

 , Chancui Deng 2, Ranzun Zhao 2,*
, Chancui Deng 2, Ranzun Zhao 2,*
1 The First Clinical Institute, Zunyi Medical University, 563000 Zunyi, Guizhou, China
2 Department of Cardiology, Affiliated Hospital of Zunyi Medical University, 563000 Zunyi, Guizhou, China
Abstract
Lipoprotein(a) (Lp(a)) is an established independent risk factor for atherosclerotic cardiovascular disease, particularly in the development of high-risk coronary plaques (HRPs). Elevated Lp(a) contributes to lipid accumulation, vascular inflammation, and plaque instability, primarily through oxidized phospholipids that promote monocyte adhesion and foam cell formation. Genetic studies have identified variants in the LPA gene as major determinants of Lp(a) levels, with higher concentrations consistently associated with adverse cardiovascular outcomes. Intravascular imaging techniques, such as optical coherence tomography and intravascular ultrasound, along with coronary computed tomography angiography (CCTA), have confirmed strong correlations between elevated Lp(a) and increased plaque burden, lipid-rich necrotic cores, and thin fibrous caps. In addition to coronary involvement, Lp(a) is implicated in systemic atherosclerosis, contributing to peripheral artery disease, cerebrovascular disease, and calcific aortic stenosis. Although conventional lipid-lowering therapies exert minimal effects on Lp(a), novel treatments such as proprotein convertase subtilisin/kexin type 9 inhibitors and RNA-targeted agents offer promising approaches to mitigating Lp(a)-mediated risk. This review summarizes current insights into the pathophysiological role of Lp(a) in HRP formation and progression, integrating evidence from genetic, mechanistic, and imaging studies, while highlighting emerging therapeutic strategies. Nonetheless, continued research is essential to enhance our understanding of Lp(a)-driven plaque vulnerability and to inform precision-targeted cardiovascular prevention.
Keywords
- lipoprotein(a)
- high-risk coronary plaques
- mechanisms
- atherosclerosis
- therapeutic strategies
Coronary high-risk plaques (HRPs) are considered the most critical potential lesions associated with acute coronary syndrome (ACS). Studies have indicated that patients with HRPs are 2 to 4 times more likely to experience future cardiovascular events than those without such plaque [1]. Both domestic and international guidelines consistently emphasize that achieving target levels of low-density lipoprotein (LDL-C) is the primary therapeutic goal for these HRPs [2]. However, even when LDL-C treatment reaches its target, residual cardiovascular risk remains significant, involving factors such as residual cholesterol risk, elevated triglycerides, impaired high-density lipoprotein (HDL-C) function, oxidative stress, inflammation, and metabolic factors like diabetes, insulin resistance, and obesity [2].
In the past few years, lipoprotein(a) (Lp(a)), a large glycoprotein attached to a low-density lipoprotein-like particle, has attracted increasing attention from researchers [3]. Multiple studies have indicated that elevated Lp(a) levels are an independent and heritable causal risk factor for atherosclerotic cardiovascular disease (ASCVD) [4]. Lp(a) plays a central role in lipid metabolism and is strongly associated with the development of atherosclerosis (AS) and HRPs. Furthermore, elevated Lp(a) levels can predict early-onset atherosclerotic vascular disease and influence coronary heart disease (CHD) risk in hypercholesterolemic patients [4]. Many authoritative consensus statements recommend incorporating Lp(a) into global cardiovascular risk assessment [5]. Therefore, grasping the role of Lp(a) in the development and progression of HRPs holds significant clinical value for reducing major adverse cardiovascular events (MACEs). This article provides a systematic review of the current understanding of Lp(a), its role, and related mechanisms in HRP formation, as well as relevant basic and clinical research, therapeutic strategies, and challenges.
Lp(a) was first identified and named in 1963 by Norwegian geneticist Kare Berg and Mohr J [6]. It is a liver-derived lipoprotein similar in structure to LDL; however, unlike LDL, it covalently binds to a unique apolipoprotein (a) (apo(a)) subunit via disulfide bonds [7]. Its lipid core mainly consists of cholesterol esters and triglycerides, while its outer layer includes phospholipids, free cholesterol, and apoB-100. Research indicates that Lp(a) plasma levels are primarily regulated by genetic factors, with significant variation among individuals (ranging from 1 to 200 mg/dL in the general population). Additionally, Lp(a) concentrations differ across racial/ethnic groups, and the relationship between Lp(a) levels and cardiovascular disease risk may vary by ethnicity [8]. But the UK Biobank paper by Amit Khera demonstrates that although levels of Lp(a) differ by ancestry, the relationship to ASCVD is the same when the data are plotted as a percentage of the population [9].
The pathophysiological roles of Lp(a) are mainly driven by its apo(a) subunit. The LPA gene, located on chromosome 6 at 6q2.6-2.7, encodes apo(a), which shares significant homology with plasminogen [10]. Apo(a) contains multiple kringle domains, including genetically variable kringle IV type 2 (KIV2) repeats, which significantly affect Lp(a) levels [11]. The number of KIV2 repeats correlates with higher Lp(a) levels and greater cardiovascular risk. Variations in apo(a) size, driven by KIV2 repeat variation, contribute to its genetic diversity [12]. Additionally, oxidized phospholipids (OxPL) bind with KIV10, influencing apo(a) structure and its functional consequences on Lp(a) metabolism [13]. Changes in the number of KIV2 structures inversely correlate with liver production rates, and larger isomers exhibit negative correlations with plasma Lp(a) concentrations, this is likely due to extended intracellular processing that results in enhanced degradation of larger isomers [14].
The quantity of kringle domains in an individual is determined by the genetic information inherited from each parent. Single-nucleotide polymorphisms (SNPs), which represent single-nucleotide changes at specific locations within a gene, contribute to genetic specificity and polymorphism [15]. Two large mendelian randomization studies have demonstrated that polymorphic variants of the LPA gene (e.g., rs3798220 and rs10455872) are directly strongly linked to increased Lp(a) levels and a higher risk of CHD, providing genetic support for the causal relationship between Lp(a) and CHD pathogenesis, suggesting that SNPs in the LPA gene play a crucial role in the formation of HRPs [11, 16].
Lp(a) is a key contributor to the onset and progression of AS, functioning
similarly to LDL-C as an initiating factor [4]. Lp(a) migrates to the arterial
wall, accumulating beneath the endothelium and forming lipid streaks that
initiate atherosclerosis [17]. Notably, due to its high lipophilicity, Lp(a)
accumulates more extensively than LDL-C. Within the arterial wall, Lp(a)
predominantly concentrates in the extracellular intima and subintima, where it
anchors via interactions between its lipoprotein structure and lysine binding
sites of apolipoprotein [13]. Lp(a) enhances inflammation through OxPLs and
promotes vascular lipid deposition [13]. The latter interaction explains the
differing affinities of Lp(a) and LDL for the arterial wall, potentially
contributing to AS progression [18]. Additionally,
Lp(a) regulates inflammatory cell aggregation within the vascular wall,
facilitating AS progression. It upregulates the expression of adhesion molecules,
such as vascular cell adhesion molecule-1 (VCAM-1) in coronary artery endothelial
cells (ECs), as well as intercellular adhesion molecule-1 (ICAM-1) in human
umbilical vein ECs. This effect is partly attributed to Lp(a)’s inhibitory action
on transforming growth factor-
Lp(a), as the primary carrier of OxPLs, plays a detrimental role in AS, vascular inflammation (VI), thrombosis, and endothelial dysfunction, contributing to cardiovascular events and the development of HRPs [27]. Although OxPLs are mainly generated through LDL oxidation, they are closely associated with Lp(a) in vivo. Low levels of Lp(a) help remove OxPLs from plasma and promote their degradation via Lp-PLA2, whereas high Lp(a) levels lead to excessive OxPL accumulation in the arterial wall, promoting macrophage apoptosis and plaque necrosis [28]. Lp(a) activates inflammatory pathways and lipid deposition through OxPLs, establishing a “pro-inflammatory - pro-oxidative - pro-thrombotic” vicious cycle. Studies show that OxPLs carried by Lp(a) activate endothelial cells, induce transendothelial migration of monocytes, and promote atherosclerosis [29]. Transcriptomic analysis reveals that Lp(a) drives endothelial pro-adhesive states through PFKFB3-dependent glycolysis [20]. Elevated Lp(a) levels correlate with the distribution of pro-atherogenic monocyte subsets in patients with stable AS, and the involvement of OxPL/apoB suggests that this may represent a potential therapeutic target for cardiovascular disease [27].
Lp(a) affects plaque stability through an increase in the expression of micro-PAR and ICAM-1, which affects plaque stability and leads to monocyte binding, resulting in an enlarged lipid core and a thinner fibrous cap in the plaque. Thus, Lp(a) plays a key role in the initiation of AS and plaque instability [21]. Collectively, these proatherogenic and proinflammatory actions of Lp(a) accelerate plaque progression and destabilization, as illustrated in Fig. 1. These molecular insights form the mechanistic foundation for understanding imaging manifestations of Lp(a)-related plaque vulnerability, which will be discussed in the following section.
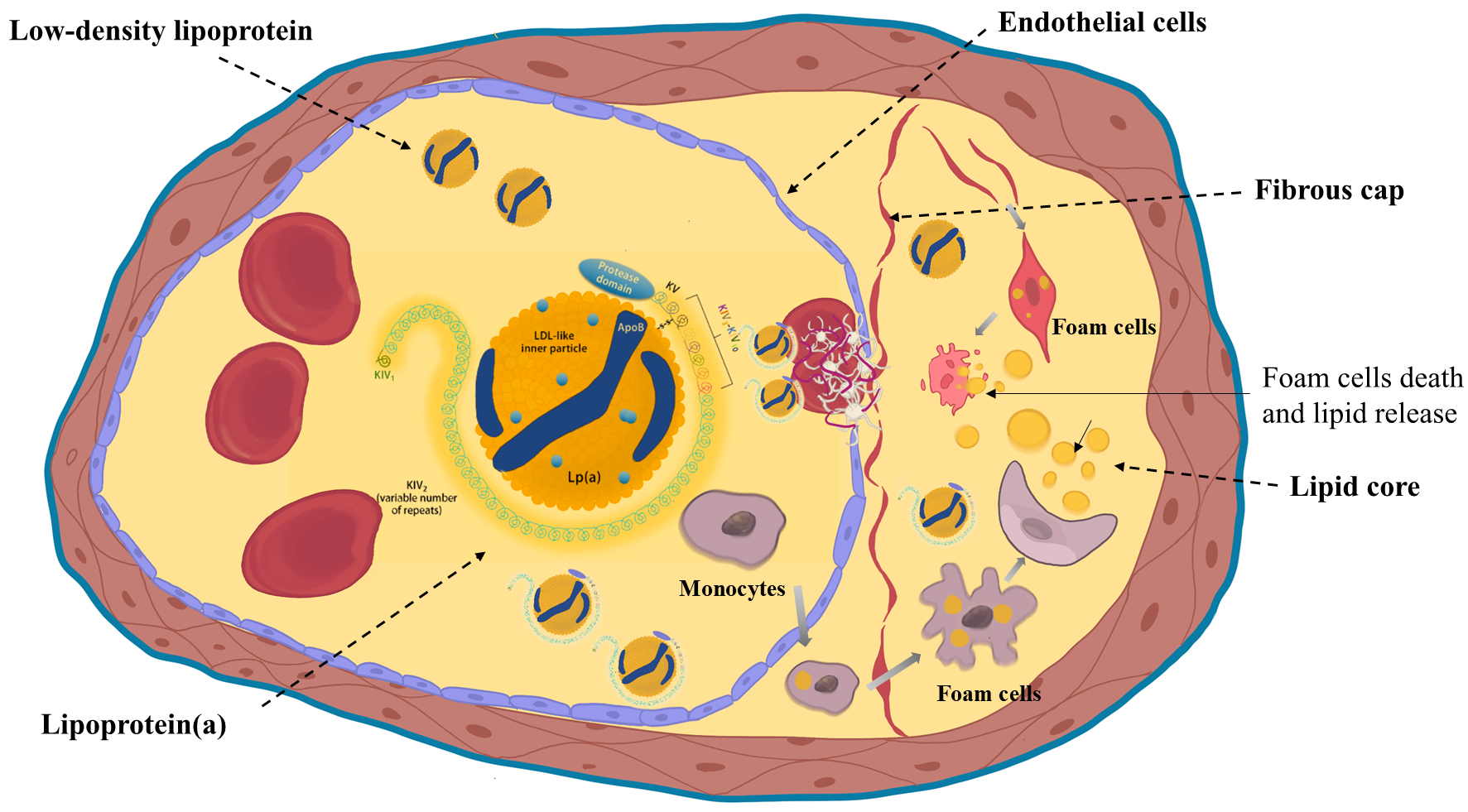
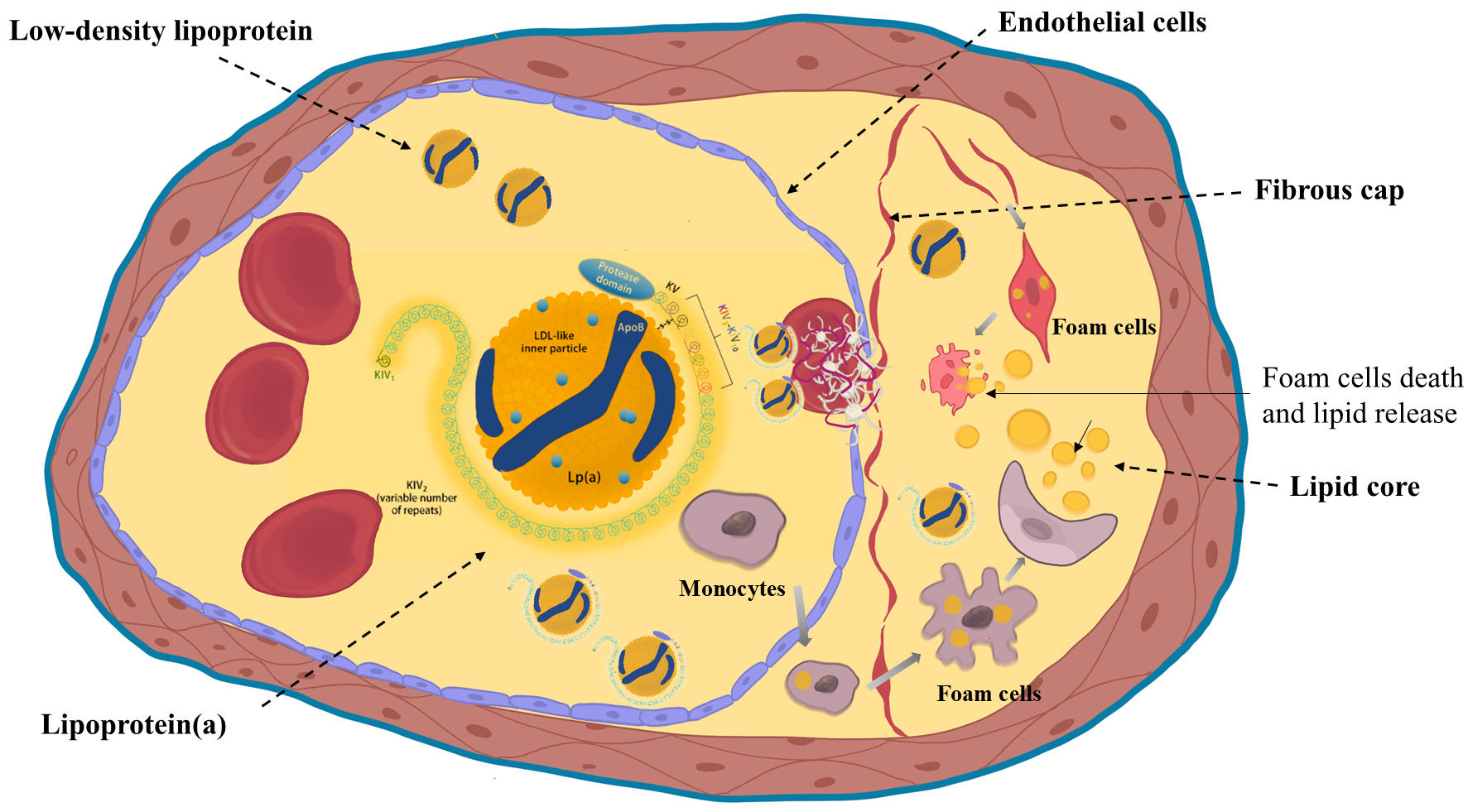 Fig. 1.
Fig. 1.
Structure of lipoprotein(a) and its atherogenic mechanisms. Lipoprotein(a) (Lp(a)) consists of an LDL-like particle and apo(a) rich in kringle domains. It promotes endothelial dysfunction, macrophage-mediated inflammation, and foam cell formation, contributing to atherogenesis. Created with Procreate. KIV, kringle IV; LDL, Low Density Lipoprotein; KV, kringle V; ApoB, apolipoprotein B.
Post-mortem studies have shown that plaque rupture is the main pathological change in acute MI, though the mechanism of transition from chronic coronary syndrome to ACS remains unclear. The concept of “vulnerable plaque”, introduced in 1994 by Muller et al. [30], refers to plaques at high risk of rupture despite not causing significant stenosis. Key features include large lipid cores, thin fibrous caps, and inflammation. Rupture of such plaques can induce thrombosis, leading to MACEs, including myocardial infarction (MI) or stroke. In 2003, Naghavi et al. [31] defined vulnerable plaques as “thrombogenic plaques with a high probability of progression to culprit plaques”. As clinical understanding advances, the term “high-risk plaque” has replaced “vulnerable plaque”, now encompassing not only plaque fragility but also systemic risk factors: vulnerable blood (hypercoagulable state due to imbalance between coagulation and fibrinolysis) and vulnerable myocardium (electrophysiological instability) [32]. This conceptual evolution marks a paradigm shift in clinical diagnosis and treatment from “local plaque intervention” to “systemic risk assessment”.
The advancement of plaque risk assessment has been driven by imaging
innovations. Coronary angiography, once the primary risk indicator, showed that
60% of acute coronary events arise from the rupture of non-severe stenotic
plaques, revealing the limitations of purely anatomical assessments [33].
Intravascular ultrasound (IVUS) and optical coherence tomography (OCT) have
revealed the in vivo microscopic characteristics of HRPs, including
lipid core
The instability of coronary HRPs is primarily influenced by local and systemic inflammation, blood flow shear stress, and matrix metalloproteinase activity [35]. HRPs include thin-cap fibroatheromas (TCFA) and calcified nodules, both of which are linked to plaque rupture, erosion, and coronary events. Plaque progression is characterized by VSMC apoptosis, matrix degradation, angiogenesis, arterial remodeling, fibrous cap rupture and thrombosis, as well as necrosis and calcification. Importantly, plaque vulnerability is a dynamic rather than a static phenomenon. With optimal medical therapy, as many as 75% of HRPs may stabilize over time, losing their high-risk characteristics. Conversely, in a certain proportion of patients, stable plaques may evolve into morphologically more fragile plaques. Studies have shown that inflammation is a crucial factor in both plaque development and rupture, especially through the infiltration of macrophages and T cells [35].
Coronary plaques continuously evolve through processes such as plaque hemorrhage, erosion, or rupture, which is a dynamic process. However, almost all technical methods for early identification of vulnerable plaques perform vascular imaging at a single time point. Even if the imaging modality can identify HRPs, it does not imply that treating a single plaque, such as placing a stent within such a plaque, can reduce subsequent morbidity and mortality. The ISCHEMIA (N Engl J Med 2020) study included 5179 patients with stable CHD. The findings suggested that traditional interventional treatment was not more effective in reducing MACEs when compared to drug therapy alone, implying that local stent implantation does not impact the systemic atherosclerotic process [36]. Steven Nissen pointed out that the concept of waiting for the rupture of a specific vulnerable plaque is overly simplistic. In fact, treatments proven to reduce coronary events are systemic, such as lipid-lowering therapy, antiplatelet therapy, and anti-inflammatory therapy. This suggests that the treatment concept focused on a single “vulnerable plaque” is no longer suitable. The essence of treatment lies in influencing the various factors involved in plaque development, and the comprehensive implementation of integrated intervention measures is of greater significance.
Lp(a) plays a key role in the development of high-risk coronary plaques by
promoting atherosclerosis, thrombosis, inflammation, and plaque instability. A
meta-analysis of 18 studies involving 23,105 asymptomatic patients (average age
55.9 years, 46.4% female) showed that elevated Lp(a) levels were significantly
associated with the risk of coronary artery calcification (CAC). Specifically,
for every 1 mg/dL increase in Lp(a), the risk of CAC
In addition, IVUS imaging results have shown that even after LDL-C levels have
been reduced to below 70 mg/dL or extremely low to
High levels of Lp(a) (
Research on high-position coronary artery plaques has advanced significantly. The integration of basic and clinical studies has facilitated early identification and intervention strategies for these plaques. Furthermore, Lp(a), as an important biomarker, warrants further exploration regarding its role in the formation of non-coronary HRPs.
Imaging Modalities-Based Insights into the Association Between Lipoprotein(a) and Coronary Plaque Characteristics. Integrating findings from OCT, IVUS, and CCTA studies provides structural validation of Lp(a)-driven plaque vulnerability (Table 1, Ref. [42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52]).
| Study names | Diagnosis of enrollment | Column count and grouping | Research methods | Content of follow-up | Results |
| Yuan X, et al. [42] | Patients with 125 DES-ISR lesions | High Lp(a): ( |
No intervention | ISNA and TCFA incidence; ROC AUC | Lp(a) identified as an independent predictor of ISNA (OR = 1.054, p |
| Low Lp(a): ( |
Retrospective, single-center observational study | ||||
| Erlinge D, et al. [43] | MI within 4 weeks, all culprit lesions successfully treated with PCI | N = 865 | PCI or NIRS + IVUS | Clinical tracking; no formal imaging or MACE | Lp(a) was uniquely associated with focal vulnerable plaques, while TC, LDL-C, non-HDL-C, and triglycerides showed no significant associations. These findings suggest Lp(a) contributes to plaque instability rather than plaque burden. |
| Low Lp(a): |
follow-up at 1, 6, 12 months, and annually (median 3.7 years) | ||||
| Intermediate Lp(a): 75–125 nmol/L | |||||
| High Lp(a): | |||||
| Fathieh S, et al. [44] | Adults from the BioHEART undergoing CCTA for suspected CAD | N = 1718 | Cross-sectional study; no longitudinal follow-up | CACS; Gensini Score; Plaque morphology (calcified, non-calcified, mixed) | Elevated Lp(a) ( |
| Low Lp(a): | |||||
| High Lp(a): | |||||
| Niccoli G, et al. [45] | Consecutive patients with ACS and obstructive CAD | CCTA (n = 500) | No intervention | Coronary lesion extent (Sullivan score, Bogaty score), plaque characteristics (lipid radian, TCFA) | Prevalence of lipid plaque in the High Lp(a) |
| OCT (n = 51) | Cross-sectional study | Lipid radian | |||
| High Lp(a): |
Baseline assessment | TCFA proportion | |||
| Yu MM, et al. [46] | Patients with stable chest pain (CCTA examination) | Derivation cohort: prospectively enrolled (n = 5607) Validation cohort: contemporaneous retrospective enrolled (n = 1122) | No intervention | Primary endpoint: fatal or nonfatal MI | High Lp(a) levels were independently linked to MI risk (HR 1.91, p |
| High Lp(a): Lp(a) |
Observational analysis | ||||
| Median follow-up 8.2 years (Q1–Q3: 7.2–9.3 years) | |||||
| Mszar R, et al. [47] | Eligibility criteria: Asymptomatic adults (40–65 years old) | N = 1795 | No intervention | Coronary CAC |
Elevated levels of Lp(a) were independently linked to the presence of any coronary plaque: (OR = 1.40, 95% CI: 1.05–1.86) |
| Baseline CCTA was performed. Not receiving any therapy for lowering lipids | High Lp(a): Lp(a) |
Cross-sectional analysis | The maximum stenosis was | ||
| Low Lp(a): Lp(a) |
A single baseline assessment | ||||
| O’Toole T, et al. [48] | Stable chest pain and no known coronary artery disease (data on Lp(a) measurements obtained on CTA). | N = 1815 | No intervention | Coronary stenosis ( |
Elevated Lp(a) showed an independent association with obstructive CAD (OR = 1.40, p |
| High Lp(a): Lp(a) |
Secondary analysis | ||||
| Low Lp(a): Lp(a) |
Short-term follow-up | ||||
| LDL-C stratification: | |||||
| Kaiser Y, et al. [49] | Patients with advanced stable CAD. | N = 191 | No intervention | Assessment of plaque type (total, low-attenuation, calcified, non-calcified) | The plaque with low attenuation (necrotic core) in the High Lp(a) showed significant annual progression |
| High Lp(a): Lp(a) |
Longitudinal observation | For every 50 mg/dL of Lp(a), necrotic core progression | |||
| Low Lp(a): Lp(a) |
Baseline and 12-month follow-up | ||||
| Berman AN, et al. [50] | Individuals aged |
N = 16,419 | No intervention | MACEs are defined as non-fatal MI, nonfatal ischemic stroke, coronary revascularization, and cardiovascular mortality. | Lp(a) independently predicts MACE in both primary and secondary prevention, with distinct risk thresholds for each group. |
| Based on Lp(a) percentile groups | Retrospective cohort study Median follow-up: 11.9 years (IQR: 6.2–14.4 years) | ||||
| Baseline ASCVD with a history of ASCVD or not | |||||
| Nurmohamed NS, et al. [51] | Patients with suspected CHD underwent baseline and repeat CCTA after 10 years. | N = 267 | Repeated CCTA imaging follow-up | Plaque volume percentage change, LDNP, PCAT | Percentage of plaque volume in the high Lp(a) group |
| High Lp(a): | |||||
| Wang X, et al. [52] | Patients undergoing CCTA | N = 1618 | No intervention | Unstable plaque: based on CCTA image features | The hLp(a)/NLR+ (both elevated Lp(a) and NLR) group had the highest risk of ASCVD (OR = 2.39, p |
| High Lp(a): plasma Lp(a) |
Cross-sectional analysis of a single assessment | Incidence of unstable plaque | |||
| High NLR: NLR |
DES-ISR, drug-eluting stent in-stent restenosis; ISNA, in-stent neoatherosclerosis; TCFA, thin-cap fibroatheromas; ROC, Receiver Operating Characteristic; AUC, Area Under the Curve; MI, myocardial infarction; PCI, percutaneous coronary intervention; NIRS, near-infrared spectroscopy; IVUS, intravascular ultrasound; MACE, major adverse cardiovascular event; TC, Serum total cholesterol; LDL-C, low-density lipoprotein; HDL-C, high-density lipoprotein; CCTA, coronary computed tomography angiography; CAD, Coronary artery disease; CACS, Coronary Artery Calcium Score; ACS, acute coronary syndrome; OCT, Optical Coherence Tomography; LAP, low-attenuation plaque; CAC, coronary artery calcification; HRPs, high-risk coronary plaques; ASCVD, atherosclerotic cardiovascular disease; CHD, coronary heart disease; LDNP, Low-density non-calcified plaque; PCAT, Pericardial adipose tissue; NLR, Neutrophil to Lymphocyte Ratio.
OCT, with its high resolution (10–20 µm), enables precise visualization
of hallmark features of vulnerable plaques, such as thin fibrous caps, lipid
cores, macrophage infiltration, and neovascularization. In a study involving 125
drug-eluting stent in-stent restenosis (DES-ISR) lesions, Yuan et al.
[42] found that patients with elevated Lp(a) levels (
In a subanalysis of the PROSPECT II study, Erlinge et al. [43] employed
near-infrared spectroscopy and intravascular ultrasound (NIRS-IVUS) to evaluate
coronary plaque characteristics in 865 patients post-myocardial infarction.
Although Lp(a) was not associated with overall plaque burden, higher Lp(a) levels
were significantly linked to focal high-risk features, such as max LCBI 4 mm
Coronary computed tomography angiography (CCTA) provides noninvasive plaque
characterization, including identification of calcified, non-calcified, and mixed
plaques. Several studies have highlighted strong associations between elevated
Lp(a) and morphologic features of vulnerable plaques. In the BioHEART study,
Fathieh et al. [44] reported that Lp(a)
Current evidence consistently supports a strong link between elevated Lp(a) and coronary plaque vulnerability. Lp(a) is notably associated with key high-risk plaque features detected by OCT (thin caps, lipid arcs), IVUS/NIRS (lipid-rich cores), and CCTA (mixed and low-attenuation plaques). These effects may be mediated by lipid accumulation, oxidative stress, inflammatory responses, and impaired fibrinolysis. While Lp(a)’s impact appears predominantly focal rather than diffuse, further validation is warranted. Future longitudinal studies incorporating multimodal imaging are needed to clarify the role of Lp(a) in plaque evolution and cardiovascular events. With Lp(a)-lowering therapies, such as antisense oligonucleotides and siRNA, advancing in clinical trials, their potential to stabilize high-risk plaques and reduce major adverse cardiovascular events merits close investigation.
Epidemiological studies have established that elevated Lp(a) contributes to the development of ASCVD and CAVD [53]. In patients with advanced stable CHD, high Lp(a) is associated with the rapid progression of low-attenuation plaques (necrotic core) in coronary arteries [49]. Lp(a) levels vary across racial groups, as shown in the INTERHEART study, which found that while elevated Lp(a) is linked to a higher risk of MI, it has a particularly strong effect in Latinos and South Asians, but a weaker association in Arabs and Africans [54]. The BiomarCaRE study, involving seven European cohorts with 56,804 participants followed for up to 24 years, also highlighted regional differences in Lp(a) levels. Elevated Lp(a) is strongly associated with an increased risk of coronary events and overall CVD, particularly in diabetic patients, providing a basis for targeting high-risk populations with Lp(a)-based therapies [55]. Thus, Lp(a) serves as a crucial target for therapeutic intervention in a significant portion of high-risk individuals.
International guidelines have yet to reach a consensus on Lp(a) testing
strategies and risk thresholds. The 2023 American College of Cardiology (ACC) and
American Heart Association (AHA) guidelines recommend Lp(a) screening for
patients with familial ASCVD, considering levels
High-risk individuals benefit from statin therapy, with the 2025 ACC/AHA
guidelines for ACS emphasizing intensified LDL-C management. All patients should
start high-intensity statins (atorvastatin 40–80 mg/d), targeting LDL-C
Lp(a) is a well-established independent risk factor for cardiovascular diseases. Among the lipid-lowering drugs currently in clinical use, only a limited number exhibit the added benefit of reducing Lp(a), which is often regarded as a supplementary effect when lowering LDL-C. While specific therapies targeting Lp(a) are under investigation, the only Food and Drug Administration (FDA)-approved treatment, lipoprotein apheresis (LA), remains accessible only to select patient populations in a few countries.
A pooled study of seven randomized controlled trials (RCTs) (n = 29,069) showed that while statin treatment reduced LDL-C by 39%, Lp(a) levels remained stable. Residual risk was dose-dependently related to Lp(a), confirming its independent predictive value in statin-treated populations, and supporting the development of Lp(a)-lowering therapies [60]. The JUPITER trial (n = 9612) further demonstrated that despite rosuvastatin significantly reducing LDL-C, Lp(a) remained an independent determinant of residual cardiovascular risk, with no racial differences, highlighting the need for targeted interventions to reduce ASCVD risk post-statin treatment [61]. Notably, a meta-analysis (n = 5256) found that high-intensity statins, such as atorvastatin, can increase Lp(a) levels, suggesting that statins may exacerbate Lp(a)-related cardiovascular residual risk via a dose-dependent mechanism [62]. Patients with high Lp(a) levels may need to avoid excessive intensification of statin therapy, highlighting the need for targeted intervention to optimize lipid management strategies.
Studies indicate that elevated Lp(a) levels, independent of LDL-C, significantly increase atherosclerotic risk [63]. Genetic prediction analyses reveal a linear dose-response relationship between Lp(a) and cardiovascular risk: a 10 mg/dL reduction in Lp(a) lowers CHD risk by 5.8% (OR = 0.942), while a 10 mg/dL decrease in LDL-C reduces risk by 14.5% (OR = 0.855). Equivalence analysis suggests that lowering Lp(a) by about 101.5 mg/dL provides the same risk reduction as decreasing LDL-C by 38.67 mg/dL (1 mmol/L). Furthermore, the Lp(a)-CHD risk relationship remains unaffected by statins, PCSK9 inhibitors, and ezetimibe’s impact on LDL-C. Clinically meaningful benefits require a significant reduction in Lp(a) (around 100 mg/dL). Even with effective LDL-C reduction, elevated Lp(a) levels continue to be linked with a higher risk of CVD, likely through mechanisms independent of traditional lipid-lowering pathways [64]. The UK Biobank study (n = 385,917) found that reducing Lp(a) by 50 mg/dL lowers peripheral arterial (PAD) risk (HR = 0.73) and venous thromboembolism risk (HR = 0.95), with no synergistic effect from LDL-C control or lifestyle interventions [65]. These results suggest that Lp(a)-targeted therapies can complement current cardiovascular risk management and support the need for broader Lp(a) testing to identify high-risk patients [66].
Drugs aimed at reducing LDL-C, including PCSK9 inhibitors, show significant
potential for lowering Lp(a). In the FOURIER trial (n = 25,096), evolocumab
reduced Lp(a) by 26.9% and decreased MACE risk by 23% in patients with high
baseline Lp(a) levels, independent of LDL-C [67]. Post hoc analyses from the
FOURIER and ODYSSEY OUTCOMES trials consistently demonstrated that PCSK9
monoclonal antibodies (mAbs) significantly reduce plasma Lp(a) levels by
approximately 20% to 30%, leading to a reduction in CVD risk independent of
LDL-C lowering. Notably, this effect was more pronounced in patient subgroups
with higher baseline Lp(a) levels and greater reductions in Lp(a) [68, 69].
Another RCT (NCT03570697) involving NSTEMI patients showed that evolocumab
combined with statins significantly reduced LDL-C and improved coronary artery
plaque stability, as evidenced by increased minimum fibrous cap thickness,
significant plaque volume regression, decreased macrophage index, and reduced
maximum lipid arc [70]. A retrospective analysis evaluated the effect of
evolocumab on plaque stabilization in patients with varying baseline Lp(a)
levels. In those with elevated Lp(a) (
A healthy lifestyle is the primary measure for preventing ASCVD and plays a crucial role in regulating blood lipid [56, 59]. However, Lp(a) concentration is mainly determined by genetic factors and is less influenced by exercise and diet [72]. Weight loss, a relatively high intake of saturated fatty acids, wine consumption, and vigorous exercise appear to correlate with reduced plasma Lp(a) levels. This effect is more pronounced in patients with higher baseline Lp(a) levels. In contrast, regular moderate physical activity does not seem to significantly influence plasma Lp(a) concentrations [73]. Therapeutic lifestyle changes (TLCs) are effective for CVD prevention. Lp(a) levels are negatively correlated with dietary saturated fatty acids (SFA) intake, suggesting SFA affects Lp(a) through epigenetic regulation of lipid metabolism. Healthy lifestyle indicators, such as fish intake, body mass index (BMI), whole grain intake, and reduced sodium and sugar intake, contribute to better Lp(a) management [74]. Regular moderate-intensity exercise improves lipoprotein metabolism but has no significant effect on Lp(a). High-intensity weight-bearing training may increase Lp(a), though its clinical significance remains unclear. Given the synergistic risks of LDL-C and Lp(a), lifestyle adjustments to lower LDL-C and increase HDL-C are recommended. Moderate aerobic exercise and lifestyle changes, such as controlling blood pressure, normalizing blood glucose, quitting smoking, and reducing alcohol intake, enhance cardiovascular health. Although the precise influence of lifestyle on Lp(a) levels is still unclear, an 8-year cohort study revealed during follow-up that a healthy lifestyle was significantly correlated with a lower risk of cardiovascular disease, irrespective of Lp(a) concentrations [75].
Given the limited efficacy of conventional therapies in managing Lp(a)-related residual risk, the need for targeted Lp(a)-lowering strategies has become increasingly urgent. Recently, several RNA-based therapeutics (e.g., Pelacarsen, Olpasiran, SLN360) have entered clinical trials, showing marked reductions in Lp(a) levels and preliminary evidence of safety and benefit.
Antisense oligonucleotides (ASOs) are short, single-stranded DNA sequences that
can hybridize with target mRNA to form ASO-RNA duplexes. Through the mechanism of
steric hindrance and ribonuclease H-mediated degradation of the RNA strand, ASOs
modulate the expression of specific molecules. Early-generation ASO drugs
targeting Lp(a) clearance included ISIS-APO(a)Rx and IONIS-APO(a)Rx. Pelacarsen,
as a second-generation IONIS-APO(a)Rx drug, exhibits enhanced durability and
stability [76]. Results from a Phase IIb trial demonstrated that monthly
subcutaneous injections of 80 mg over 6 months reduced median Lp(a) levels by
80%, with 98% of patients achieving Lp(a) levels below 50 mg/dL and showing
favorable safety profiles. The global Phase III trial (NCT04023552), enrolling
8300 patients with cardiovascular disease and Lp(a)
Olpasiran, a GalNAc-conjugated siRNA molecule, inhibits Lp(a) particle assembly
by degrading apo(a) mRNA and blocking LPA gene expression. Phase I
trials showed dose-dependent reductions in Lp(a) of
Lepodisiran is a long-acting GalNAc-conjugated siRNA that degrades apo(a) mRNA via the RNA-induced silencing complex (RISC) after entering liver cell nuclei, thereby inhibiting Lp(a) synthesis. A first-in-human study showed that a single 608 mg dose reduced Lp(a) by 97% at 48 weeks, with reductions maintained at 40%–50% through week 48. There were no serious adverse events during the study period, and the drug was well-tolerated. A phase III trial is underway in China to evaluate its efficacy in reducing MACE in ASCVD patients with elevated Lp(a) [79].
SLN360, a highly selective siRNA, significantly reduced Lp(a) by 96% after a single 600 mg injection, with effects lasting over 6 months. The drug was well tolerated, with no serious adverse events reported. It demonstrated potent and durable inhibition of Lp(a), leading to a phase III trial for high-risk ASCVD patients [80].
Muvalaplin (LY3473329) is the first oral drug targeting Lp(a) assembly. It inhibits Lp(a) formation by disrupting the interaction between apo(a) and apoB. Phase II trials showed that daily 30–100 mg doses for 12 weeks reduced Lp(a) by 65%, significantly lowering levels in high-risk cardiovascular patients [81].
Additionally, Lipoprotein apheresis (LA) has become an important therapeutic
option for patients with drug-resistant angina and elevated Lp(a), providing
benefits such as improved myocardial perfusion, reduced atherosclerotic burden,
and enhanced exercise capacity [82]. The use of LA varies across countries; in
Spain, the UK, and Japan, it is restricted to patients with homozygous or
heterozygous familial hypercholesterolemia (HoFH/HeFH), whereas in Germany and
the US, its indications have been extended to include drug-resistant
Lp(a)-elevated CVD [83, 84]. Despite these advancements, LA’s clinical
application remains limited by its invasiveness (requiring 1–2 sessions per
week, lasting 2–4 hours each), high cost (
Clinical studies have shown that LDL-C levels are inversely associated with mortality risk in AMI patients, especially those with high inflammatory risk, a phenomenon known as the “lipid paradox”. Statins not only lower LDL-C but also exert anti-inflammatory effects, which may mitigate the lipid paradox. These findings emphasize the need for personalized lipid-lowering strategies based on inflammatory status [85].
The OCEAN(a)-DOSE trial (n = 53) showed significant variability in Lp(a)
measurements (CVi = 10%) among ASCVD patients with Lp(a)
The structural diversity of Lp(a) influences its particle size, which in turn affects measurement outcomes. Variability in the number of KIV-2 repeat sequences within apo(a) leads to substantial differences in particle size [87]. Furthermore, due to molecular weight differences among apo(a) isoforms, there is no fixed conversion factor between mass-based units (mg/dL) and particle-based units (nmol/L) [88]. Traditional immunoassays may underestimate small-particle isoforms and overestimate large-particle isoforms due to cross-reactivity or misrecognition of repetitive structural motifs. Additionally, discrepancies in calibrator isoform composition across commercial assay kits contribute to poor inter-assay comparability, particularly in populations with elevated Lp(a) levels [89].
Current strategies and guidelines addressing these challenges include the use of isoform-insensitive assays that target non-KIV-2 regions of apo(a), such as the KV domain, to ensure each Lp(a) molecule is detected only once. The WHO/International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) recommends the use of assays calibrated with the WHO/IFCC reference material (SRM 2B), including ELISA or turbidimetric methods [90]. However, fully isoform-independent commercial assays are not yet available, and users should verify whether the manufacturer specifies “isoform insensitivity” in the product documentation. Unit selection is also critical: in clinical settings, particle concentration expressed in nmol/L is preferred over mass concentration in mg/dL, as cardiovascular risk correlates more closely with Lp(a) particle number. Due to the structural heterogeneity of Lp(a), thresholds derived from different studies (e.g., 50 mg/dL vs. 125 nmol/L) cannot be directly interchanged and must be interpreted according to their original measurement units.
The management strategy for HRPs targeting Lp(a) primarily includes: ① Lipid management: combined use of statins and ezetimibe, with PCSK9 inhibitors if necessary. However, statins may increase plasma Lp(a) levels, warranting investigation into their impact on residual cardiovascular risk. ② Family screening: first-degree relatives should undergo Lp(a) testing. ③ Anti-thrombotic therapy: aspirin may be considered for high-risk individuals [62]. ④ Imaging monitoring: regular assessment of atherosclerotic plaque progression optimizes Lp(a)-targeted high-risk coronary plaque management strategies. Recent advances in Lp(a)-lowering therapies, including antisense oligonucleotides and siRNA-based approaches, offer promising potential for reducing residual cardiovascular risk (Table 2).
| Drug | Mechanism | Route | Key Trial | Peak efficacy | Frequency | Safety profile | Phase III status |
| Pelacarsen | Second-gen ASO (IONIS-APO(a)Rx) | Subcutaneous | Phase IIb + HOR IZON | 80% (median) | Monthly | Favorable, no SAEs | NCT04023552, completion 2025 |
| Olpasiran | GalNAc-conjugated siRNA | Subcutaneous | OCEAN(a)-DOSE (Phase II) | Every 12 weeks | Mild injection site pain | NCT05581303, completion Dec 2026 | |
| Lepodisiran | Long-acting GalNAc-siRNA | Subcutaneous | First-in-human | 97% at 48 weeks | Single dose for 48 weeks | Well tolerated | Phase III ongoing (China) |
| SLN360 | Highly selective siRNA | Subcutaneous | First-in-human | 96% (single 600 mg dose) | No SAEs reported | Phase III initiated (high-risk ASCVD) | |
| Muvalaplin | Oral small molecule (apo(a)-apoB disruptor) | Oral | Phase II | 65% (12 weeks) | Daily | Well tolerated | Phase III has not yet been initiated |
ASO, Antisense oligonucleotide.
Future research should focus on elucidating the functional mechanisms of Lp(a) in coronary HRPs, including its associations with plaque instability, hemodynamic changes, and intercellular interactions. Simultaneously, by integrating high-resolution imaging technologies and multi-omics analyses, the correlations between Lp(a) levels and plaque structural and functional indicators should be explored to refine measurement methodologies and enhance risk assessment models. The efficacy of combining Lp(a)-targeted therapies (e.g., ASOs, RNA interference) with conventional treatments (lipid-lowering, anti-inflammatory, and antithrombotic therapies) should be evaluated. Additionally, further investigation is needed on the impact of statin-induced Lp(a) elevation on residual cardiovascular risk. It is essential to integrate existing treatment modalities to assess their long-term clinical benefits and safety, thereby providing robust evidence-based support for reducing the risk of coronary events.
In summary, Lp(a) holds great potential as a key biomarker and therapeutic target for coronary HRPs. Advances in omics, big data, and personalized medicine are expected to improve risk assessment, treatment strategies, and outcomes, reducing acute coronary event incidence.
In summary, converging evidence from genetic, mechanistic, and intravascular imaging studies has firmly established lipoprotein(a) as both a biomarker and a causal mediator of coronary plaque vulnerability. Elevated Lp(a) promotes lipid accumulation, inflammatory activation, and prothrombotic changes within the arterial wall, leading to accelerated plaque progression and destabilization. Advanced imaging modalities, including OCT, IVUS, and CCTA, have provided in vivo validation that high Lp(a) levels are strongly associated with thin-cap fibroatheromas, enlarged lipid cores, and features of neoatherosclerosis—linking molecular abnormalities to structural plaque instability.
Despite contemporary lipid-lowering therapies, the residual cardiovascular risk attributable to elevated Lp(a) remains substantial. These insights highlight the necessity of integrating molecular characterization with plaque imaging to enable precise risk stratification and tailored therapeutic interventions. Such integration will be crucial for transforming our understanding of Lp(a)-driven atherogenesis into clinically actionable strategies.
SF, RZ, and CD conceptualized the research. Literature searches were conducted on SF and CD. Data organization was carried out by SF and CD. The initial draft was written by SF. CD and RZ reviewed and edited the draft. RZ provided resources for this review. Supervised studies by RZ and CD. All authors have contributed to the editing and revision of the manuscript, read and approved the published version, and agreed to be responsible for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.