



 , Matteo Morello 2,3, Paola Pastena 4, Marco Bernardi 5, Luigi Spadafora 5, Stefano Cacciatore 6,7, Francesco Perone 8, Giuseppe Caminiti 9,10, Pierre Sabouret 11,12, Arturo Cesaro 13,14, Francesca Maria Di Muro 15,16,†, Michele Golino 2,17,*,†
, Matteo Morello 2,3, Paola Pastena 4, Marco Bernardi 5, Luigi Spadafora 5, Stefano Cacciatore 6,7, Francesco Perone 8, Giuseppe Caminiti 9,10, Pierre Sabouret 11,12, Arturo Cesaro 13,14, Francesca Maria Di Muro 15,16,†, Michele Golino 2,17,*,†
1 Department of Systems Medicine, University Tor Vergata, 00133 Rome, Italy
2 Robert M. Berne Cardiovascular Research Center and Department of Medicine, University of Virginia, Charlottesville, VA 22908, USA
3 Department of Molecular and Translational Medicine, University of Brescia, 25121 Brescia, Italy
4 Division of Cardiology, Department of Pediatrics, Columbia University Irving Medical Center, New York, NY 10032, USA
5 Department of Medico-Surgical Sciences and Biotechnologies, Sapienza University, 00185 Latina, Italy
6 Department of Geriatrics, Orthopedics, and Rheumatology, Università Cattolica del Sacro Cuore, 00168 Rome, Italy
7 Fondazione Policlinico Universitario “Agostino Gemelli” IRCCS, 00168 Rome, Italy
8 Cardiac Rehabilitation Unit, Rehabilitation Clinic “Villa delle Magnolie”, Castel Morrone, 81020 Caserta, Italy
9 Department of Human Science and Promotion of Quality of Life, San Raffaele Open University, 00166 Rome, Italy
10 Cardiology Rehabilitation Unit, IRCCS San Raffaele, 00166 Rome, Italy
11 Heart Institute, Boulevard de l'Hôpital, ACTION Study Group-CHU Pitié-Salpétrière Paris, 75013 Paris, France
12 Collège National des Cardiologues Français (CNCF), 75014 Paris, France
13 Department of Translational Medical Sciences, University of Campania “Luigi Vanvitelli”, 80138 Naples, Italy
14 Division of Cardiology, A.O.R.N. “Sant’Anna e San Sebastiano”, 81100 Caserta, Italy
15 Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA
16 Cardiology Unit, Cardiovascular and Thoracic Department, University Hospital San Giovanni di Dio e Ruggi d’Aragona, 84131 Salerno, Italy
17 Pauley Heart Center, Virginia Commonwealth University, Richmond, VA 23298, USA
†These authors contributed equally.
Abstract
Vitamin D is a key regulator of calcium and phosphorus homeostasis; meanwhile, the dietary absence of vitamin D represents the most common nutritional deficiency worldwide. The discovery of vitamin D receptors and conversion enzymes within the cardiovascular system has fueled growing interest in the potential roles of vitamin D beyond bone health. Indeed, preclinical studies have suggested that vitamin D might regulate vascular tone and exert antifibrotic and anti-remodeling effects on the myocardium. Furthermore, a deficit in vitamin D has been associated with an increased risk of hypertension, atherosclerosis, and heart failure. These findings have prompted several interventional studies to investigate whether vitamin D supplementation can mitigate cardiovascular risk. However, current evidence regarding the cardiovascular benefits of vitamin D intake remains inconsistent and inconclusive. This review aims to provide a comprehensive overview of the “good”, the “bad”, and the “unknown” aspects of the relationship between vitamin D and cardiovascular disease.
Graphical Abstract
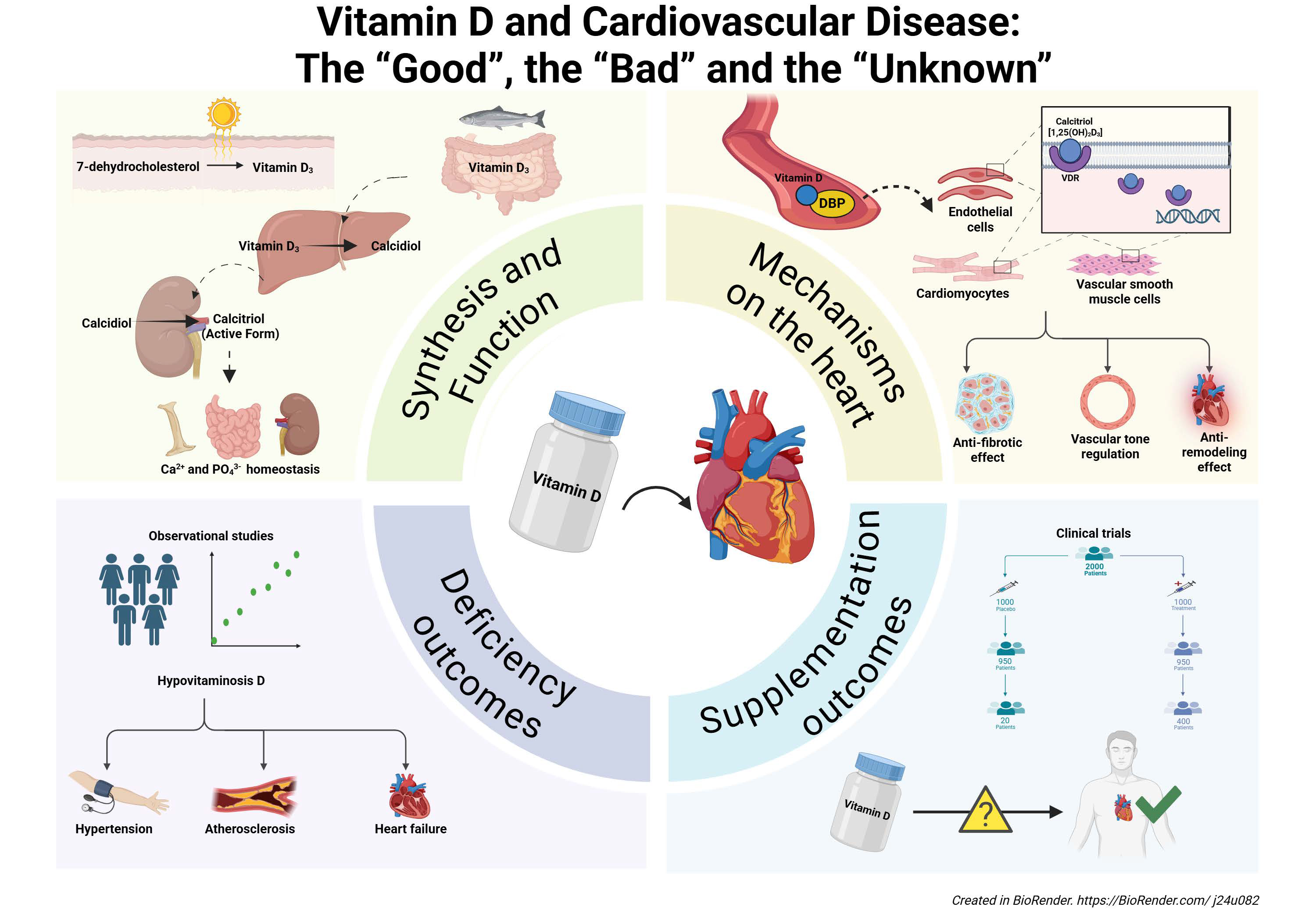
Keywords
- vitamin D deficiency
- cardiovascular diseases
- hypertension
- heart failure
- atherosclerosis
Despite significant advances in the prevention and treatment of cardiovascular disease (CVD), one person in the United States dies from heart disease or stroke every 34 seconds [1, 2]. This alarming statistic underscores the urgent need to identify novel, modifiable risk factors beyond traditional targets such as hypertension, hyperlipidemia, or diabetes. Among emerging candidates, Vitamin D, a fat-soluble nutrient historically associated with bone homeostasis, has garnered increasing attention for its potential role in CVD health.
Originally discovered in the context of rickets, vitamin D has long been recognized as a cornerstone of calcium and phosphate metabolism [3]. However, the subsequent identification of vitamin D receptors (VDRs) and associated metabolic enzymes widely expressed across the cardiovascular tissues, including cardiomyocytes, vascular smooth muscle cells, and endothelial tissue, have led to the hypothesis that vitamin D may exert pleiotropic effects on key pathophysiological mechanisms such as vascular tone regulation, myocardial remodeling, inflammation, fibrosis, and atherogenesis [4, 5, 6].
Vitamin D deficiency, or hypovitaminosis D, is still considered the most prevalent nutritional deficiency worldwide. It affects about one billion people, with high prevalence in older adults and those with limited sun exposure or darker skin pigmentation [7, 8]. In addition to its well-established role in bone health and calcium and phosphorus homeostasis, low vitamin D levels may increase the risk of several CVDs, such as hypertension, coronary artery disease, and heart failure (HF) [9, 10, 11]. This knowledge has raised the possibility that vitamin D could represent a low-cost and widely accessible tool for cardiovascular risk reduction.
Several interventional studies have reported conflicting results and no consistent cardiovascular benefits. Even a large meta-analysis has failed to establish a definitive role for vitamin D supplementation in cardiovascular risk reduction [12]. In this review, we examined the “good”, the “bad”, and the “unknown” of the relationship between vitamin D and cardiovascular health.
Vitamin D exists in two dietary forms: D3 (cholecalciferol) and D2 (ergocalciferol),
which differ slightly in structure [13]. Vitamin D metabolism begins from the
synthesis of 7-dehydrocholesterol, which undergoes two sequential hydroxylations:
the first, by hepatic 25-hydroxylase, produces 25-hydroxyvitamin D (25(OH)D), and
the second, by 1-
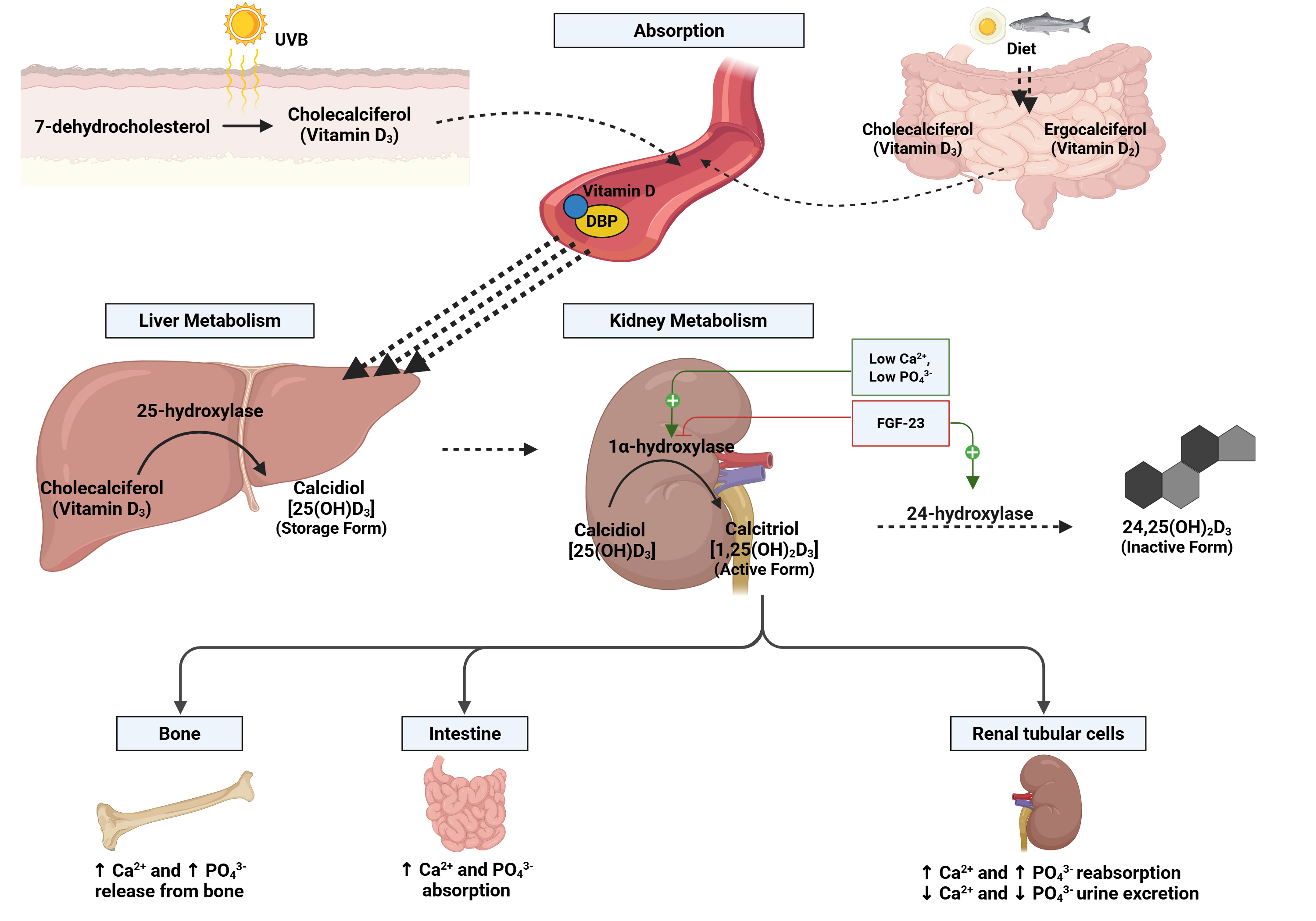 Fig. 1.
Fig. 1.
Vitamin D metabolism pathway. Vitamin D metabolism begins with
its production in the skin (under ultraviolet B (UVB) light, as cholecalciferol
or Vitamin D3) or its intake from food (as cholecalciferol, Vitamin D3, or
ergocalciferol, Vitamin D2). After absorption, Vitamin D binds to a transport
protein (DBP) and travels to the liver. There, it is converted into calcidiol
[25(OH)D3], the storage form, by the enzyme 25-hydroxylase. In the kidneys,
calcidiol is converted into calcitriol [1,25(OH)2D3], the active form,
through 1
Both circulating 25(OH)D and 1,25(OH)2D are mainly bound to vitamin D
binding protein (DBP) and albumin; however, the half-life of circulating 25(OH)D
(10–20 days) is higher than that of 1,25(OH)2D (10–20 hours), due to the
higher affinity for DBP of the former [14, 15]. Circulating 1,25(OH)2D is
tightly regulated by parathyroid hormone (PTH) and fibroblast growth factor-23
(FGF-23) to maintain plasma calcium and phosphate within their physiological
ranges [7, 16]. Calcitriol binds the vitamin D receptor (VDR), inducing a
conformational change that leads to hetero-dimerization with the retinoid X
receptor (RXR) and translocation of this complex into the nucleus, where it binds
to the promoter region of more than 200 target genes [17]. Although 25(OH)D is
the preferred biomarker of vitamin D level due to its longer half-life, there is
still no universal consensus on the optimal threshold values. Most guidelines
define deficiency as a serum concentration below 30 nmol/L. In contrast,
sufficiency is variably defined, ranging from
Vitamin D plays an active role in cardiovascular physiology, primarily mediated by the expression of its receptors and activating enzymes in cardiomyocytes, endothelial cells, and vascular smooth muscle cells [19]. Preclinical studies have shown that VDR-null mice exhibit increased left ventricular mass, elevated atrial natriuretic peptide levels, and dysregulation of cardiac metalloproteinases and fibroblasts. These alterations promote fibrotic extracellular matrix deposition, leading to ventricular dilatation and impaired electromechanical coupling [20, 21, 22, 23, 24, 25].
In endothelial cells, VDR activation regulates vascular endothelial growth factor expression, influences calcium influx, and modulates the vascular endothelium-dependent tone [26, 27]. In VDR-deficient mice, endothelial nitric oxide (NO) synthase is reduced by more than 50%, and acetylcholine-induced aortic relaxation is considerably impaired [28, 29]. The increased renin expression and renin-angiotensin-aldosterone system (RAAS) activation have been suggested as additional mechanisms, as observed in Fig. 2 [30]. Therefore, in hypertensive rats, chronic treatment with 1,25(OH)2D showed to reduce reactive oxygen species (ROS) levels and cyclooxygenase-1 (COX-1) expression with beneficial effects on blood pressure [31].
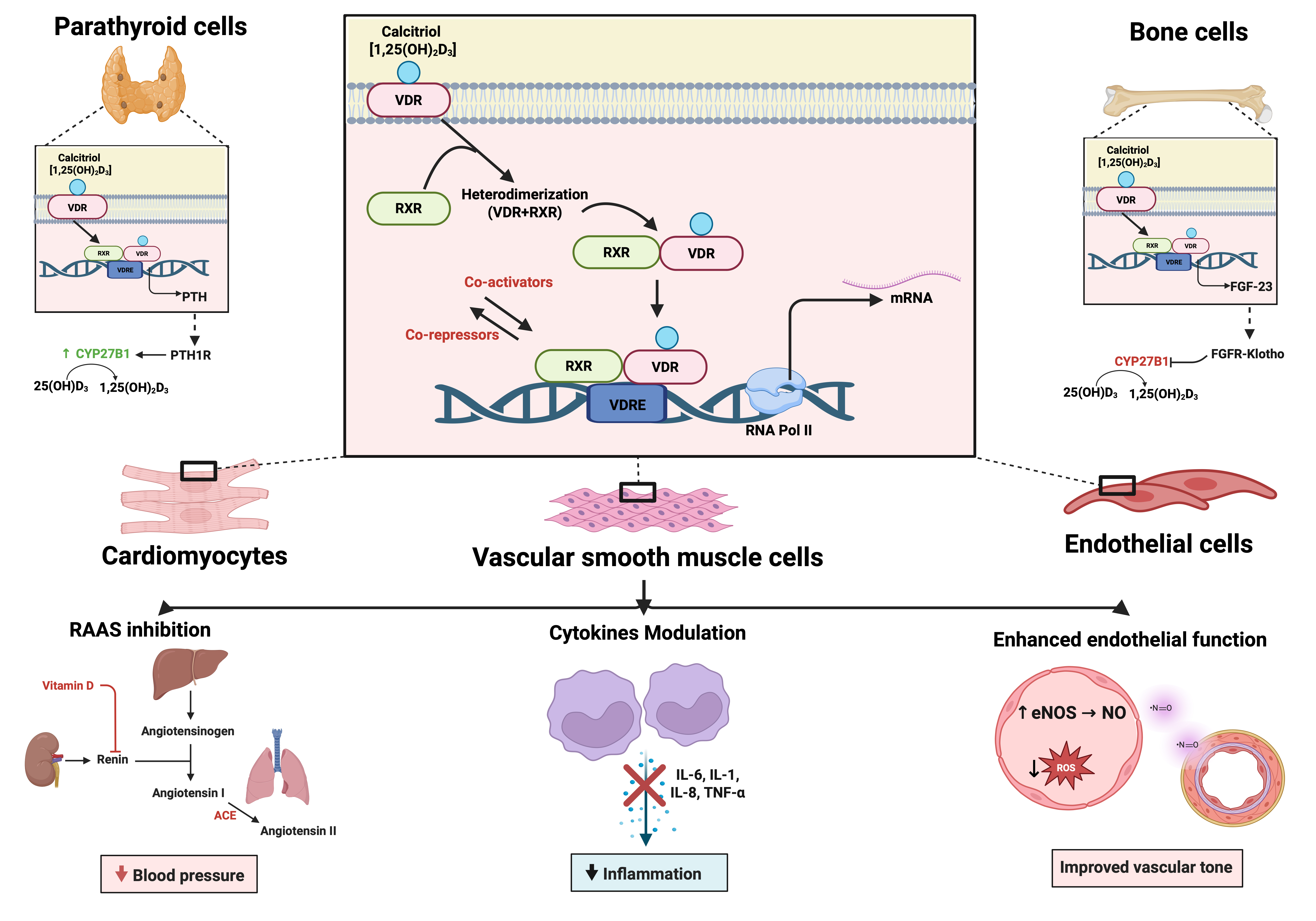 Fig. 2.
Fig. 2.
Mechanisms of Vitamin D on the cardiovascular system. The
active form of Vitamin D (calcitriol or 1,25(OH)2D3) binds to the Vitamin D
receptor (VDR), which heterodimerizes with the retinoid X receptor (RXR). This
complex binds to vitamin D response elements (VDREs) in target genes, modulating
transcription via co-activators and co-repressors. The effects are cell-type
specific: in parathyroid cells, Vitamin D downregulates parathyroid hormone (PTH)
expression; in bone cells, it promotes FGF-23 production; and in cardiovascular
cells-including cardiomyocytes, vascular smooth muscle cells, and endothelial
cells-it mediates beneficial effects via: inhibition of the
renin-angiotensin-aldosterone system (RAAS), downregulation of pro-inflammatory
cytokines (IL-6, IL-1, IL-8, TNF-
Vitamin D also exerts significant anti-inflammatory effects, modulating both
innate and adaptive immune responses. It suppresses proinflammatory cytokines
such as interleukin (IL)-6, tumor necrosis factor-alpha (TNF-
Beyond its broad anti-inflammatory effects, vitamin D plays a key role in
modulating the pathogenesis of atherosclerosis. It influences monocyte activity
and the regulation of matrix metalloproteinases (MMPs). Specifically, vitamin D
has been shown to reduce the expression of TNF-
Nakagawa et al. [43] demonstrated that 1,25(OH)2D downregulates MMP-2 and MMP-9 expression in cultured cells, stabilizing atherosclerotic plaques and reducing the risk of rupture, thrombosis, and lumen obstruction. Vitamin D also reduces cholesterol accumulation in macrophages and inhibits low-density lipoprotein (LDL) uptake within atheromatous plaques [44]. Furthermore, it modulates thrombogenic activity by regulating thrombomodulin and tissue factor expression in monocytes, thereby affecting platelet aggregation and coagulation potential [45].
Vitamin D improves endothelial function by upregulating endothelial NO synthase
(eNOS) through phosphoinositide 3-kinase/protein kinase B (PI3K/AKT)-dependent
pathways, enhancing NO production and reducing endothelial oxidative stress [46].
Furthermore, vitamin D suppresses
NF-
In apolipoprotein E-deficient mouse models, active vitamin D administration reduced the number of atherosclerotic lesions, decreased macrophage infiltration, and limited CD4+ T-cell accumulation in the aortic sinus. Oral calcitriol further attenuated atherosclerosis by promoting the induction of regulatory T cells and immature dendritic cells with tolerogenic properties [49].
Experimental evidence also indicates that vitamin D enhances insulin secretion
and sensitivity by modulating both pancreatic and inflammatory pathways [50]. The
discovery of VDRs in pancreatic
In vitro studies using rat insulinoma-derived
These findings suggest that vitamin D may contribute to glycemic control via
direct effects on pancreatic
The role of vitamin D in cardiovascular health is complex and, at times,
controversial. While vitamin D deficiency is associated with adverse outcomes,
excessive levels may also have harmful effects. One major concern involves the
potential link between vitamin D supplementation and vascular calcification.
Vascular calcification results from the deposition of calcium phosphate crystals
within arterial walls, reducing arterial compliance and increasing cardiovascular
risk. Preclinical studies have reported that supraphysiological vitamin D levels
can induce vascular calcification in animal models [55, 56]. For instance, rats
administered high doses of vitamin D showed significant arterial stiffness and
aortic calcification [57]. Similarly, vitamin D and calcium supplementation
promoted vascular calcification in pseudoxanthoma elasticum mouse models [58].
Notably, such vascular remodeling appeared reversible upon reduction of vitamin D
levels [57]. Human data also support this association. Case reports and small
cohort studies have described metastatic arterial calcifications and soft-tissue
calcifications in patients with hypervitaminosis D, hypercalcemia, and extremely
elevated 25(OH)D levels (e.g.,
Large-scale RCTs, including patients with pre-hypertension or stage I
hypertension, further reinforced these findings. A six-month study comparing
daily high-dose (4000 IU) versus low-dose (400 IU) cholecalciferol found no
significant difference in 24-hour systolic blood pressure (–0.8 vs –1.6 mm Hg;
p = 0.71) [68]. Likewise, an Austrian RCT of 188 hypertensive patients
receiving 25(OH)D
Large-scale randomized controlled trials underscore this lack of clinical benefit (Table 1, Ref. [72, 73, 74]). VINDICATE (VItamiN D treatIng Patients with Chronic heArT failurE) [72], VITAL (VITamin D and omegA-3) [73], and VIDA (Vitamin D Assessment Study) [74], trials failed to demonstrate meaningful reductions in cardiovascular outcomes with vitamin D supplementation. The VINDICATE trial, which focused on patients with systolic HF, evaluated 4000 IU/day of vitamin D3 for 12 months. Although improvements in left ventricular structure and function were seen, no gains were observed in functional capacity, as measured by the six-minute walk test, suggesting limited clinical impact despite structural changes [72]. The VITAL trial [73] enrolled over 25,000 middle-aged and older adults and randomized them to receive vitamin D3 (2000 IU/day) and/or omega-3 fatty acids. After more than six years of follow-up, no significant reductions were observed in rates of myocardial infarction, stroke, or cardiovascular death when compared with placebo. In the ViDA trial conducted in New Zealand, participants received 100,000 IU/month of vitamin D3 or placebo for approximately three years. This regimen also did not reduce the incidence of cardiovascular events, including myocardial infarction, angina, or stroke [74].
| Study | Year | Study design | Population | Intervention | Outcomes | Main findings |
| VINDICATE (VitamIN D treatIng patients with Chronic heArT failurE) study [72] | 2016 | Randomized, double-blind, placebo-controlled trial | n = 229 adults with HFrEF and vit D deficiency | Daily vitamin D3 (4000 IU) vs. placebo for 12 months | Primary endpoint: 6MWT distance. Secondary endpoints: LVEF, left ventricular dimensions (LVEDD, LVESD), left ventricular volumes (LVEDV, LVESV), renal function, serum calcium, PTH levels | No improvement in 6MWT. ↑ LVEF by 6.1%, ↓ LV dimensions. Safe, no hypercalcemia or renal harm. |
| VITamin D and OmegA-3 TriaL (VITAL) [73] | 2019 | Randomized, double-blind, placebo-controlled trial | n = 25,871 adults, |
Daily vitamin D3 (2000 IU) vs. placebo for 5.3 years | Major cardiovascular events (myocardial infarction, stroke, cardiovascular mortality), total cancer incidence, cancer mortality, all-cause mortality | No reduction in major CVD (HR 0.97), total cancer (HR 0.96), or mortality. ↓ Cancer mortality after excluding first 2 years (HR 0.75). |
| Vitamin D Assessment (ViDA) study [74] | 2020 | Randomized, double-blind, placebo-controlled trial | n = 5110 adults 50–84 yrs | Monthly high-dose vitamin D3 (100,000 IU) vs. placebo for a median of 3.3 years | Primary endpoints: CVD, acute respiratory infections, fractures, falls, total cancer incidence. Secondary outcomes: statin persistence, lung function, BMD, arterial function. | No effect on CVD (HR 1.02), fractures, falls, or cancer. Improved statin adherence (HR 1.15; p = 0.02), better lung function in ever-smokers (+57 mL FEV1; p = 0.03), and enhanced arterial function in vitamin D–deficient individuals (p = 0.03). |
Studies are ordered by year of publication. Abbreviations: 6MWT, 6-Minute Walk
Test; BMD, Bone Mineral Density; CVD, Cardiovascular Disease; FEV1, Forced
Expiratory Volume in 1 Second; HF, Heart Failure; HFrEF, Heart Failure with
Reduced Ejection Fraction; HR, hazard ratio; LVEDD, Left Ventricular End-Diastolic Diameter; LVEDV,
Left Ventricular End-Diastolic Volume; LVEF, Left Ventricular Ejection Fraction;
LVESD, Left Ventricular End-Systolic Diameter; LVESV, Left Ventricular
End-Systolic Volume; LVSD, Left Ventricular Systolic Dysfunction; PTH,
Parathyroid Hormone; RCT, Randomized Controlled Trial; ViDA, Vitamin D Assessment
study;
A persistent uncertainty in the relationship between vitamin D–and cardiovascular disease lies in the inconsistency across the available evidence. Meta-analyses by Parker et al. [75], Zittermann et al. [76], and Gaksch et al. [77] report inverse associations between circulating vitamin D levels and cardiovascular risk or all-cause mortality. Parker et al. [75] found that individuals with the highest vitamin D levels had 43% lower odds of cardiometabolic disorders (odds ratio (OR) 0.57, 95% confidence interval (CI): 0.48–0.68); Zittermann et al. [76] observed a nonlinear reduction in all-cause mortality with optimal 25(OH)D concentrations around 75–87.5 nmol/L; and Gaksch et al. [77], using pooled individual data from over 26,000 participants, showed significantly higher mortality risk at levels below 30 nmol/L compared to the reference range of 75–100 nmol/L. However, these findings contrast sharply with the results from RCTs, which have not consistently demonstrated the clinical benefits of vitamin D supplementation. In particular, the meta-analysis by Barbarawi et al. [12] found no significant reduction in major adverse cardiovascular events among vitamin D-treated patients, and Bjelakovic et al. [78] reported only a minor all-cause mortality benefit, exclusively linked to vitamin D3 and not D2 or active analogs. These discrepancies may be attributed to methodological differences, confounding factors such as physical activity, sun exposure, comorbidities, and baseline 25(OH)D levels. Notably, neither VITAL [73] nor ViDA [74] stratified participants by baseline vitamin D status, which may have diluted any effect in individuals with profound deficiency.
In VITAL, for instance, only a small subset of ~500 participants had 25(OH)D levels below 25 nmol/L [73]. Additionally, ethnic disparities may contribute to inconsistent findings. For example, individuals with darker skin pigmentation often have lower serum 25(OH)D levels due to reduced cutaneous synthesis, yet the clinical relevance of this biochemical deficiency remains debated [79, 80]. In VITAL, over 20% of participants were African American, a group that tends to have lower vitamin D levels but may be less susceptible to its adverse skeletal or cardiovascular consequences, possibly due to differences in vitamin D-binding protein polymorphisms and tissue-level vitamin D responsiveness [81]. Thus, any potential benefit may be restricted to severely deficient individuals underrepresented in these trials. Future trials may need to stratify by ethnicity, baseline deficiency, and genetic polymorphisms to more accurately identify responders to vitamin D supplementation.
Vitamin D status is heavily influenced by non-nutritional variables, making causal inference complex. Physical activity strongly correlates with higher serum 25(OH)D levels, possibly through enhanced lipolysis and release from adipose stores [82, 83, 84, 85, 86]. However, this association may be exercise-specific: while continuous combination training increased serum vitamin D, endurance training did not show similar effects [85]. Sun exposure, the major endogenous source of vitamin D, introduces further bias. Individuals with higher outdoor activity levels not only have greater vitamin D production but also tend to have lower baseline cardiovascular risk, potentially confounding associations between vitamin D and CV outcomes. The heterogeneity in dosing regimens, supplementation duration, and study populations further limits comparability across trials and the generalizability of results. Many trials enrolled elderly, institutionalized, or comorbid individuals whose high disease burden may have masked subtle cardiovascular benefits from vitamin D repletion.
There is no consensus on optimal serum 25(OH)D thresholds for cardiovascular
protection. The Institute of Medicine (IOM) recommends daily intakes of 400–800
IU primarily for skeletal health but notes that current evidence is insufficient
to support recommendations for cardiovascular outcomes [87]. Meanwhile, others
suggest that 1500–2000 IU/day may be necessary to maintain optimal serum levels
(
To complement interventional evidence, several high-quality observational
studies have consistently reported an inverse association between serum 25(OH)D
levels and cardiovascular outcomes. Large prospective cohorts such as the Third
National Health and Nutrition Examination Survey (NHANES III) [105], the
Ludwigshafen Risk and Cardiovascular Health Study (LURIC) study [106], and the
Framingham Offspring Study [9] demonstrated that individuals with lower 25(OH)D
concentrations had significantly higher risks of all-cause or cardiovascular
mortality. Specifically, Melamed et al. [105] found that the lowest
quartile of 25(OH)D (
| First Author [Reference] | Year | Population (N) | Study design | Outcomes |
| Melamed, ML [105] | 2008 | 13,331 US adults (NHANES III) | Prospective | Lowest quartile of 25(OH)D ( |
| Dobnig, H [106] | 2008 | 3258 patients referred to coronary angiography (LURIC cohort) | Prospective | Lowest quartiles of 25(OH)D (medians 7.6 & 13.3 ng/mL) associated with increased all-cause mortality (HR up to 2.08; 95% CI 1.60–2.70) and cardiovascular mortality (HR up to 2.22; 95% CI 1.57–3.13). |
| Wang, TJ [9] | 2008 | 1739 participants from the Framingham Offspring Study, free of cardiovascular disease at baseline | Prospective | Lowest 25(OH)D levels ( |
| Pilz, S [107] | 2008 | 3299 patients referred for coronary angiography | Cross-sectional with longitudinal follow-up | Severe vitamin D deficiency ( |
| Acharya, P [108] | 2021 | 20,025 U.S. Veterans with baseline 25(OH)D |
Retrospective, case-control | Patients who achieved |
| Simon, J [109] | 2024 | 86 acute ischemic stroke patients | Prospective | 25(OH)D deficiency ( |
| Candemir, B [110] | 2025 | 120 obese patients (BMI |
Retrospective | Lowest 25(OH)D levels ( |
Studies are ordered by year of publication. Abbreviations: BMI, body mass index; CI, confidence interval; CV, cardiovascular; HR, hazard ratio; MI, Myocardial Infarction; LURIC, Ludwigshafen Risk and Cardiovascular Health Study; NHANES III, Third National Health and Nutrition Examination Survey (1988–1994); NIHSS, National Institutes of Health Stroke Scale; SYNTAX, Synergy Between PCI With TAXUS and Cardiac Surgery.
Several high-quality trials have recently been completed and contribute valuable
insights into the cardiovascular effects of vitamin D supplementation (Table 3,
Ref. [111, 112, 113, 114, 115]). The VITAL trial [111], which enrolled over 25,000 U.S.
adults without prior cardiovascular disease, showed that daily supplementation
with 2000 IU of vitamin D3 did not reduce the incidence of major cardiovascular
events (myocardial infarction, stroke, cardiovascular mortality) compared to
placebo. The VITAL Rhythm substudy [112] focused on atrial fibrillation and
similarly found no overall reduction in AF incidence, although a subgroup
analysis suggested a possible benefit in Black participants. The VITAL Heart
Failure (VITAL HF) substudy [116], which evaluated HF outcomes, reported no
significant effect of vitamin D3 on incident HF. Other recently completed studies
include: the DO-HEALTH trial [113], which found no benefit on major adverse cardiovascular events (MACE) or
hypertension, although omega-3 supplementation improved lipid profiles; the D2d
trial [114], which showed no significant reduction in MACE, but a small
improvement in atherosclerotic cardiovascular disease (ASCVD) risk score; the D-Health Trial [115], which reported no
reduction in CVD incidence or mortality with monthly high-dose vitamin D3, but
observed that baseline vitamin D deficiency was associated with higher
cardiovascular risk. Finally, the COSMOS trial (NCT02422745) explored
cardiovascular outcomes using a factorial design involving cocoa extract and
multivitamins; although completed, cardiovascular-specific results are still
under analysis. These trials underscore the current limitations of vitamin D
interventional research: despite strong mechanistic and observational data, large
RCTs have yet to confirm clear cardiovascular benefits. One possible explanation
lies in the heterogeneity of study designs, including substantial variability in
baseline vitamin D status, dosing regimens (daily vs. bolus), treatment duration
(weeks vs. years), and inconsistent thresholds for what constitutes sufficiency
or deficiency. In many cases, participants were enrolled regardless of their
vitamin D levels, potentially diluting the benefit among those who were already
replete. Most trials did not stratify or tailor therapy based on vitamin D
deficiency, nor did they incorporate biomarkers to identify those most likely to
benefit. For example, individuals with severe deficiency (
| Study | ClinicalTrials.gov Identifier or Reference | Start year | Study design | Population | Intervention/Exposure | Primary endpoint(s) | Secondary endpoint(s) | Major findings |
| Recently Completed | ||||||||
| VITAL | [111] | 2010 | Interventional; Randomized, placebo-controlled trial | Adults aged |
Daily vitamin D3 (2000 IU) and/or marine omega-3 fatty acids (EPA 465 mg + DHA 375 mg) vs. placebo for a median of 5.3 years | Major cardiovascular events (myocardial infarction, stroke, cardiovascular mortality), invasive cancer | Coronary revascularization, death from invasive cancer, death from any cause | No significant reduction in the incidence of major cardiovascular events (MI, stroke, CV death) or cancer with vitamin D3 (2000 IU/day) supplementation vs. placebo in the general population. |
| VITAL Rhythm | [112] | 2012 | Interventional, Randomized, double-blind, placebo-controlled trial | Adults aged |
Daily vitamin D3 (2000 IU) and/or marine omega-3 fatty acids (EPA 460 mg + DHA 380 mg) vs. placebo for a median of 5.3 years | Incident AF | AF subtypes (paroxysmal vs. persistent AF); sudden cardiac death; ECG changes | No reduction in incident atrial fibrillation with vitamin D3 (2000 IU/day) or omega-3 fatty acids. |
| DO-HEALTH | [113] | 2012 | Randomized, placebo-controlled trial | Elderly (mean age 74), 61.7% women | Vitamin D3 (2000 IU/day) |
Hypertension, MACE, lipid profile | Lipid biomarkers, BP, physical activity | No benefit on MACE; omega-3 improved lipids |
| D2d | [114] | 2013 | Randomized, placebo-controlled trial | Adults with prediabetes (n = 2423) | Vitamin D3 (4000 IU/day) vs. placebo | MACE, ASCVD risk score | BP, lipids, hs-CRP, ASCVD risk factors | No MACE reduction; small benefit in ASCVD risk score |
| D-Health Trial | [115] | 2014 | Randomized, placebo-controlled trial | 21,315 adults aged 60–84 years in Australia, without known vitamin D deficiency | Monthly oral vitamin D3 (60,000 IU) vs. placebo for 5 years | CVD incidence and mortality | All-cause mortality | No CVD reduction; deficiency linked to higher CVD risk |
| Ongoing | ||||||||
| TARGET-D | NCT02996721 | 2017 | Interventional; Randomized, Open-Label, Parallel Assignment | Patients with a history of MI and vitamin D deficiency | Standard of care vs. individualized vitamin D3 supplementation to achieve 25(OH)D |
Death, myocardial infarction, heart failure hospitalization, and CVA | NA | Pending |
| INVITE | NCT02925195 | 2017 | Interventional; Randomized, Double-blind, Parallel Assignment, placebo-controlled trial | 1600 Adults from the Multi-Ethnic Study of Atherosclerosis (MESA) study | Daily vitamin D3 (2000 IU) vs. placebo in 3:1 ratio for 16 weeks | To identify genetic polymorphisms, clinical characteristics, and biomarkers that modify the biologic response to vitamin D3 treatment | Change in blood pressure, in urine calcium concentrations and serum calcium concentrations | Pending |
| VINDICATE-MI | NCT03086746 | 2018 | Prospective cohort | Adults ( |
Baseline vitamin D levels | Left ventricular remodeling ( |
Vitamin D, Vitamin D binding protein and PTH levels | Pending |
| Vitamin D3 supplementation (4000 IU daily) vs. placebo | ||||||||
| VINDICATE 2 | NCT03416361 | 2023 | Interventional; Randomized, Quadruple-Blind, Parallel Assignment | Adults ( |
4000 IU Vitamin D3 (chewable tablets, 2 per day) vs. placebo | Time to death or first hospitalization for heart failure (24 months) | Total mortality, cost-effectiveness (ICER for vitamin D), change in patient quality of life (EQ5D-5L) | Pending |
Studies are ordered by year of publication. Abbreviations: AF, Atrial fibrillation; ASCVD, Atherosclerotic Cardiovascular Disease; BP, Blood Pressure; CHF, Chronic heart failure; CVA, Cerebrovascular accident; CVD, Cardiovascular disease; DHA, Docosahexaenoic acid; ECG, Electrocardiogram; EQ5D-5L, EuroQol 5-Dimension 5-Level questionnaire (measure of health-related quality of life); EPA, Eicosapentaenoic acid; HF, Heart failure; hs-CRP, High-Sensitivity C-Reactive Protein; ICER, Incremental cost-effectiveness ratio; LVEF, Left ventricular ejection fraction; LVESVi, Left ventricular end-systolic volume index; LVSD, Left ventricular systolic dysfunction; MACE, Major Adverse Cardiovascular Events; MI, Myocardial infarction; NA, Not Applicable; SHEP, simple home-based exercise program; RCT, Randomized Controlled Trial; STEMI, ST-elevation myocardial infarction.
To address these gaps, several ongoing studies are focusing on more personalized and targeted approaches. Trials such as INVITe (NCT02925195) are ongoing and aim to uncover genetic and metabolic predictors of individual responses to vitamin D. Others, like TARGET-D (NCT02996721) and VINDICATE 2 (NCT03416361), are selectively enrolling participants with documented deficiency and high cardiovascular risk, aiming to clarify whether supplementation is beneficial in those who are most likely to respond. Pediatric populations are also being investigated. The Vitamin D and Vascular Health in Children (NCT01797302) trial assesses vascular function in obese children and evaluates the effects of daily supplementation (600–2000 IU) over six months, while Low vs. Moderate to High Dose Vitamin D for Prevention of COVID-19 (NCT04868903) explores optimal dosing in infants. In acute care settings, the VIOLET trial (NCT03096314) tests whether a single high dose (540,000 IU) of vitamin D3 could reduce mortality in critically ill, vitamin D–deficient patients. Additional studies are exploring metabolic and structural outcomes, such as glycemic control in children with type 1 diabetes (NCT05141968) and cardiac remodeling following myocardial infarction or in HF, such as in VINDICATE-MI (NCT03086746). Collectively, these trials aim to address key knowledge gaps regarding optimal dosing strategies, the most responsive target populations, and the true efficacy of vitamin D in cardiovascular prevention and therapy. Their results may help reconcile the current discrepancies between observational and interventional evidence and determine whether vitamin D can play a meaningful role in cardiovascular health.
Vitamin D remains a compelling yet enigmatic player in cardiovascular health. The Good includes its anti-inflammatory, antifibrotic, and vasoprotective properties. The Bad highlights concerns surrounding the potential adverse effects of over-supplementation and the unmet expectations in large RCTs. Finally, the Unknown lies in the persistent gap between association and causation, complicated by confounding variables, heterogeneous populations, and inconsistencies in dosing regimens. As ongoing large-scale trials unfold, there is cautious optimism that great clarity will emerge, revealing whether vitamin D is a silent bystander or a modifiable contributor to cardiovascular disease prevention and management.
VDR, vitamin D receptor; DBP, vitamin D binding protein; PTH, parathyroid hormone; FGF-23, fibroblast growth factor-23; RXR, retinoid X receptor; ROS, reactive oxygen species; COX-1, cyclooxygenase-1; IL, interleukin; TNF-
DMG, FMDM and MG conceived and designed the review, and drafted the initial version of the manuscript. PP created the figures. MM, PP, MB, LS, SC, FP, GC, PS and AC provided clinical expertise and revised the tables and the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Francesco Perone is serving as Guest Editor of this journal. We declare that Francesco Perone had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Ruan Kruger.
This manuscript was prepared with the assistance of artificial intelligence tools, including ChatGPT-4o (OpenAI, San Francisco, CA, USA) and Grammarly (Grammarly Inc., San Francisco, CA, USA), following established best practices (Biondi-Zoccai G, editor. ChatGPT for Medical Research. Torino: Edizioni Minerva Medica; 2024). Figures were created using BioRender.com. The authors have thoroughly reviewed, edited, and approved the final content, and they take full responsibility for the accuracy, integrity, and intellectual contributions of the work. All ethical standards and guidelines governing the use of artificial intelligence in research have been fully respected.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.