


1 Department of Cardiovascular Surgery, Zhongshan Hospital, Fudan University, 200032 Shanghai, China
2 Shanghai Medical College, Fudan University, 200032 Shanghai, China
3 Department of Cardiovascular Surgery, Guizhou Provincial People’s Hospital, 550002 Guiyang, Guizhou, China
†These authors contributed equally.
Abstract
Acute Stanford type A aortic dissection (ATAAD) is a life-threatening cardiovascular emergency that demands prompt and accurate diagnosis due to a high associated mortality. Although imaging remains the diagnostic gold standard, the limited accessibility and time sensitivity of this technique underscore the need for reliable serum biomarkers. D-dimer is the most widely used biomarker, offering high sensitivity; however, the limited specificity of using D-dimer has prompted a search for novel biomarkers with greater diagnostic precision. Interestingly, proteoglycans (PGs) are essential constituents of the extracellular matrix (ECM) and have emerged as promising candidates for ATAAD, as PGs are released into the circulation during medial degradation, a defining histological feature of ATAAD. Moreover, emerging evidence suggests that specific PGs exhibit favorable specificity and stability, potentially enabling distinction between ATAAD and other acute cardiovascular syndromes. Additionally, in contrast to D-dimer, which is rapidly cleared within 8 hours, certain PGs, such as aggrecan, remain stable for up to 72 hours, offering an advantage for detecting ATAAD in patients presenting beyond the early acute phase. This review summarizes the potential of aortic PGs as biomarkers of medial degeneration and circulating PGs as serum diagnostic markers of ATAAD. Future research is warranted to establish PGs as clinically reliable biomarkers, with the potential to enhance current diagnostic frameworks and support an earlier, more accurate identification of ATAAD.
Graphical Abstract
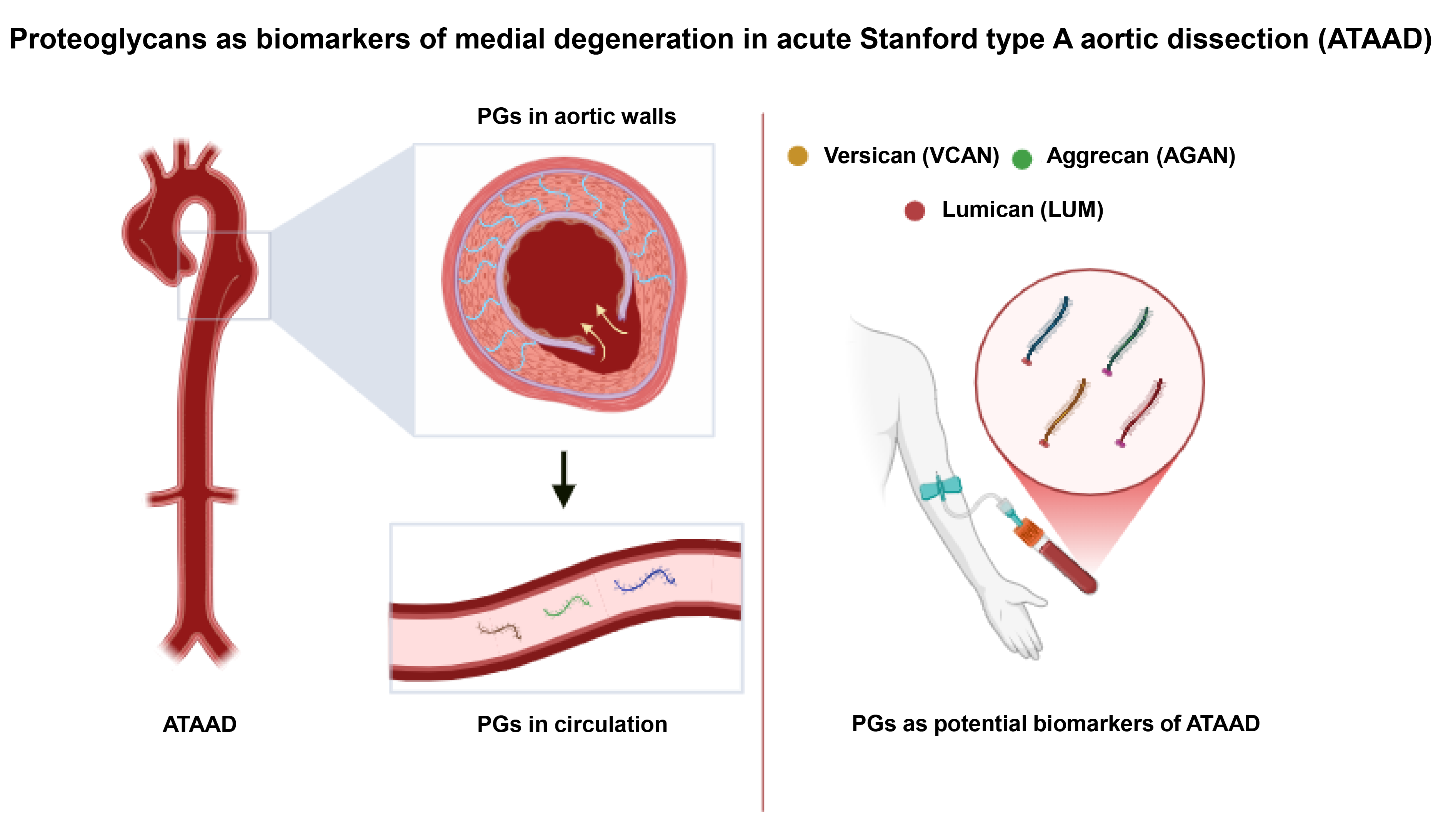
Keywords
- acute Stanford type A aortic dissection
- thoracic aortic dissection
- serum biomarker
- proteoglycans
- aggrecan
- versican
- lumican
Thoracic aortic dissection is a catastrophic cardiovascular emergency caused by intimal tearing and false lumen formation [1]. Briefly, it is classified into Stanford type A aortic dissection (ATAAD), which involves the ascending aorta, and otherwise Stanford type B. The antegrade and retrograde propagation of ATAAD may trigger multiple complications (i.e., acute aortic regurgitation, myocardial ischemia, cardiac tamponade, stroke, or organ malperfusion) and even aortic rupture and death [1, 2]. Open surgical repair remains the guideline-recommended therapy for ATAAD [1]. Despite advances in imaging and surgical procedures, untreated ATAAD still has a mortality rate of 40–50% within the first 48 hours [3].
Accurate and timely diagnosis is essential for early surgical interventions of ATAAD. The primary symptoms are often chest pains, which may also be considered as acute coronary syndrome (ACS) or pulmonary embolism (PE) [3, 4], thereby contributing to diagnostic delays or even misdiagnosis [5]. When ATAAD involves coronary arteries, the patients may display syndromes of ACS with ST-segment elevation on the electrocardiogram and abnormal myocardial enzymes. This may further enhance the challenge for distinguishing ATAAD and ACS. Notably, approximately 80% of misdiagnosed ATAAD cases are initially mistaken for ACS, followed by the inappropriate administration of thrombolytic therapy and thereby worsened outcomes [6]. Current diagnosis of ATAAD relies on imaging modalities such as computed tomography angiography (CTA), magnetic resonance angiography (MRA), and transesophageal echocardiography (TEE) [1]. Although these techniques offer high diagnostic accuracy, they are inherently constrained by several factors including limited accessibility, time-consuming patient-transferring [7], ionizing radiation exposure [8], and contrast-induced nephropathy risk [9]. Typical ATAAD is marked by a dilated aortic root, ascending aorta or aortic arch with an intimal flap. However, conventional computed tomography (CT) scans may miss highly suspected ATAAD patients with atypical CT images [10, 11]. These limitations emphasize the necessity for developing highly sensitive and specific biomarkers to enhance the early diagnosis of ATAAD.
Proteoglycans (PGs), essential ECM components comprising a core protein linked to glycosaminoglycan (GAG) chains, contribute to aortic structural integrity, cellular signaling and ECM remodeling [12, 13, 14]. In recent years, PGs have garnered attention as potential biomarkers for ATAAD. On one side, acute aortic tearing leads to the destruction of normal aortic walls and the release of PGs into the blood circulation. On the other hand, dissection formation triggers vascular remodeling marked by the accumulation of PGs. This review compares the potential of specific PGs as biomarkers for ATAAD. Future progress of biomarker research of PGs may provide novel insights into early diagnosis of ATAAD in the emergency.
Despite their diagnostic value, currently available biomarkers for ATAAD present several limitations that restrict their standalone diagnostic utility. Intimal tearing is the defining anatomic feature of ATAAD, triggering thrombosis in the false lumen. D-dimer is a fibrin degradation product released during fibrinolysis and is a classical biomarker of thrombosis [15]. Several meta-analyses consistently report high pooled sensitivities for D-dimer in ATAAD, typically ranging from 95% to 98%, making it a valuable rule-out test in patients with low clinical suspicion [16, 17]. However, its clinical utility is constrained by poor specificity ranging from 42% to 70%, given its elevation in other thrombotic and inflammatory conditions, including ACS and PE [15, 18, 19], deep vein thrombosis [20, 21, 22] and disseminated intravascular coagulation [15].
In emergency departments, where patients often present with nonspecific symptoms, D-dimer’s specificity is frequently compromised due to the high prevalence of alternative conditions that elevate D-dimer, such as PE, deep vein thrombosis (DVT), ACS, and systemic inflammation [18, 19, 20, 21, 22, 23]. In contrast, in specialized cardiovascular centers where pre-test probability is higher, the positive predictive value of D-dimer improves, though specificity remains moderate.
Patient-specific factors further affect D-dimer’s diagnostic performance. In
elderly patients, age-related increases in baseline D-dimer levels reduce
specificity, necessitating the use of age-adjusted cutoffs (e.g., age
Moreover, D-dimer’s short biological half-life (
Given these limitations, the 2015 American College of Emergency Physicians
issued a level C recommendation against using D-dimer as a standalone test for
ATAAD. Integration with clinical decision tools, such as the Aortic Dissection
Detection Risk Score (ADD-RS), has been shown to improve diagnostic accuracy by
combining D-dimer testing with structured risk assessment. For example, the
combination of ADD-RS
Medial degeneration is the defining histological feature of ATAAD including vascular smooth muscle cell (VSMC) damage, extracellular matrix (ECM) degradation, remodeling and vascular inflammation [30]. VSMC damage leads to release of cellular proteins into circulation. Smooth muscle myosin heavy chain showed a 20-fold increase in the ATAAD patients. However, it is elevated only 3–6 hours after onset and creates a limited time window for use in the emergency department [31]. BB-isozyme of creatine kinase of the muscle, another product of VSMC injury, is elevated in the ATAAD patients but has a short time course and lacks specificity [32]. ECM supports aortic integrity, elasticity, and strength. ECM degradation and disorganization are remarkable hallmarks of ATAAD. Elastin, a major component of aortic ECM, showed elevations in the ATAAD patients. Nevertheless, only a less than 2-fold increase was observed over normal controls, making reliable clinical use questionable [33]. Matrix metalloproteinases (MMPs), particularly MMP-9, are involved in ECM degradation and are elevated in ATAAD, but their levels overlap with other vascular pathologies [34]. Although vascular inflammation is a key process in ATAAD, it may exist in other cardiovascular disorders and lacks sufficient specificity. Soluble suppression of tumorigenesis-2 (sST2), a cytokine receptor involved in the cardiovascular inflammation, showed promising diagnostic performance in a Chinese study (sensitivity 99.1%, specificity 84.9% at a 34.6 ng/mL cut-off) [35], yet these results were not replicated in European cohorts due to high inter-individual variability [36]. Other markers (such as the neutrophil-to-lymphocyte ratio) have been evaluated, yet none consistently outperform D-dimer in accuracy or reproducibility [37]. Taken together, these limitations highlight the unmet need for novel biomarkers that offer both high sensitivity and specificity, maintain stability across disease phases, and remain consistent across diverse populations.
Proteoglycans are intricate macromolecules consisting of a core protein attached by one or more GAG chains [38]. GAGs are negatively charged polysaccharides composed of repeating disaccharide units, one of which is an amino sugar (i.e., N-acetylglucosamine or N-acetylgalactosamine) and the other is a hexuronic acid (i.e., glucuronic acid or its epimer iduronic acid) [39]. The sulfation and carboxylation of these units confer a strong polyanionic character to GAGs, enabling proteoglycans to bind cations such as Na+ from the interstitial fluid to preserve electroneutrality. This property allows them to generate Donnan osmotic pressure, thereby retaining water and maintaining tissue hydration and compressibility [40].
Up to now, over 90 PGs subtypes have been identified with the advances in glycoproteomic techniques [41] with approximately 20 identified in the blood vessels [12]. PGs can be broadly categorized based on their GAG components, core protein characteristics and cellular locations [12] as shown in Table 1. GAG components include chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS), heparan sulfate (HS) and hyaluronan (HA) [40]. These GAGs are covalently attached to specific types of core proteins to form 4 major categories, including chondroitin sulfate proteoglycans (CSPGs) (i.e., versican and aggrecan), dermatan sulfate proteoglycans (DSPGs) (i.e., decorin and biglycan), keratan sulfate proteoglycans (KSPGs) (i.e., lumican and fibromodulin), heparan sulfate proteoglycans (HSPGs) (i.e., syndecan and perlecan) and hyaluronan-associated proteoglycans (HAPG) [39, 42]. According to cellular localization, PGs are classified into intracellular PGs (i.e., serglycin), cell-surface PGs (i.e., syndecans and glypicans) and ECM PGs. ECM PGs can further be subdivided into small leucine-rich proteoglycans (SLRPGs) such as decorin and biglycan, and hyalectans like aggrecan and versican [39]. The diversity of PGs allows them to fulfill a wide range of biological functions in various tissues and organs, including vasculatures.
| Classification based on GAG components, subclassification based on core proteins | ||||||
| CSPG | DSPG | KSPG | HSPG | HAPG | ||
| Aggrecan (ACAN) | Decorin (DCN) | Lumican (LUM) | Syndecan | SDC1 | ||
| Versican (VCAN) | Biglycan (BGN) | Fibromodulin (FMOD) | SDC2 | |||
| Neurocan (NCAN) | Mimecan (OGN) | SDC3 | ||||
| Brevican (BCAN) | SDC4 | |||||
| NG2 (CSPG4) | Glypican | GPC1 | ||||
| Thrombo-modulin (THBD) | GPC2 | |||||
| Chondroitin sulfate proteoglycan 4 (CSPG4) | GPC3 | |||||
| Inter-alpha-trypsin inhibitor heavy chain 1 (ITIH1) | GPC4 | |||||
| Inter-alpha-trypsin inhibitor heavy chain 2 (ITIH2) | GPC5 | |||||
| Inter-alpha-trypsin inhibitor heavy chain 5 (ITIH5) | GPC6 | |||||
| Serglycin (SRGN) | Perlecan | HSPG2 | ||||
| Agrin | AGRN | |||||
| Testican | SPOCK1 | |||||
| SPOCK2 | ||||||
| SPOCK3 | ||||||
| N-Tes | ||||||
| Classification based on cellular localization | ||||||
| Intracellular PG | Cell-surface PG | ECM PG | ||||
| Serglycin | Syndecans | SLRPG | Hyalectans | |||
| Glypicans | Decorin (DCN) | ACAN | ||||
| Biglycan (BGN) | VCAN | |||||
PG, proteoglycan; CSPG, chondroitin sulfate proteoglycan; HSPG, heparan sulfate proteoglycan; ECM, extracellular matrix; SLRPG, small leucine-rich proteoglycan; DSPG, dermatan sulfate proteoglycan; KSPG, keratan sulfate proteoglycan; HAPG, hyaluronan-associated proteoglycans; SPOCK, SPARC/osteonectin, CWCV and Kazal-like domain proteoglycan.
PGs perform indispensable roles in maintaining ECM structure and function, contributing to vascular integrity and homeostasis. PGs play a pivotal role in organizing vascular ECM to ensure vessel elasticity and adapt to mechanical forces [38, 39]. PGs are direct space-filling molecules that regulate hydration, maintain tissue turgor through Gibbs-Donnan osmotic pressure, and provide mechanical resilience [43]. PGs may also help organize the structural framework of ECM by interacting with other ECM components. For instance, the core protein of decorin interacts with collagen fibrils to regulate their organization and diameter, enhancing tensile strength [39]. Similarly, versican interacts with hyaluronan to form hydrophilic aggregates that confer compressive resistance and are essential for vascular integrity under pulsatile flow [43].
Beyond their structural roles, PGs are central to cell signaling processes that
influence vascular remodeling, inflammation, and endothelial function [12]. The
negatively charged GAG chains bind to growth factors, cytokines, and chemokines,
thereby modulating their local bioavailability and activity. PGs facilitate the
sequestration and presentation of transforming growth factor-beta (TGF-
The dysregulation of PGs is closely associated with vascular diseases. Alterations in the expression, composition, and distribution of PGs disrupt the ECM’s structural integrity, rendering blood vessels susceptible to mechanical stress and pathological remodeling [46, 47]. Under normal conditions, PGs constitute a minor component of the ECM, with tightly regulated interactions that maintain vessel architecture and mechanical properties. However, PGs often increase in the vascular diseases and are frequently accompanied by changes in GAG sulfation patterns [12]. In atherosclerosis, proteoglycans (i.e., versican) bind lipoproteins and promote lipid retention and the formation of atheromatous plaques [48]. PGs also modulate inflammatory responses. For example, decorin and biglycan contribute to macrophage recruitment and foam cell formation [49]. In hypertension and pulmonary arterial hypertension, PGs (i.e., glypican and decorin) worsen disease progression by interacting with endothelial cells and VSMCs to influence vascular calcification, fibrosis, and thrombosis [50, 51, 52].
Unlike intracranial or abdominal aortic aneurysms, which rarely exhibit PG accumulation, ATAAD is uniquely characterized by significant pools of PGs within regions of medial degeneration [53, 54]. These soluble fragments, reflecting the molecular disarray of the aortic wall, hold considerable promise as serum biomarkers for detecting and monitoring ATAAD. Excessive PG aggregation VSMC damage, disrupts elastic fiber organization, and fragments collagen, collectively weakening the tensile and elastic integrity of the aortic wall [55, 56, 57].
Beyond structural disruption, PG degradation products act as bioactive signaling molecules. Cleavage by MMPs and A disintegrin-like and metalloprotease domain with thrombospondin type 1 motifs (ADAMTS) family proteases yields soluble PG fragments that function as damage-associated molecular patterns (DAMPs), activating innate immune pathways and promoting macrophage and neutrophil recruitment [43, 47, 58, 59]. These immune cells further secrete proteases and reactive oxygen species, amplifying ECM injury. Non-proteolytic mechanisms, including hyaluronidase activity and macrophage-mediated release, also contribute to PG dissemination into circulation [41].
Importantly, PG synthesis itself is upregulated in response to mechanical stress and inflammatory signaling, driving maladaptive ECM remodeling [60]. Computational and biomechanical models confirm that PG-rich pools create local stress concentrations sufficient to sever elastic lamellae, impair mechanotransduction pathways critical for ECM homeostasis, and destabilize wall architecture [2, 43, 60, 61, 62]. Together, these processes leave the vessel wall structurally weakened and primed for dissection and rupture.
Beyond their general accumulation in ATAAD, aggrecan and versican, two large aggregating CSPGs, play central roles in driving the molecular and structural changes that underlie medial degeneration [60, 63]. Both molecules possess large, highly sulfated glycosaminoglycan chains that attract water and alter tissue biomechanics by generating osmotic swelling pressures, but their impact extends beyond passive matrix disruption. Through regulated turnover, proteolytic processing, and ECM interactions, aggrecan and versican shape VSMC phenotype, inflammatory signaling, and aortic medial integrity. These multifaceted functions position them as both contributors to wall destabilization and mediators of pathways central to ATAAD pathogenesis.
Most found in cartilaginous tissues [64], aggrecan (ACAN) is abnormally
deposited in the thoracic aortic media during ATAAD [60, 63]. Its highly sulfated
chondroitin and keratan sulfate GAG chains create highly hydrated microdomains
that increase interlamellar spacing and compromise elastin–collagen load sharing
[55, 57, 59]. Histological analyses have also demonstrated that ACAN colocalizes
with elastic fiber breaks and apoptotic VSMCs [64]. Proteolytic cleavage of
aggrecan by ADAMTS enzymes (e.g., ADAMTS-4, ADAMTS-5) generates neoepitopes that
act as DAMPs, activating toll-like receptors (TLR2, TLR4) on immune cells and
promoting secretion of interleukin-1
Aberrant aggrecan accumulation has been consistently documented in thoracic aortic dissection (TAD) tissue, where it colocalizes with elastic lamellar breaks and VSMC apoptosis [60, 63, 65, 66]. Its long chondroitin and keratan sulfate chains create highly hydrated domains that expand interlamellar spacing and compromise elastin-collagen load sharing [14, 39, 47, 66]. Balanced aggrecan turnover depends on the activity of MMPs and ADAMTS proteases, specifically ADAMTS-5 activity [46, 66]. Mice lacking ADAMTS-5 exhibit impaired aggrecan cleavage, abnormal proteolytic fragment profiles, and ascending aortic anomalies as a result of ACAN accumulation [46, 65, 67]. In human TAD, upregulation of ADAMTS-1 and ADAMTS-4 further underscores dysregulated proteoglycanase activity [68]. Beyond mechanics, aggrecan peptide produced as a result of MMP and ADAMTS cleavage sites act as DAMPs, engaging toll-like receptors on immune cells to stimulate pro-inflammatory cytokine release [69, 70, 71].
While much of this work has been conducted in the context of arthritic pain and cartilage degeneration, the underlying signaling mechanisms remain relevant to vascular pathology. In particular, activation of TLR4 has been shown to promote a pro-inflammatory VSMC phenotype and to drive cellular proliferation during vascular remodeling and atherogenesis [72, 73]. Taken together, these findings support a mechanistic link by which aggrecan-derived DAMPs may contribute to VSMC dysfunction and the progression of medial degeneration in ATAAD.
VCAN, a large chondroitin sulfate PG highly expressed in vascular tissue, also plays a prominent role in medial degeneration through its effects on ECM organization, VSMC behavior, and inflammation [12, 39, 60, 74]. In ATAAD, VCAN is enriched in areas of VSMC dropout, where it disrupts pericellular adhesion, alters fibronectin and type I collagen organization, and weakens medial tensile strength [61, 74, 75]. Its binding to hyaluronan produces bulky pericellular matrices that interfere with integrin signaling, promote VSMC detachment, and impair nutrient diffusion [74, 76, 77]. Proteolysis by ADAMTS enzymes generates versikine, a bioactive fragment that promotes apoptosis, leukocyte adhesion, cytokine release, and MMP expression [74, 78, 79, 80].
Versican accumulation is also mechanistically linked to phenotypic modulation of
VSMCs. VSMC phenotypic switching is a central feature of medial degeneration,
characterized by the downregulation of contractile proteins such as
SM22
Aggrecan and versican act as both structural disruptors and signaling hubs. Their excessive accumulation imposes mechanical stress and lamellar delamination, while their proteolytic fragments amplify inflammation and promote VSMC dysfunction. By fostering phenotypic switching and impairing ECM homeostasis, these proteoglycans establish a self-reinforcing cycle of degeneration that predisposes the thoracic aorta to dissection and rupture [60, 81]. Their restricted association with thoracic aortic medial degeneration also underpins their translational potential as biomarkers: circulating aggrecan and versican fragments are detectable in acute dissection and may provide diagnostic specificity relative to other acute aortic syndromes [60, 63, 65].
HSPGs are multifunctional components of the vascular ECM that play integral roles in structural integrity, cell signaling, and mechanical homeostasis [39, 87]. Comprising a protein core modified with HS GAG chains, HSPGs are positioned to respond to both biomechanical and biochemical stimuli in the vessel wall. Among them, perlecan and SDC-1 have emerged as particularly relevant to the medial degeneration of ATAAD. These PGs not only influence the mechanical properties of the aorta but may also serve as candidate biomarkers for disease risk or activity [88, 89].
Perlecan, encoded by HSPG2, is a large HSPG broadly expressed across vascular and avascular tissues, where it localizes predominantly to basement membranes and the apical surface of cells [39]. Within the thoracic aortic wall, it is enriched at the interface of elastin and fibrillin-1 fibers in the medial layer, supporting elastic lamellae organization and matrix integrity [90]. It contributes to elastic lamellae organization, supports endothelial nitric oxide signaling [91], and maintains the contractile phenotype of VSMC [92]. The significance of perlecan in ATAAD was demonstrated in a vascular-specific perlecan knockout mouse model, in which spontaneous thoracic aortic dissection occurred with notable frequency, approximately 15% at 10 weeks and up to 38.9% by 50 weeks of age [88]. On the cellular level, perlecan deficiency was associated with significant downregulation of VSMC contractile markers (Acta2, MYH11), with preserved expression of Myocd, suggesting a phenotypic drift away from the contractile state. Notably, MMP activity (MMP-2 and MMP-9) was not elevated, indicating that ECM degradation was not enzymatically driven but structurally preconditioned. Given its role in maintaining elastic fiber maturity and VSMC phenotype, perlecan represents a mechanistically grounded risk determinant for medial degeneration and dissection.
However, its largely tissue-restricted localization limits its feasibility as a circulating biomarker. Instead, perlecan shows greater promise as a tissue-level marker of structural integrity or as a candidate genetic modifier in syndromic or familial aortopathies [93]. In Marfan syndrome mouse models, lower HSPG2 expression is associated with more severe cardiovascular and skeletal manifestations, indicating that variation in HSPG2 can influence disease severity [93]. In the context of syndromic or familial aortopathies, immunohistochemical or transcript-based assessment of perlecan in aortic tissue could provide insights into structural vulnerability before dissection occurs. Moreover, rare variants in HSPG2 have been reported in human connective tissue and skeletal disorders, and expression levels of HSPG2 correlate with severity in animal models. These observations indicate that HSPG2 acts not only as a structural ECM component but also as a genetic modifier influencing susceptibility to medial degeneration and dissection. Assessing perlecan in aortic tissue may therefore provide a means of preclinical ATAAD risk evaluation, supporting more precise patient stratification and informing decisions on the optimal timing of prophylactic surgery.
SDC-1 is another transmembrane HSPG expressed by VSMCs and endothelial cells,
involved in modulating inflammation [94, 95, 96], ECM-cell interactions, and
extracellular remodeling [94, 97, 98]. Although most of the existing literature
focuses on its role in aneurysmal thoracic aorta, its regulation in ATAAD-prone
tissues and relevance to dissection biology is increasingly appreciated [89].
Syndecan-1 expression was significantly elevated in the aneurysmal thoracic
aorta: 3.6-fold at the mRNA level and 2.3-fold at the protein level relative to
healthy aortas. This pattern of upregulation suggests that SDC-1 is a component
of the medial remodeling response, potentially reflecting VSMC activation under
hemodynamic stress. However, mechanistic interrogation in a murine model designed
to induce dissection through
Biglycan and decorin, archetypal class I SLRPs, share conserved structural
domains including leucine-rich repeats and chondroitin/dermatan sulfate
glycosaminoglycan side chains, allowing them to bind collagen, modulate
TGF-
These mechanistic roles, coupled with evidence of their proteolytic release during vascular injury, raise the possibility that biglycan and decorin may serve as measurable serum biomarkers of acute aortic pathology. Soluble biglycan has been identified as a DAMP, capable of activating TLR2 and TLR4 signaling and promoting sterile inflammation [102, 103, 104], a hallmark of acute aortic syndromes. Elevated circulating levels of biglycan have been documented in a range of inflammatory and cardiovascular conditions, including atherosclerosis [105, 106], pulmonary hypertension [107] and chronic liver disease [108], suggesting its potential as a nonspecific indicator of vascular ECM remodeling. Decorin may behave similarly, although data on its serum dynamics in aortic pathology are still limited.
However, several challenges currently impede the clinical translation of these molecules as biomarkers. Neither biglycan nor decorin is specific to the aorta or to dissection, and both are altered in a variety of systemic inflammatory and fibrotic states. No human studies have yet investigated the utility of biglycan or decorin as serum biomarkers for ATAAD. Most existing findings derive from tissue analyses or animal models, and the lack of standardized serum assays further limits clinical translation. Consequently, while these molecules have compelling mechanistic links to medial degeneration, their value as circulating biomarkers for acute aortic dissection remains entirely theoretical, highlighting a clear gap in human-focused research.
Among the promising candidates, aggrecan and versican, two large aggregating CSPGs, have demonstrated significant potential [60, 63, 109]. Their diagnostic value arises from their physiological roles in maintaining ECM integrity and their pathological accumulation and release during aortic wall remodeling and degeneration. In the aorta, aggrecan and versican localize to the tunica media, contributing to its mechanical properties and ability to withstand hemodynamic stress [39]. Aggrecan and versican contribute distinct yet complementary roles to vascular biology. Aggrecan, traditionally associated with cartilage, maintains tissue hydration, and resists compressive forces through its extensive GAG chains [39]. Although its expression in the vasculature was initially underappreciated, proteomic studies have confirmed its presence in the aorta, where it contributes to ECM stability [110]. Versican, ubiquitously expressed across tissues such as the adult brain and large vessels, regulates VSMC adhesion, proliferation, and migration [39, 111].
The elevated levels of aggrecan and versican have been shown to distinguish
ATAAD from other conditions. In a pivotal study by König et al.
[63], serum plasma levels of aggrecan were systematically evaluated in ATAAD
through a two-phase design, first using a pilot cohort and then validating the
findings in an expanded population. The results of the initial phase revealed a
significant elevation in plasma aggrecan levels in ATAAD patients, with
concentrations four to five times higher than those in both control groups.
Specifically, the mean plasma aggrecan level in ATAAD patients (n = 14) was
50.16
Aggrecan demonstrates high diagnostic specificity for ATAAD, with serum levels unaffected by age, sex, aortic root involvement, or dissection extent. Its levels also remain unchanged in non-dissecting ascending aortic aneurysms, excluding its utility as a screening marker for chronic aneurysmal disease. The significance of these findings is further amplified by the results of receiver operating characteristic (ROC) curve analysis, which determined an optimal diagnostic threshold of 14.3 ng/mL. At this level, aggrecan demonstrated a sensitivity of 97% and a specificity of 81%, far exceeding the specificity of widely used biomarkers such as D-dimer (53%) [112]. The high specificity is essential in the emergency, where misdiagnosis may result in inappropriate treatments that exacerbate patient outcomes. Moreover, aggrecan’s diagnostic utility is enhanced by its stability. Aggrecan levels remain elevated for up to 72 hours post-symptom onset, providing a wide diagnostic window even in delayed presentations—a common challenge in ATAAD diagnosis [113].
Although aggrecan demonstrates better performance than markers D-dimer, its specificity and sensitivity remain inferior to high-sensitive troponin assays used in myocardial infarction. [Additionally, the diagnostic accuracy reported by König et al. [63] is based on a relatively small cohort of 33 non-consecutive, hemodynamically stable ATAAD patients, all with Stanford type A dissections. While the cohort included both sexes in nearly equal proportions and reflected common cardiovascular risk factors such as hypertension, hyperlipidemia, diabetes, obesity, and nicotine use, these features limit the generalizability of the findings to broader, more diverse patient populations, including those with type B dissections or differing comorbidity profiles.] As a major component of cartilage, aggrecan is also prone to degradation in osteoarthritis [69], raising concerns about diagnostic confounding, especially in broader clinical populations.
The diagnostic value of versican is revealed by proteomic studies using liquid chromatography–tandem mass spectrometry (LC-MS/MS) Elevation of versican and its proteolytic fragments, such as the DPEAAE neoepitope, a versican-specific cleavage product, was consistently detected in ATAAD aortic tissue and blood circulation [60]. Immunohistochemical analyses revealed intense versican staining across multiple lamellar units in the tunica media, with the highest concentrations in regions of medial degeneration. The release of versican fragments into the bloodstream during ECM remodeling provides an additional layer of diagnostic utility, potentially enabling real-time monitoring of disease activity and progression.
Unlike aggrecan, versican is also elevated in non-dissected aneurysmal thoracic aorta. In a transcriptomic study conducted by our team, versican-positive myofibroblasts were identified as critical intermediates in the pathological transition of VSMC from a contractile to a synthetic phenotype [61]. Besides, versican presents similar challenges due to its involvement in various pathological conditions, further limiting disease specificity. For example, ATAAD may be secondary to thoracic aortic dilation and aneurysm. The elevation of versican in aneurysmal thoracic aorta limits its utility in distinguishing ATAAD and thoracic aortic aneurysms (TAAs). Future efforts to improve specificity could focus on characterizing the unique versican degradome in ATAAD. Quantifying specific neoepitopes, such as those generated by ADAMTS proteases versus other proteinases (e.g., MMPs), may reveal a fragment signature unique to acute dissection, providing an actionable strategy for biomarker development.
SLRPs are a family of ECM molecules that play diverse roles in tissue homeostasis, from collagen fibrillogenesis to the regulation of inflammation and signal transduction [114, 115, 116]. Among these molecules, lumican (LUM), a class II SLRP, has garnered attention for its involvement in the vascular remodeling and aortic wall homeostasis, particularly in the context of ATAAD [39]. LUM plays a role in the ECM architecture by controlling collagen fibril assembly and by interacting with other matrix constituents to uphold vascular stability [117, 118, 119]. In the context of ATAAD, heightened proteolytic activity cleaves ECM components, releasing LUM into the circulation and thereby linking structural aortic degeneration to elevated serum levels of this molecule [120, 121, 122, 123]. Such dysregulation of LUM demonstrates its potential utility as a biomarker for early diagnosis, stratification of disease severity, and prognostic evaluation in ATAAD.
Initial proteomic investigations into serum alterations in ATAAD first
highlighted LUM’s diagnostic potential [122, 123]. Using isobaric tags for
relative and absolute quantification (iTRAQ), researchers identified LUM as a
significantly elevated ECM protein in ATAAD patients compared to both healthy
controls and AMI patients. Serum concentrations of LUM in ATAAD patients (2.66
While moderate in isolation, LUM’s diagnostic accuracy improved significantly
when combined with complementary biomarkers, such as peptidase inhibitor 16
(PI16) and fibrinogen-like protein 1 (FGL1) [122]. The selection of PI16 and FGL1
to complement Lumican in a multimarker panel was driven by a targeted proteomic
discovery process. These candidates were identified through a systematic
screening approach using iTRAQ and label-free proteomics on serum from ATAAD
patients, which revealed both PI16 and FGL1 as significantly upregulated proteins
[122]. Their pathobiological interactions are distinct yet synergistic in the
context of ATAAD pathogenesis. PI16, a peptidase inhibitor, is postulated to
modulate inflammatory and proteolytic processes that contribute to the
degradation of the aortic wall’s structural integrity by inhibiting MMPs and
consequently driving the accumulation of PGs [124, 125]. Conversely, FGL1 is
implicated in dysregulated hemostasis and enhances TGF-
The validity of this combinatorial approach is supported by the panel’s significantly enhanced AUC of 0.780, with sensitivity and specificity values of 67.5% and 76.7%, respectively [122]. This demonstrates superior discriminatory power compared to any single marker alone, as the integration of biomarkers from complementary pathways yields greater diagnostic specificity and a more robust reflection of the disease’s complex biology. This successful combination establishes a blueprint for integrating other emerging biomarkers (e.g., ACAN, sST2, etc.) into even more comprehensive panels. This could lead to the development of a highly accurate, rapid biomarker assay that not only aids in diagnosis but also helps stratify rupture risk and guide personalized therapeutic strategies, fundamentally improving ATAAD management.
Beyond initial diagnostic associations, subsequent research explored how
circulating LUM levels may relate to clinical severity in ATAAD, correlating
serum LUM levels with vascular involvement using multi-slice computed tomography
angiography (MSCTA) [120]. Elevated LUM levels were observed in ATAAD patients
(2.32
As research progressed, attention also turned to LUM’s potential role in
predicting clinical outcomes and disease prognosis. In a 2021 study of 58
patients undergoing surgical repair for ATAAD, Hsu et al. [126]
demonstrated that elevated preoperative serum LUM concentrations, quantified via
enzyme-linked immunosorbent assay (ELISA), were significantly associated with an
increased risk of prolonged mechanical ventilation (
Collectively, these findings highlight the complementary diagnostic potential of different PGs in ATAAD. A comparative summary of their vascular roles, serum alterations, and diagnostic performance is provided in Table 2 (Ref. [44, 60, 61, 63, 64, 66, 80, 88, 89, 91, 92, 94, 95, 96, 99, 101, 102, 108, 111, 114, 116, 120, 122]).
| Proteoglycan | Type/Family | Key roles in vasculature | Serum elevation in ATAAD | Diagnostic performance | Specificity for ATAAD | Limitations | References |
| Aggrecan | CSPG/Hyalectan | ECM hydration, load resistance, structural integrity | Markedly elevated (up to 10 |
Sensitivity: 97% Specificity: 81% (cutoff: 14.3 ng/mL) |
High; low in STEMI and aneurysm | Confounding from cartilage diseases (e.g., osteoarthritis); not useful for screening chronic aneurysms | [60, 63, 64, 66] |
| Versican | CSPG/Hyalectan | VSMC migration, ECM remodeling, signaling | Elevated in ATAAD and aneurysm tissues | No standardized serum cutoff; detected in LC-MS/MS and IHC studies | Moderate; expression is also high in cancer/fibrosis | Lacks ATAAD-specific signature; needs neoepitope or fragment-level profiling | [60, 61, 80, 111] |
| Lumican | SLRP (Class II) | Collagen fibrillogenesis, ECM integrity | Elevated in ATAAD vs. controls and AMI | AUC alone: 0.636 AUC (3-marker panel): 0.780 |
Moderate; reflects ECM remodeling | Limited sensitivity when used alone; better in multimarker panels | [114, 116, 120, 122] |
| Perlecan | HSPG (Basement Membrane) | Elastic lamellae organization, VSMC phenotype maintenance | No direct serum data; increased risk in KO mice | Not applicable | Low; best studied at tissue/genetic level | Not suitable as a serum biomarker; may be better as a structural or genetic marker | [88, 91, 92] |
| Syndecan-1 (SDC-1) | HSPG (Transmembrane) | Inflammatory modulation, ECM-cell interaction | Upregulated in aneurysmal thoracic tissue; serum data lacking | No validated serum performance data | Low; overexpressed in many vascular injuries | Currently lacks diagnostic value; may reflect stress response rather than disease cause | [89, 94, 95, 96] |
| Biglycan/Decorin | SLRP (Class I) | Collagen crosslinking, TGF- |
Evidence from animal/tissue studies only | Not validated in serum | Low; altered in various systemic diseases | Limited human data; no established serum assay or kinetics in ATAAD | [44, 99, 101, 102, 108] |
AUC, area under the receiver operating characteristic curve; AMI, acute
myocardial infarction; ATAAD, acute type A aortic dissection; CSPG, chondroitin
sulfate proteoglycan; ECM, extracellular matrix; HSPG, heparan sulfate
proteoglycan; IHC, immunohistochemistry; KO, knockout (mice); LC-MS/MS, liquid
chromatography–tandem mass spectrometry; SLRP, small leucine-rich proteoglycan;
STEMI, ST-elevation myocardial infarction; TGF-
PGs are emerging as promising serum biomarkers for ATAAD, reflecting underlying ECM disruption. Elevated levels of aggrecan, versican, and lumican in affected patients suggest these molecules may offer diagnostic value beyond traditional markers such as D-dimer. Their greater specificity could facilitate more accurate differentiation of ATAAD from other acute cardiovascular syndromes, particularly when incorporated into tiered diagnostic algorithms or used in conjunction with high-sensitivity assays.
ATAAD may derive from both normal and aneurysmal thoracic aorta with different ages, genders and severity, which emphasizes the heterogeneity of ATAAD. For example, chronic aortic dilation and aneurysm lead to progressive ECM remodeling and PGs accumulation. Future research should aim to clarify the pathophysiological role of PGs in ATAAD by exploring their differential expression across previous aortic diameters, age groups, disease severity, and dissection stages. Characterizing these dynamics may improve risk stratification and inform stage-specific biomarker development.
In parallel, investigating PG metabolites particularly those produced by ADAMTS-mediated cleavage could uncover novel diagnostic or therapeutic targets [46]. In parallel, investigating PG metabolites, particularly those produced by ADAMTS-mediated cleavage, could reveal novel diagnostic or therapeutic targets. Key ADAMTS isoforms implicated in PG degradation in ATAAD include ADAMTS-1, ADAMTS-4, and ADAMTS-5 [41, 46, 67, 68]. ADAMTS-1 and ADAMTS-4 cleave versican, contributing to ECM remodeling, while ADAMTS-5 (aggrecanase-2) targets aggrecan and is elevated in thoracic aortic aneurysm and dissection tissues. Understanding the roles of these isoforms could guide future studies on PG-based biomarkers and potential therapeutic interventions.
Correlating tissue expression with serum levels and clinical outcomes will be essential for validating PG utility as circulating biomarkers. Integrating PGs with established markers like D-dimer may further enhance diagnostic precision, but this approach requires confirmation through large, multicenter studies before translation into clinical practice. Harnessing the diagnostic and prognostic power of PGs could mark a turning point in ATAAD care, transforming current paradigms through precision-driven, evidence-based strategies.
PGs are emerging as biologically meaningful serum biomarkers for TAD, directly reflecting ECM disruption rather than systemic fibrinolysis alone. Their release into circulation during medial degeneration provides a mechanistic link between molecular pathology and measurable serum signals, distinguishing them from established but nonspecific markers such as D-dimer.
Among the leading candidates, aggrecan and versican capture osmotic and structural perturbations within the aortic wall, while lumican reflects collagen remodeling and disease severity. Collectively, these molecules demonstrate superior specificity compared to conventional biomarkers and offer the potential to differentiate TAD from acute coronary syndromes or pulmonary embolism, conditions that frequently confound diagnosis. Emerging evidence also suggests prognostic applications, with circulating proteoglycan levels correlating with disease extent and postoperative outcomes.
Nevertheless, significant challenges remain before clinical translation can be achieved. The pathways governing proteoglycan release and clearance are incompletely understood, and current assays lack standardization across platforms. Large, prospective, multicenter studies are needed to define diagnostic thresholds, validate reproducibility, and evaluate performance across diverse populations. Furthermore, their integration into multimarker panels alongside D-dimer may provide the greatest diagnostic yield, balancing sensitivity with enhanced specificity.
In sum, proteoglycans hold promise to advance precision diagnostics in TAD, bridging molecular pathology with clinical practice. Their successful validation could transform current diagnostic frameworks, enabling earlier detection, improved risk stratification, and targeted perioperative management, ultimately improving patient survival and long-term outcomes.
JC and LW designed the study. GB, WS, KH, GF, YH and ZW performed the literature research and data collection. GF and WS wrote the manuscript. JC and WS revised the manuscript. All authors contributed to the editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
Our works are sponsored by the National Natural Science Foundation of China (No.82200526), the Shanghai Rising-Star Program (No.23QB1400900), the Shanghai “Rising Stars of Medical Talents” Youth Development Program (No.SHWRS2023-62), the Shanghai Oriental Elite Talent Program (2023) and the Clinical Research Fund of Shanghai Municipal Health Commission (No.20224Y0286).
The authors declare no conflict of interest. Jinmiao Chen is serving as one of the Editorial Board members of this journal. We declare that Jinmiao Chen had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Carmela Rita Balistreri.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.