
it is important to note that the absence of LVH does not rule out ATTR-CA, as
upto 10% of ATTR-CA patients may have normal wall thickness, particularly in the
early stages of the disease [40]. Atrial enlargement, often bilateral, is a
common feature in ATTR-CA together with near-normal sized ventricles, which
represents increased LV preload secondary to increased myocardial stiffness—a
hallmark of restrictive physiology. Advanced diastolic dysfunction (grade
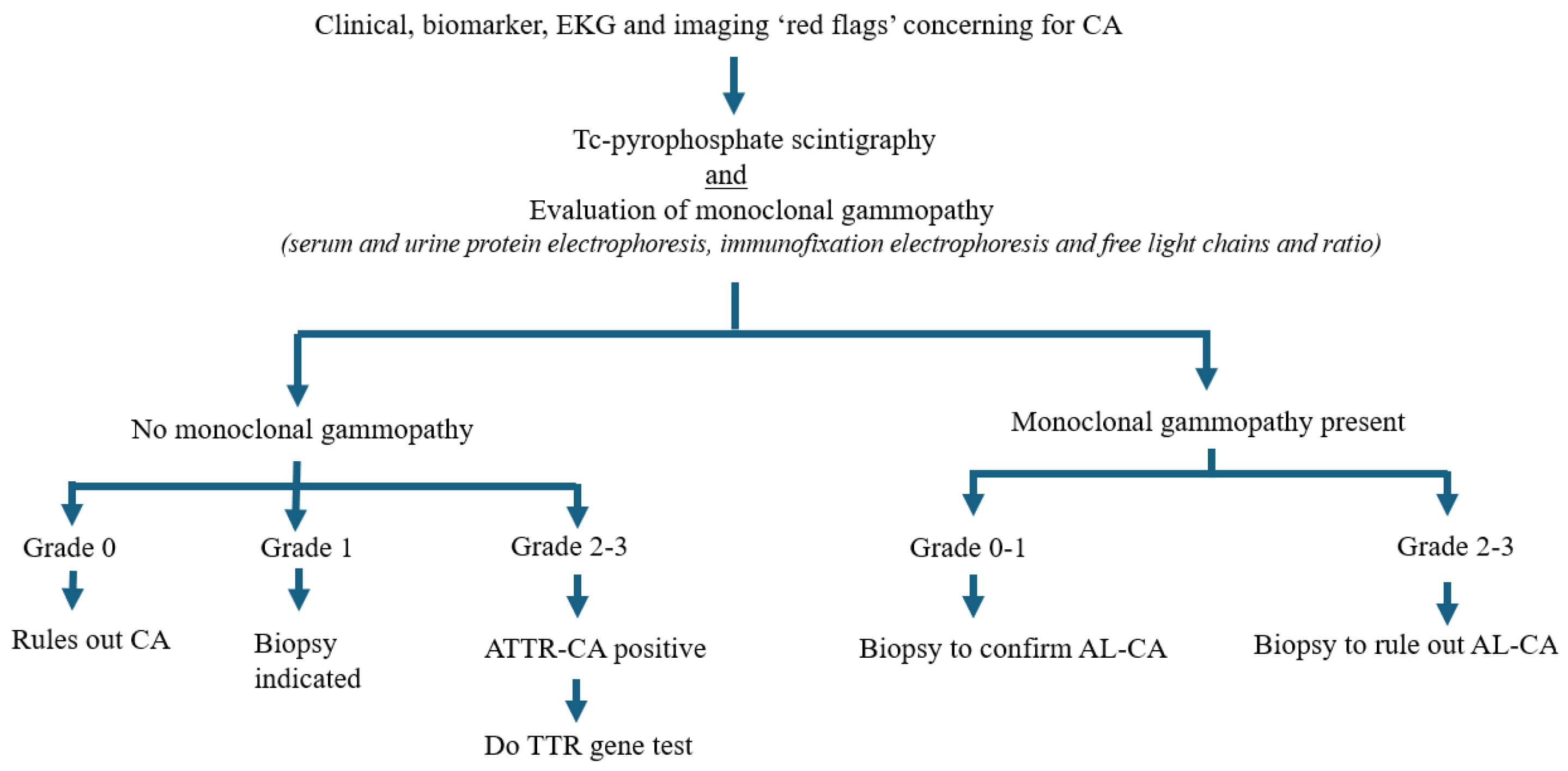
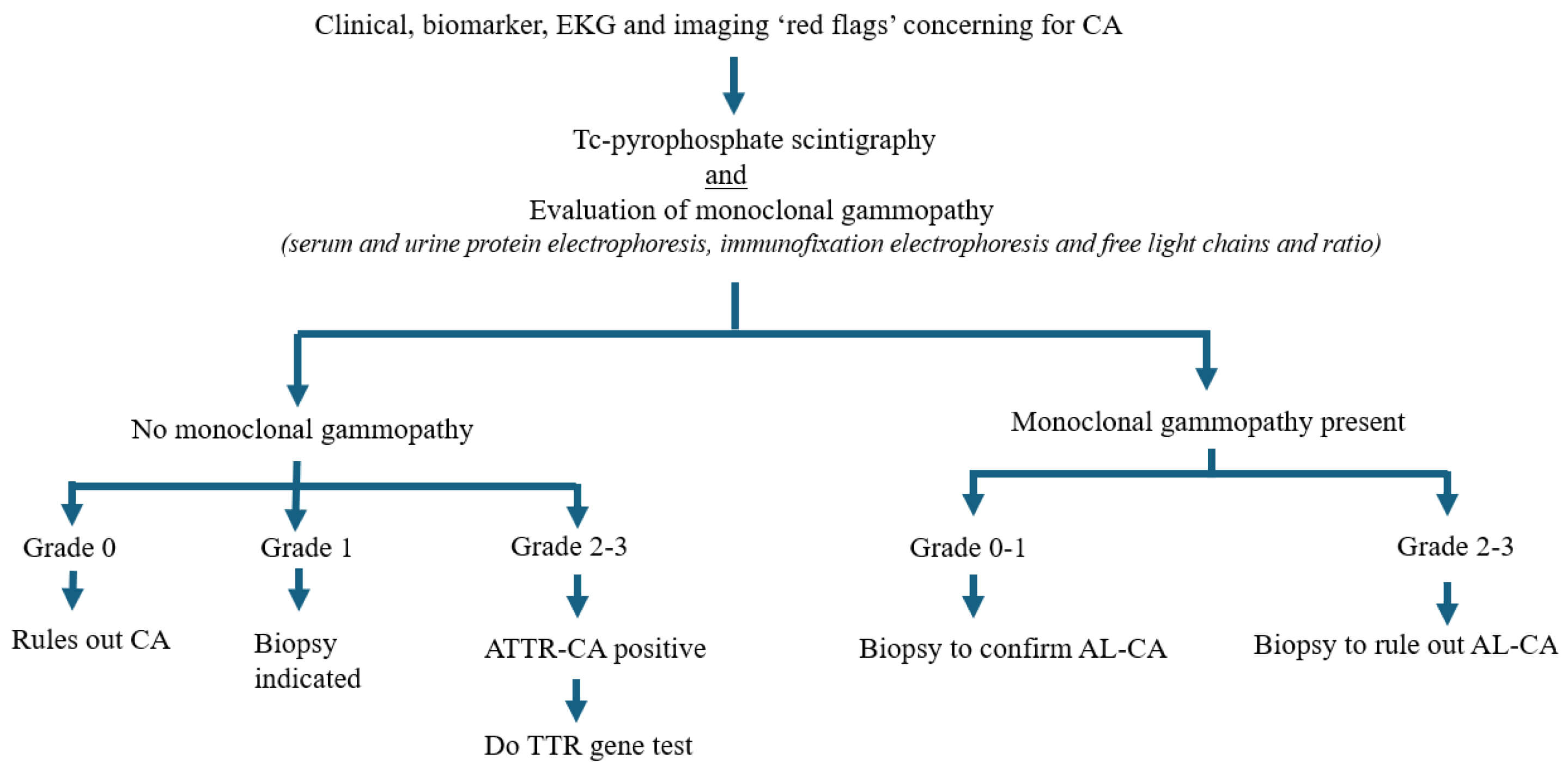 Fig. 3.
Fig. 3.
Algorithmic flowchart demonstrating screening and diagnosis of cardiac amyloidosis. Red flag features prompt nuclear scintigraphy, which is always performed in conjunction with serum evaluation of monoclonal gammopathy to rule out light chain amyloidosis. CA, cardiac amyloidosis; AL-CA, light chain cardiac amyloidosis; ATTR-CA, transthyretin cardiac amyloidosis; TTR, transthyretin; EKG, electrocardiogram.
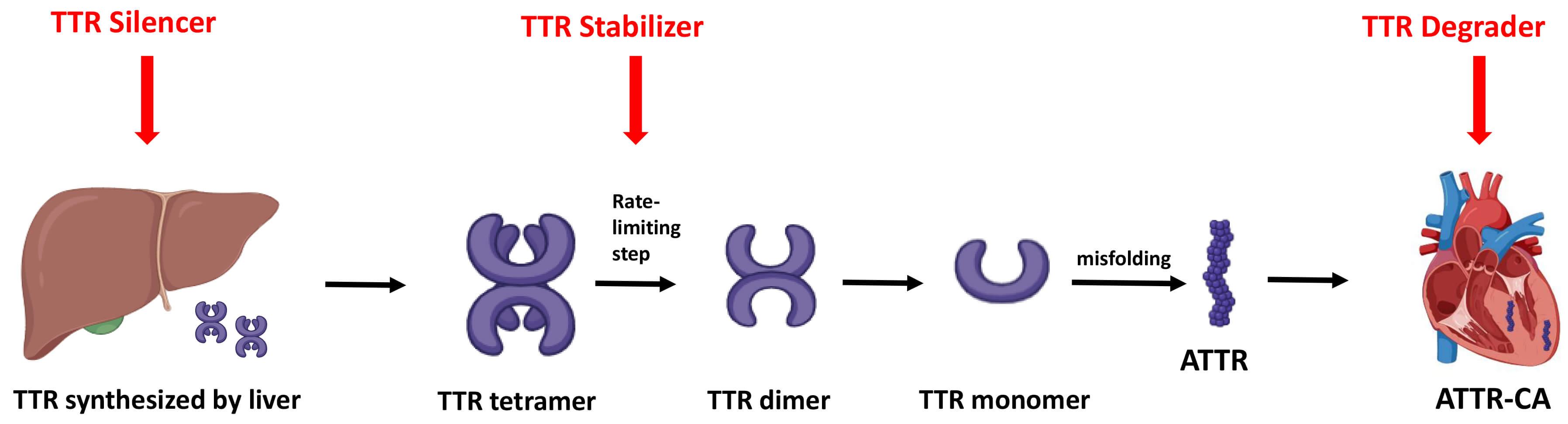
The treatment of ATTR-CA in HF involves a two-pronged approach. Firstly, mitigation of congestion, initiation of GDMT, and management of any co-existing arrhythmia. Secondly, initiation of amyloid-specific treatments aimed at directly stabilizing transthyretin, and/or inhibiting fibril formation (Fig. 4).
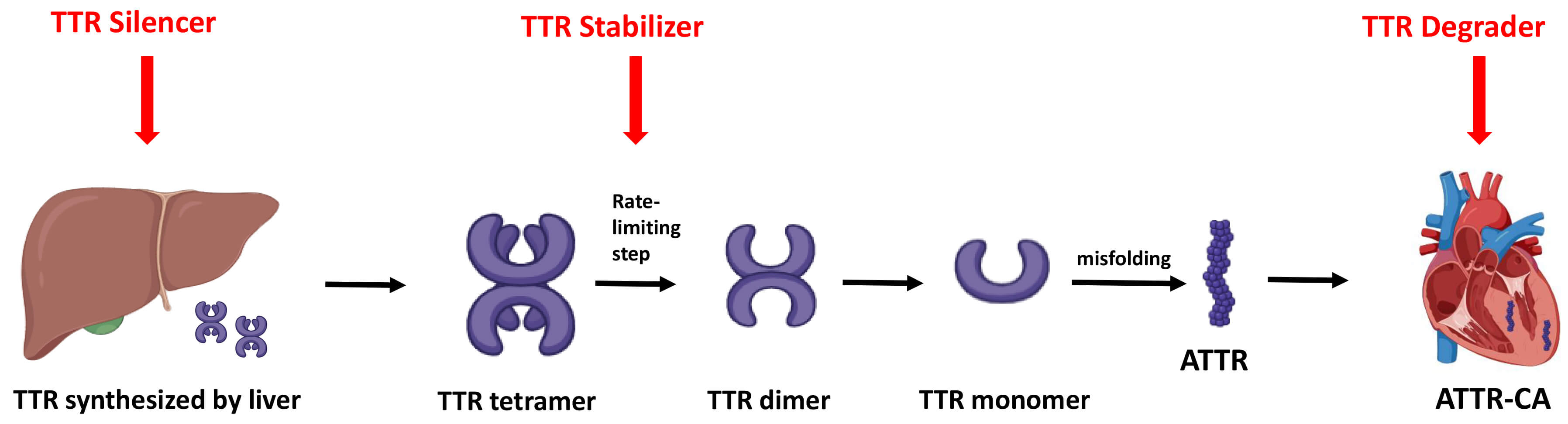 Fig. 4.
Fig. 4.
Mechanisms of amyloid-specific therapies. Overview of the three main therapeutic strategies for ATTR-CA: gene silencers reduce hepatic TTR production, stabilizers prevent tetramer dissociation, and degraders promote clearance of misfolded or deposited amyloid fibrils. TTR, transthyretin; ATTR, transthyretin amyloid fibril; ATTR-CA, transthyretin cardiac amyloidosis.
Fluid retention is a major cause of symptoms, poor quality of life, and adverse outcomes in patients with ATTR-CA and HF. Loop diuretics are mainstay therapy for symptomatic relief, helping to mitigate congestion and maintain adequate preload. While furosemide is the most commonly used agent, torsemide and bumetanide have higher potency and bioavailability [43]. The choice of a loop diuretic should be individualized and tailored to the severity of symptoms, cost and patient preferences. It is worth noting that the cardiac output in ATTR-CA is dependent on higher filling pressures given the restrictive physiology [44], thereby increasing the risk of organ hypoperfusion with aggressive diuresis. Therefore, cautious diuresis is crucial. Serum creatinine and estimated glomerular filtration rate should be closely monitored while diuresis is instituted, especially with intravenous medications while inpatient. In cases of severe fluid retention unresponsive to loop diuretics alone, metolazone can be employed intermittently at doses of 5 mg or 10 mg [45]. Metolazone is a thiazide-like diuretic that primarily inhibits sodium reabsorption in the distal convoluted tubule, thereby enhancing diuresis when used synergistically with loop diuretics. Adjunctive therapy with spironolactone, a mineralocorticoid receptor antagonist, is another potential strategy. In addition to providing a diuretic effect, spironolactone offers the benefit of mitigating hypokalemia—a common electrolyte disturbance associated with loop diuretics.
Diuretic use also carries prognostic significance. Diuretic dose and New York Heart Association (NYHA) functional class have been shown to independently predict mortality in ATTR-CA and, when incorporated into existing staging systems Mayo and UK risk scores [28, 33], significantly improve their prognostic accuracy [46]. In a study comprising 309 consecutive ATTR-CA patients, higher diuretic dose at diagnosis (per 1 mg/kg increase) was independently associated with increased all-cause mortality (adjusted HR 1.43, 95% CI 1.06–1.93) [46]. Incorporating diuretic dose and NYHA class into existing Mayo and UK risk scores improved prognostic discrimination (area under the curve (AUC) up to ~0.80) while maintaining calibration.
Patients with ATTR-CA were excluded from large-scale HF trials that formed the
basis of current GDMT pillars in HFrEF [47, 48, 49, 50, 51, 52, 53, 54, 55], and therefore it is unknown
whether conventional HF medications that have substantial benefits in patients
with non-amyloid HF may also benefit in those with ATTR-CA. Patients with ATTR-CA
appear to respond differently to neurohormonal (NH) blockade with
angiotensin-converting enzyme inhibitors (ACEi)/angiotensin receptor blockers
(ARBs), beta-blockers, and mineralocorticoid receptor antagonists (MRAs), as
compared with other patients with HF. This is thought to result from poor
hemodynamic tolerance due to an altered pressure–volume relationship, where
stroke volume is relatively fixed and ventricular–vascular coupling may be
impaired. Evidence from small studies has been inconsistent—some indicate that
low doses of these medications are generally well tolerated, while others suggest
they are poorly tolerated and may even lead to worse outcomes [56, 57]. The
absence of large-scale clinical trials contributes to this ongoing knowledge gap.
Consequently, multiple consensus guidelines recommend avoiding the use of
Betablockers are one of the important pillars of GDMT. In ATTR-CA, patients may
experience intolerance to betablockers because of the restrictive nature of the
disease, whereby they have a low, fixed stroke volume and therefore rely on
relatively higher heart rates to maintain adequate cardiac output. A reduction of
heart rate may negatively impair cardiac output, as well as potentially blunt the
chronotropic response needed to augment cardiac output during exercise.
Exacerbation of pre-existing orthostatic hypotension, worsening fatigue and,
occasionally, syncope due to bradyarrhythmia can be seen in ATTR-CA with the
institution of betablockers. While discontinuation of beta-blockers is seen in up
to one-third patients, majority are able to continue, and few may require dose
reduction. A recent large-scale observational study comprising 2371 patients with
ATTR-CA who were followed up on GDMT for 28 months found use of beta-blockers to
be associated with lower mortality among patients with LVEF
ACEi/ARBs/angiotensin receptor-neprilysin inhibitor (ARNi) have traditionally been believed to be poorly tolerated in ATTR-CA. In the setting of overt restrictive physiology, afterload reduction may predispose to hypotension without improving stroke volume. ACEi/ARB/ARNi can potentially exacerbate hypotension due to amyloid-associated autonomic dysfunction in ATTR-CA. In addition, some patients with ATTR-CA may not be suitable candidates for ACEi/ARB/ARNi due to pre-existing advanced kidney disease, which is not uncommon in ATTR-CA and may or may not be directly related to the disease itself. Observational studies have shown that ACEi/ARBs do not appear to confer a mortality benefit in ATTR-CA [60], although more robust studies are needed to confirm this. However, existing data also suggest that, just like beta-blockers, ACEi/ARBs are not as poorly tolerated as previously thought, and about two-thirds of patients with ATTR-CA tend to tolerate them once initiated. Data on ARNi remain very limited. We recommend the use of ACEi/ARBs in patients with ATTR-CA in the absence of hypotension or worsening kidney function. Careful monitoring of blood pressure, kidney function, and potassium levels is required.
MRAs are thought to be third pillar of conventional HFrEF therapies.
Observational studies have suggested a potential survival benefit from MRAs in
ATTR-CA. In a large-scale observational study from the UK National Amyloidosis
Centre, involving over 2300 patients with ATTR-CA, MRAs demonstrated favorable
tolerability and potential survival benefit [60]. After a median follow-up of
approximately 28 months, MRA therapy was discontinued in only 7.5% of patients,
compared with discontinuation rates of 21.7% for
Sodium-glucose cotransporter 2 inhibitors (SGLT2is) have been shown to improve
the outcome of patients with HF, but patients with ATTR-CA have been excluded
from all phase III trials on empagliflozin and dapagliflozin [61, 62, 63]. A recent
multicenter observational study included 220 patients receiving SGLT2i therapy
(mean age: 77 years; mean LVEF: 46%), who were compared to 220 propensity
score–matched controls, and found that SGLT2i treatment was generally well
tolerated (4.5% discontinuation rate) [64]. Over a median follow-up of 28
months, SGLT2i use was associated with reduced all-cause mortality (hazard ratio
[HR] 0.57; 95% confidence interval [CI] 0.37–0.89; p = 0.010),
cardiovascular mortality (HR 0.41; 95% CI 0.24–0.71; p
While the discussion of GDMT for HF in the context of ATTR-CA is clinically
important, it is crucial to acknowledge that much of the available evidence is
derived from observational studies, registries, or retrospective analyses. These
studies, while valuable for hypothesis generation and real-world insight, are
inherently limited by confounding factors and potential selection bias. For
example, patients who are prescribed
Atrial fibrillation is the most common rhythm disturbance in ATTR-CA [65]. Rhythm control strategy is often preferred as these patients are often unable to tolerate rate control therapies [66]. Data on catheter ablation is still limited. In addition, ATTR-CA patients with atrial fibrillation are at a significantly higher risk of stroke than non-amyloid patients with atrial fibrillation [38]. Anticoagulation may be considered irrespective of CHA2DS2-VASc [38]. Direct oral anticoagulants are safe to use as an alternative to vitamin K antagonists [67]; however, caution is warranted in the presence of significant renal or hepatic impairment, which may influence drug metabolism and bleeding risk [68].
Bradyarrhythmias are more prevalent in ATTRwt-CA than ATTRv-CA [69]. Pacemaker implantation, when indicated, can help provide symptomatic relief but does not confer mortality benefit [69]. Progression of conduction system disease is common and often leads to increased right ventricular pacing burden with time. Therefore, cardiac resynchronizing therapy (biventricular therapy) is often considered a better and safer option in these patients [70]. The role of prophylactic pacemaker implantation is controversial.
Ventricular arrhythmias are common in ATTR-CA [71]. Data on the efficacy of implantable cardioverter-defibrillators (ICDs) for primary or secondary prevention in ATTR-CA are limited to small-scale observational studies. While universal ICD placement in all patients with ATTR-CA remains controversial, selected patients—particularly those with a history of sustained ventricular arrhythmias—may derive benefit, especially in the context of secondary prevention [72, 73, 74, 75, 76, 77, 78].
In patients with ATTRv with polyneuropathy, several gene-silencing therapies
have shown efficacy in randomized controlled trials. Eplontersen, in the
NEUROTTRansform trial (n = 144), reduced serum TTR by 81.7%, stabilized
neuropathy progression, and improved quality of life over 66 weeks
(p
Among TTR stabilizers, tafamidis remains the only widely approved agent based on robust phase III evidence. In the ATTR-ACT trial (n = 441), tafamidis significantly reduced all-cause mortality and cardiovascular hospitalizations over 30 months in patients with ATTR-CA, while also slowing functional decline and preserving quality of life [59]. Acoramidis, evaluated in the ATTRibute-CM trial (n = 632), demonstrated a significant benefit on a hierarchical composite outcome (mortality, cardiovascular hospitalizations, NT-proBNP, and 6-minute walk distance), with a favorable safety profile and a win ratio of 1.8 compared to placebo [84]. While not yet widely adopted, diflunisal, a nonsteroidal anti-inflammatory drug with TTR-stabilizing properties, has shown beneficial effects in small observational studies and early trials, particularly in slowing neuropathy progression in ATTRv with predominant neuropathy, though its use is limited by safety concerns (e.g., renal and gastrointestinal toxicity) and lack of large-scale randomized controlled trial (RCT) data. Looking ahead, newer stabilizers may offer enhanced binding and clinical benefit, and combination trials with silencers may shape future treatment paradigms [85].
Emerging TTR degraders, which aim to target and remove existing amyloid fibrils rather than simply inhibiting their formation or production, represent a promising therapeutic avenue in ATTRCA. Among these, the combination of doxycycline—a tetracycline antibiotic—with tauroursodeoxycholic acid (TUDCA) has gained experimental traction. In animal models of ATTR, this duo effectively reduced both fibrillar and nonfibrillar deposits, and in a small phase II openlabel study, the combination stabilized cardiac and neurologic disease in most treated patients over one year with an acceptable safety profile [86]. Encouragingly, a larger randomized phase III trial comparing doxycycline/TUDCA plus supportive therapy versus supportive care alone (NCT03481972) is currently underway [87]. Meanwhile, a broader class of antiamyloid approaches is under active investigation, including monoclonal antibodies like PRX004 (now NNC6019) and ALXN2220 (formerly NI006), designed to directly bind and facilitate clearance of TTR deposits; both are being evaluated in Phase II/III trials in ATTRCA patients [87]. Taken together, these strategies offer hope for therapeutic removal of established amyloid, potentially complementing existing stabilizers and silencers in the management of ATTRCA.
Despite growing momentum toward proactive family screening in ATTRv-CA, current discourse continues to underrepresent the critical roles of genetic counseling and cascade testing. This underrepresentation is significant, given the critical role of early identification in altering disease trajectory. Genetic counseling provides essential support by educating at-risk relatives about inheritance patterns, clinical implications, and testing options, while also guiding interpretation of variants. Family screening enables the detection of preclinical disease and timely intervention. A recent multinational evaluation of the 2021 ESC consensus recommendations underscores its importance: among 159 asymptomatic relatives across 10 European centers, 25% were already diagnosed with ATTRv-CA at baseline, and 13% of these had no red flag abnormalities on standard ECG, echocardiography, or biomarkers [88]. Additionally, 9.4% developed ATTRv-CA during follow-up, demonstrating the value of serial screening. The ESC criteria showed a high negative predictive value (97%), supporting their clinical utility, while male sex and rare TTR variants were associated with greater disease risk. These findings reinforce the need for structured genetic counseling and systematic screening protocols to ensure early diagnosis and enable timely therapeutic intervention—particularly important in an era of emerging disease-modifying treatments.
ATTR-CA has emerged as a significant and underrecognized contributor to HF, affecting both HFpEF and HFrEF populations. Advances in imaging and greater clinical awareness have revealed a higher community prevalence than previously appreciated, with ATTR-CA often mimicking other cardiac conditions and evading timely diagnosis. While conventional GDMT for HF offers symptomatic and mortality benefits, these agents have traditionally been thought to be poorly tolerated in ATTR-CA due to the low, fixed stroke volume and autonomic dysfunction. Recent observational data, however, suggest that some patients—particularly those with systolic dysfunction—can tolerate and potentially benefit from beta-blockers, ACEi/ARBs, and MRAs with careful monitoring. Most importantly, amyloid-specific therapies have revolutionized the management of ATTR-CA. Stabilizers such as tafamidis have demonstrated robust mortality and morbidity benefits, while gene silencers offer additional disease-modifying potential. As therapeutic options expand, early recognition and subtype identification will be critical to optimizing outcomes. Broader screening strategies and continued research into tailored treatment approaches will be essential in improving the care of patients with this complex and increasingly treatable form of HF.
SB: Conception, project supervision, literature review, drafting the manuscript, figure preparation, final review; MH: literature review, drafting the manuscript, final review; AM: literature review, drafting the manuscript, final review. All authors contributed to the conception and editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.