


1 Department of General Medicine, First Affiliated Hospital of Xinjiang Medical University, 830011 Urumchi, Xinjiang, China
2 Department of Pharmacy, First Affiliated Hospital of Xinjiang Medical University, 830011 Urumchi, Xinjiang, China
Abstract
Ventricular remodeling in right heart failure is a complex pathological process involving interactions between multiple mechanisms. Overactivation of the neuro-hormonal pathways, activation of the oxidative stress response, expression of cytokines, apoptosis of cardiomyocytes, and alterations of the extracellular matrix (ECM) are among the major mechanisms involved in the development of ventricular remodeling in right heart failure. These mechanisms are involved in ventricular remodeling, such as myocardial hypertrophy and fibrosis, leading to the deterioration of myocardial systolic and diastolic function. A deeper understanding of these mechanisms can help develop more effective therapeutic strategies in patients with right heart failure (RHF) to improve patient survival and quality of life. Despite the importance of ventricular remodeling in RHF, there are a limited number of studies in this field. This article explores in-depth historical and current information about the specific mechanisms in ventricular remodeling in RHF, providing a theoretical rationale for recognizing its importance in health and disease.
Keywords
- right heart failure
- ventricular remodeling
- autonomic nervous system
- cytokines
- extracellular matrix
Right heart failure (RHF) [1] is one of the most serious outcomes of pulmonary hypertension (PAH) and leads to ventricular remodeling, which in turn affects the normal function of the heart. The free wall of the right ventricle (RV) is thinner than that of the left ventricle (LV), which is usually between 3 and 5 millimeters thick in adults, and the RV is about one-third to one-sixth smaller in mass than the LV. Cardiomyocytes in the RV are about 15% smaller than those in the LV; however, the collagen content of the RV is increased, by about 30% [2]. Ventricular remodeling is a complex biological process involving the interaction of multiple mechanisms. Myocardial remodeling is a broad definition for any change in the heart’s structure and function, which can be categorized into physiological and pathological types. Physiological myocardial remodeling is a beneficial adaptive process, which results in decreased wall stress, increased cardiac pump function, and improved vascularization. Early pathologic myocardial remodeling serves to reduce ventricular wall stress and temporarily protect cardiac pump function but eventually progresses to heart failure and death. Pathological myocardial remodeling is associated with cellular hypertrophy, rhabdomyolysis, glycogen accumulation, chronic oxidative stress, cell death, inflammation, fibrosis, decreased capillary density, and electrical disturbances [3]. Over-activation of the neuro-hormonal pathways, oxidative stress, cytokine expression, cardiomyocyte apoptosis, and extracellular matrix (ECM) alterations are the main pathophysiological mechanisms that comprise ventricular remodeling in RHF.
Hyperactivation of the neuro-hormonal pathways play a crucial role in the
development and progression of RHF. Overactivation of the
renin-angiotensin-aldosterone system (RAAS) [4], the sympathetic nervous system,
and neuro-hormonal pathways, such as natriuretic peptides, are collectively
involved in the complex process of ventricular remodeling, such as myocardial
hypertrophy, cardiomyocyte apoptosis, and fibrosis. In addition, overactivation
of neuro-hormonal pathways causes vasoconstriction and sodium retention, further
decreasing cardiac function. During ventricular remodeling, increased expression
of several cytokines such as tumor necrosis factor-alpha (TNF-
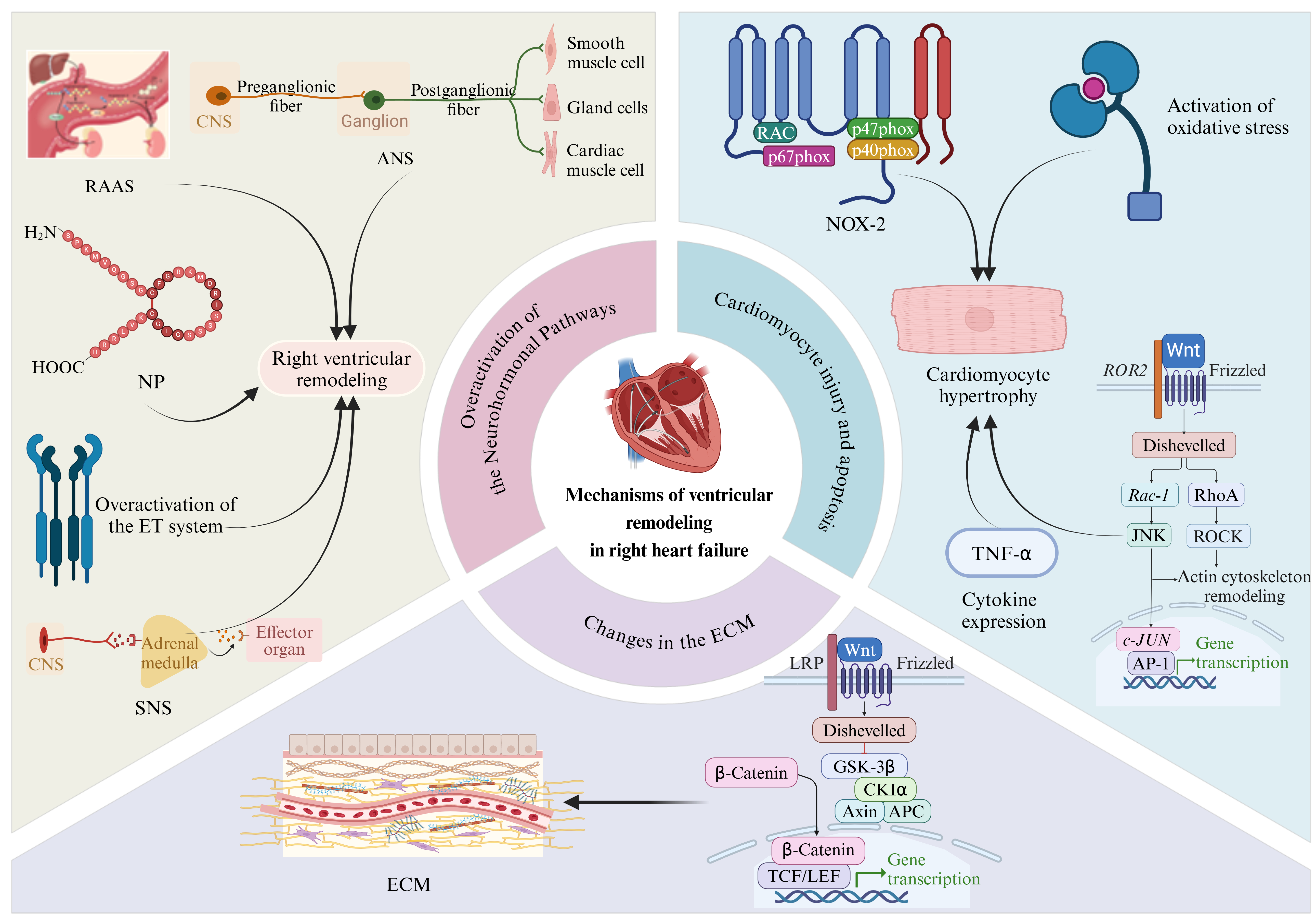 Fig. 1.
Fig. 1.
Mechanisms associated with ventricular remodeling in right heart
failure. The upper left panel shows the effect of neuro-hormonal pathway
activation on ventricular remodeling in patients with right heart failure. The
upper right panel highlights the effects of cardiomyocyte injury and apoptosis on
right ventricular remodeling. The lower part of this figure shows the effect of
extracellular matrix alterations on right ventricular remodeling. Overactivation
of the neuro-hormonal pathways: RAAS, renin-angiotensin-aldosterone system; ANS,
autonomic nervous system; NP, natriuretic peptide; ET, endothelin; SNS,
sympathoadrenal system. Cardiomyocyte injury and apoptosis: TNF-
In summary, the mechanisms responsible for ventricular remodeling in RHF include overactivation of the Neuro-hormonal Pathways, activation of oxidative stress, expression of cytokines, apoptosis of cardiomyocytes, and alterations in the ECM. There is a close interaction between these mechanisms involved in the complex biological process of ventricular remodeling in RHF. Therapeutic approaches that target these key mechanisms can help to significantly improve the clinical symptoms of RHF and effectively slow the progression of ventricular remodeling. A deeper understanding of these mechanisms will provide important guidance for developing more effective therapeutic strategies, to increase survival and improve the quality of life of patients with RHF.
The autonomic nervous system (ANS), also known as the vegetative nervous system or the involuntary nervous system, is an important part of the peripheral nervous system of vertebrates. Through the differentiation and development of the somatic nerves, a functionally independent nervous system is formed, which is responsible for regulating the internal environmental homeostasis of the body and the function of organs. The ANS and biologically active substances such as vasopressin, angiotensin, oxytocin, and cytokines play a synergistic role in the functional regulation of the cardiovascular system, working together to maintain the stability of the body’s internal environment and the ability to adapt to changes in the external environment. However, in pathological states such as heart failure and hypertension, the interactions between the ANS and these regulatory factors change significantly, interfering with the normal regulatory mechanisms of the cardiovascular system and leading to a series of imbalances in physiological and pathological processes [5].
The ANS, named after its function in regulating the activity of internal organs, vascular smooth muscle, cardiac muscle, and glands, can autonomously regulate the activity of these tissues and organs, ensuring the stability of the internal environment of the organism and adapting to changes in the external environment. The ANS is mainly comprised of three core components: the ANS, the parasympathetic nervous system, and the enteric nervous system. The scope of this paper is limited to the sympathetic and parasympathetic nervous systems to investigate their functions and regulatory mechanisms in physiological and pathological states. They synergistically regulate the activity and secretion of the body’s organs, blood vessels, smooth muscles, and glands [6]. Recently, study has shown that the regulation of cardiac neuromodulation extends beyond the central nervous system’s external influence to include a specialized, intrinsic cardiac nervous system (ICNS) [7]. This sophisticated network comprises sensory neurons, both pre-and postganglionic parasympathetic neurons, as well as biphenotypic neurons capable of releasing neurotransmitters of the sympathetic and parasympathetic systems. Together, these elements orchestrate a complex neural network crucial for the nuanced regulation of the cardiovascular system [8].
The ANS plays a pivotal role in regulating cardiac function. Specifically, it is responsible for precisely and efficiently regulating myocardial contractile force, blood pressure, and heart rhythm. Cardiac sympathetic preganglionic fibers originate from thoracic segments 1 to 6 of the spinal cord and reach myocardial tissues and coronary vessels mainly through postganglionic fibers emitted by stellate ganglionic exchange neurons. Postganglionic neurons contain the enzyme tyrosine hydroxylase to synthesize norepinephrine (NE), the main neurotransmitter of sympathetic nerves. Sympathetic activation in the body is characterized by increased circulating blood concentrations of catecholamines (CA), including amines such as NE, epinephrine, and dopamine [9]. The parasympathetic preganglionic fibers of the heart emanate from the dorsal nuclei of the vagus nerve in the medulla oblongata. After exchanging neurons in the cardiac ganglionic plexus, they innervate primarily the atria and the cardiac conduction system. Parasympathetic nerves, in turn, act to slow the heart rate and reduce myocardial contractility by releasing acetylcholine. However, there are no reports of altered parasympathetic tone in RHF in the current literature, which should receive further attention in future studies. The critical role of the ANS in modulating the ST-segment elevation phenomenon and tachyarrhythmic events in patients with Brugada syndrome has been revealed [10]. The sympathetic nervous and parasympathetic systems function antagonistically, synergistically, or independently of each other to maintain the functional balance of the autonomic effector organs and to ensure the homeostasis of the organism’s internal environment and its adaptation to changes in the external environment. Several studies have confirmed that imbalances in the ANS, that is, abnormal increases or decreases in sympathetic or parasympathetic tone, are closely associated with the onset and progression of a variety of diseases, such as arrhythmias after myocardial infarction [11], Obstructive sleep apnea [12], diabetes [13] and Parkinson’s disease [14]. Carotid pressure receptor stimulation (CBS) is a non-pharmacological autonomic modulation intervention. By analyzing the effects of CBS on wild pachydermine-induced PAH and its underlying mechanisms, it was suggested that CBS could significantly attenuate right ventricular dysfunction, improve pulmonary artery and right ventricular remodeling, and thus improve the survival rate of rats with monocrotaline-induced PAH [15]. The ANS, which influences myocardial contractility, blood pressure, and heart rhythm, is essential for controlling cardiac function. Numerous diseases are strongly linked to their imbalance, and comprehending the disease process requires a thorough grasp of the autonomic nerve system’s regulatory processes.
RAAS [4] is an important blood pressure regulatory system in the body,
consisting mainly of renin, angiotensin, and aldosterone. Renin is a protein
hydrolyzing enzyme secreted by the periglomerular cells of the glomerulus, and
when it enters the circulation, it interacts with
In the heart, the RAAS regulates cardiac function, modulates coronary artery resistance, and inhibits cardiomyocyte growth. It also significantly affects vascular structure and the operation of the coagulation system, and it can control vasodilation and contraction. The body’s ability to maintain blood pressure homeostasis and heart function depends heavily on the RAAS. Diseases, including hypertension and cardiac hypertrophy, can arise when the RAAS is not working properly. Studies have confirmed that the Janus kinase 2 (JAK2)/signal transduction and activation of transcription 3 (STAT3) signaling pathway plays a key role in regulating the expression of transforming growth factor II, collagen type I alpha 1 (COL1A1), and myosin heavy chain 7 (Myh7), which in turn participates in Ang II-induced myocardial hypertrophy and fibrosis, and ultimately promotes the development of cardiac remodeling [18]. Dehydroepiandrosterone effectively promotes the improvement of pulmonary hemodynamics, alleviates the process of pulmonary vascular remodeling, and enhances cardiac function through the inhibition of the STAT3 signaling pathway, thereby attenuating the remodeling phenomenon of the RV [19]. Given the limitations of the scope of this study, clinical trials have not been able to adequately confirm the effectiveness of RAAS inhibition therapy in patients with coronary artery disease and RHF [20]. To prevent and treat RHF, a thorough understanding of the physiology and pathological mechanisms underlying the RAAS is essential. By revealing the association between its regulatory mechanisms and the development of diseases, it is expected to provide new ideas and methods for clinical treatment, thereby improving patients’ quality of life and reducing the disease burden.
Natriuretic peptide (NP) [21] also maintains homeostasis within the cardiovascular system, with potent natriuretic, diuretic, vasodilator, and inhibitory effects on sympathetic nerve activity. There are five main types of natriuretic peptides: atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), C-type natriuretic peptide (CNP), dendroaspis natriuretic peptide (DNP), and uroguanosine [22]. ANP and BNP play a local (autocrine/paracrine) regulatory role in the heart, and another natriuretic peptide, CNP, acts as a regulator within the vascular wall. In this review, we will only discuss ANP, BNP, and CNP. Initial purification of BNP was achieved from porcine brain extracts, yet subsequent studies have shown that the heart is the primary source of circulating BNP. In addition, CNP is produced by both nerve tissue and vascular endothelial cells. The natriuretic peptide is present in ventricular septal particles and can be secreted from these particles. Its secretion depends on volume expansion and increased pressure load in the ventricles. When cardiomyocytes are stretched and stimulated, they secrete B-type natriuretic peptide protomer precursors, which are cleaved into biologically active and inactive natriuretic peptides under endonuclease. NP has several effects on the heart. First, it dilates blood vessels, causing a decrease in cardiac output and a reduction in cardiac load, with diuretic, sodium, and drainage effects. ANP and BNP exhibit classical endocrine actions in vascular smooth muscle and renal units. Second, natriuretic peptides can lower blood pressure and can also lead to a decrease in cardiac load. During overloading of cardiac function or squeezing of cardiac capacity, natriuretic peptides are activated, leading to elevated levels of natriuretic peptides, which help diagnose heart failure. There are significant associations between the levels of these two natriuretic peptides at moderate concentrations and the risk of developing structural heart disease and heart failure [23].
The three membrane-bound natriuretic peptide receptors known as natriuretic
peptide receptor-A, natriuretic peptide receptor-B (NPR-B), and natriuretic
peptide receptor-C are also included in the natriuretic peptide system [24]. The
human natriuretic peptide type A (NPPA) gene, found on chromosome 1,
region 1p36.21, encodes an ANP precursor longer than 2 kb and has three exons.
Approximately 10 kb upstream of the NPPA, the natriuretic peptide precursor (NPPB) gene produces a BNP precursor with a similar structure;
it is likely that the two genes evolved via gene duplication [25]. Brain
NPPB and N-terminal brain natriuretic peptide precursor levels must be
prioritized by healthy persons, and this is because variations in the
concentrations of these markers might indicate early cardiac responses [26]. The
NPPB gene is upregulated in response to mechanical stretching that may
occur in hypertension or other risk factors for heart failure. In addition, the
promoter of the NPPB gene contains a kinase element associated with
extracellular signaling stimulated by the RAAS, which regulates gene expression
and subsequent biological processes [27]. Upon binding to the NP receptor,
NPPB can trigger a series of biological effects, including the promotion
of diuretic and natriuretic processes, the lowering of blood pressure, and the
attenuation of the activity of the RAAS. Together, these effects help maintain
the body’s water-salt balance and cardiovascular homeostasis [28]. In the
GPx3-deficient pulmonary artery banding animal model, an exacerbation of adverse
RV remodeling was observed, accompanied by a significant increase in the levels
of CTGF, transforming growth factor-
Over-activation of the ET system manifests as by excessive endothelin (ET)
secretion, which in turn triggers excessive contraction of the cardiovascular
system and elevated blood pressure. ET [31] is a 21-amino acid peptide derived
from big ET by the action of a converting enzyme, and includes three isoforms of
ET-1, ET-2, and ET-3. Among them, ET-1 is mainly produced by vascular ET cells,
and its effects are mediated through two G protein-coupled receptors, ET-A and
ET-B, which are widely distributed in various human body tissues, especially in
cardiac tissues. ET-1 is potent vasoconstrictor, myocardial tone, and mitogenic
effects, regulates salt and water balance, and maintains vascular tone and blood
pressure stability [32]. Human cardiomyocytes also express ET. ET is mostly
expressed in healthy mesenchymal and ET cells that have been isolated from the
ventricles, ET expression in cardiomyocytes noticeably increases under
pathological conditions such as ischemic cardiomyopathy [33]. As a
multifunctional regulator, TGF-
Activation of the sympathoadrenal system (SNS) is also one of the important
mechanisms of ventricular remodeling in RHF. During activation of the SNS, the
adrenal glands secrete excessive amounts of CA, including epinephrine and
norepinephrine, to maintain cardiovascular systemic function [48]. Activation of
this system leads to an increase in the concentration of calcium ions in
cardiomyocytes and an increase in myocardial contractility, but it also leads to
apoptosis and fibrosis of cardiomyocytes, which in turn aggravates ventricular
remodeling. The SNS is a system of sympathetic nerves and the adrenal medulla
that primarily responds to emergencies. ARs can be categorized into several
subtypes, such as
The SNS influences gene expression in cardiomyocytes primarily through the
release of catecholamine neurotransmitters, which alters the structure and
function of cardiomyocytes. This process involves the activation and regulation
of several signaling pathways. These include the following pathways: (1)
Oxidative stress initiates a cascade of detrimental processes within
cardiomyocytes, including peroxidation of membrane lipids and protein
denaturation, thereby compromising cellular integrity and function. This stressor
not only accelerates cardiomyocyte death but also fuels the progression of
ventricular remodeling. Additionally, oxidative stress elicits an inflammatory
response that exacerbates necrosis of cardiomyocytes. One key manifestation of
oxidative stress in cardiomyocytes is the induction of apoptosis, a programmed
cell death process mediated by proteins such as p53 and Bax. Furthermore,
oxidative stress triggers an abnormal surge in intracellular Ca2⁺ ion
levels, resulting in calcium overload which exacerbates cardiomyocyte injury.
Pathological conditions such as myocardial ischemia, reperfusion, and
hypertension are characterized by increased levels of oxidative stress in
cardiomyocytes, primarily stemming from free radicals and mitochondrial
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity. This
oxidative stress directly impairs mitochondrial function by accentuating the
opening of the mitochondrial permeability transition pores, causing a decline in
mitochondrial membrane potential. Consequently, mitochondrial swelling and
rupture ensue, releasing cytochrome C (cyt-C) into the cytosol. This event
triggers the caspase cascade, ultimately initiating apoptosis in cardiomyocytes.
The sthdy in apoptosis signal-regulating kinase 1 (ASK1) gene-deficient mice
confirmed the critical role of the reactive oxygen species (ROS)/ASK1 pathway in
necrotic and apoptotic cell death, providing important clues to ASK1 as a
potential therapeutic target for reducing left ventricular remodeling after
myocardial infarction [54]. Inhibition of matrix metalloproteinases (MMPs)
induction and myocardial remodeling by TNF-
In summary, activation of oxidative stress has significant negative effects on
the myocardium, leading to myocardial injury and ventricular remodeling.
Therefore, therapeutic strategies targeting oxidative stress are important for
improving the symptoms of RHF. Oxidative stress damages cellular macromolecules,
including many key cardiac myosin proteins, membrane lipids, especially
cardiolipin, and mitochondrial DNA (mtDNA), ultimately leading to cardiomyocyte
death and even myocardial remodeling and dysfunction. A study by Fang et
al. [56], bromodomain-containing protein 4 (BRD4) knockdown dramatically
increased the production of nuclear factor erythroid 2-related factor-2 (Nrf2) and heme oxygenase 1
(HO-1) proteins while suppressing TLR4 and nuclear factor-kappa B (NF-
Various cytokines such as TNF-
Apoptosis of cardiomyocytes is one of the important mechanisms of ventricular remodeling in RHF. Cardiomyocyte apoptosis is the active initiation and execution of a series of ordered biochemical reactions in cardiomyocytes in response to various stimuli, leading to programmed cell death. This process leads to a decrease in the number of cardiomyocytes, which in turn triggers the destruction of myocardial tissue. Cardiomyocyte apoptosis in the peri-infarct and remote infarct zones is closely associated with ventricular dilatation and reduced ventricular systolic function after myocardial infarction, with the correlation appearing to be more pronounced in the remote infarct zone. The prolonged presence of apoptosis in the noninfarcted area allows continued death of surviving cardiomyocytes, which limits structural and functional recovery after infarction and promotes the development of ventricular remodeling. The remodeling process also involves sliding myofibers of nonischemic myocardium against each other, causing thinning of the ventricular wall and enlargement of the ventricular cavity. This compensatory response results in an increase in stroke volume and a reduction in the extent of peripheral myocardial shortening. During inter-cellular sliding, this structural rearrangement of myofiber components requires the death of individual cardiomyocytes to cause a change in position between the cells. In addition, apoptosis in the peri-infarct zone and cardiomyocytes in the remote infarct zone may play an important role in acute and chronic ventricular wall remodeling through these mechanisms. If the area of the myocardium in which hibernation occurs is large, neuroendocrine mechanisms may be triggered to initiate or accelerate the process of cardiac remodeling, resulting in an undesirable vicious cycle. Magnesium lithospermate B administration inhibited cardiomyocyte hypertrophy and decreased the expression of NADPH oxidase (NOX) (especially NOX2 and NOX4) and VPO1, which effectively prevented hypoxia-induced RV remodeling process in PAH rats by blocking the activation of NOX/VPO1 and ERK signaling pathways [66]. Mechanistic study demonstrated that the use of betaine significantly increased the expression of Rho A, Rho-associated protein kinase 1 (ROCK1), and ROCK2, which alleviated pulmonary vascular remodeling and cardiomyocyte apoptosis through the altered Rho A/ROCK signaling pathway, ultimately attenuating RHF [67].
Thus, apoptosis of cardiomyocytes is one of the important mechanisms of ventricular remodeling and profoundly impacts its development and the recovery of cardiac function. Strategies to prevent and treat cardiomyocyte apoptosis may provide new therapies to improve ventricular remodeling and cardiac function.
Another key process in RHF is the change in ECM. The ECM is responsible for
mechanical and electrical connections and serves as the structural foundation of
the heart. Excessive ECM buildup in heart failure increases ventricular stiffness
and interferes with electrical coupling, resulting in alterations in conduction,
arrhythmias, and sudden death [67]. Alterations in the ECM include an imbalance
in the proliferation and degradation of collagen fibers, leading to the
disorganization of cardiomyocytes and interstitial fibrosis, which further
affects the diastolic and contractile functions of the myocardium. Myocardial ECM
is mainly composed of collagen, matrix metalloproteinases, and cell surface
adhesion molecules, and plays a key role in maintaining cardiac shape and
transmitting intercellular signals. During ventricular remodeling, the balance
between synthesis and degradation of the cardiac ECM is disrupted. Excessive
deposition of collagen leads to stiffening of the myocardial ECM and affects the
diastolic function of the myocardium. In contrast, the activity of matrix
metalloproteinases (MMPs) is finely regulated, and their activity status directly
influences the degradation process of the ECM in the myocardium and plays an
important role in ventricular remodeling. Furthermore, through controlling the
expression of MMPs and tissue inhibitory factors, a range of growth factors and
cytokines, including TGF-
RHF ventricular remodeling is a multifaceted process encompassing numerous intricate mechanisms. Despite noteworthy advancements that have been achieved within this domain, the scope and depth of pertinent research remain limited. There is a pressing need for further exploration to broaden the understanding of this complex phenomenon and to deepen the insights gained from existing studies. Given the prevailing research focal points, we propose the adoption of the following strategies to propel the advancement of this field in a more precise and efficient trajectory: (1) using artificial intelligence technology to assist in the collation and analysis of data from patients with RHF ventricular remodeling; (2) constructing a large-scale, multicenter database of patients with RHF ventricular remodeling; and (3) combining the multi-homological data related to RHF ventricular remodeling to undertake a comprehensive study. The over-activation of neurohormones plays a central role in RHF ventricular remodeling. For example, abnormal secretion of hormones such as angiotensin and aldosterone will directly lead to the emergence of remodeling phenomena such as myocardial hypertrophy and interstitial fibrosis. However, due to the constraints and limitations of current research, evidence on the effectiveness of RAAS inhibition therapy in patient populations with right heart failure is still lacking in clinical practice. This emphasizes the urgency and importance of further in-depth research and validation of the efficacy of RAAS inhibition therapy in these patient populations. Activation of the SNS results in increased sensitivity of cardiomyocytes to CA, further promoting apoptosis and necrosis of cardiomyocytes. Under oxidative stress, the production of reactive oxygen radicals in cardiomyocytes increases, leading to cardiomyocyte injury and apoptosis and promotes the development of ventricular remodeling. Mechanisms such as aberrant expression of cytokines, remodeling of the ECM, and apoptosis of cardiomyocytes are interrelated and interact with each other, and together, they participate in the complex process of RHF ventricular remodeling. Therefore, therapeutic approaches targeting these mechanisms are important for improving the symptoms of RHF. By modulating neurohormone secretion, inhibiting SNS activation, attenuating oxidative stress, regulating cytokine expression, and improving ECM remodeling, the process of ventricular remodeling can be effectively decreased, thereby improving the prognosis and quality of life of patients with RHF. Future studies should further explore the specific details of these mechanisms to provide more effective strategies and methods for treating RHF.
DJ and YW designed the research study. DJ, RW and JW performed the research. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors are grateful to the First Affiliated Hospital of Xinjiang Medical University for their support.
This research was funded by the Xinjiang Uygur Autonomous Region Youth Science and Technology Top Talent Project-Youth Science and Technology Innovation Talent Training (2022TSYCCX0034), and National Natural Science Foundation of China Grants (82260081 and 81860096). College Students’ Innovative Entrepreneurial Training Plan Program of Xinjiang Medical University (X202310760095).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.