
 , Wolfgang Koenig 3,4,5
, Wolfgang Koenig 3,4,51 Department of Cardiology, University Heart & Vascular Center Hamburg, University Medical Center Hamburg-Eppendorf, Hamburg, 20246 Hamburg, Germany
2 German Center for Cardiovascular Research (DZHK), Partner Site Hamburg/Kiel/Luebeck, Hamburg, Germany
3 Deutsches Herzzentrum München, Technische Universität München, 80636 Munich, Germany
4 German Centre for Cardiovascular Research (DZHK), Partner Site Munich Heart Alliance, Munich, Germany
5 Institute of Epidemiology and Medical Biometry, University of Ulm, 89081 Ulm, Germany
Abstract
Familial hypercholesterolemia (FH), a condition, which is characterized by a life-long exposure to markedly elevated low-density lipoprotein (LDL) concentrations from birth, and it still remains underdiagnosed and undertreated, despite the fact that its heterogeneous form represents one of the commonest genetic disorders to date. Indeed, only 10% of all estimated affected individuals have been diagnosed worldwide and for the most of them diagnosis comes too late, when atherosclerotic cardiovascular disease (ASCVD) has already been developed. Undiagnosed and undertreated FH leads to accelerated ASCVD with a high rate of premature deaths. Recently, several novel treatment modalities have been introduced, especially for the management of severe hypercholesterolemia. Nonetheless, a substantial number of FH patients still do not achieve guideline-recommended LDL cholesterol target values. In the present review we will summarize and critically discuss pitfalls and challenges in successful diagnosis and treatment of FH.
Keywords
- familial hypercholesterolemia
- atherosclerotic cardiovascular disease
- lipid-lowering therapy
Being the most common genetic disorder to date [1], familial hypercholesterolemia (FH) remains vastly underdiagnosed and undertreated. Almost 60 years were needed from the first description of FH by the Norwegian physician Dr. Carl Müller [2] in the late 1930s until FH gained public health priority by the World Health Organization (WHO) in 1998 [3]. It took further 25 years until FH pediatric screening was recognized by the European Commission Public Health Best Practice Portal as one of the best practices in non-communicable disease prevention in 2022 [4]. Yet, challenges in FH have been unresolved for decades, despite the constantly growing scientific knowledge on its pathogenesis, recent development of novel therapeutics, and multiple efforts to overcome the existing gaps in FH care by, e.g., raising its awareness in the community. With these assumptions, it is not surprising that only 10% of all estimated affected individuals have been diagnosed worldwide [1]. Regrettably, only 2% of FH cases are diagnosed before the age of 18 years [1]. For most subjects, diagnosis occurs late in life, mostly at the age of about 45 years, often when atherosclerotic cardiovascular disease (ASCVD) has already developed [5, 6]. Thus, there is an unmet need for the identification of new index cases much earlier in their life course. Therefore, an integrated multidisciplinary approach including pediatricians, primary care physicians and clinicians in adult hospital settings is important to facilitate systematic FH screening in combination with reverse cascade screening of first degree relatives of FH patients [7, 8, 9, 10]. More importantly, even if the diagnosis seems to be certain, it does not always imply that the index patient is adequately treated. The European Atherosclerosis Society Familial Hypercholesterolemia Studies Collaboration (FHSC) global registry has impressively shown, that less than 3% of patients achieved the guideline-recommended low-density lipoprotein cholesterol (LDL-C) target values [5]. The present review summarizes our current knowledge about FH and critically discusses pitfalls and challenges in successful diagnosis and treatment of this genetic disorder.
As the name implies, FH represents an inherited disease, characterized by a life-long exposure to markedly elevated LDL-C concentrations from birth, thereby predisposing affected individuals to premature ASCVD.
For years, the “classical” form of FH has been recognized as an autosomal
co-dominant monogenic condition, which is caused by variations of genes involved
in LDL-C metabolism and clearance [11]. Among them, about 80% of genetic
variants are caused by the mutation in the LDLR gene, encoding the LDL
transmembrane receptor (LDL-R), which results in complete or partial loss of its
function (so called “null” or “defective” LDL-R variants) [11, 12]. The
remaining pathogenic variants are related to mutations within the genes encoding
the apolipoprotein B (apoB) (APOB) (5–10%), with reduced binding of
the apoB to the LDL-R or due to the gain-of-function mutation of proprotein
convertase subtilisin/kexin 9 (PCSK9) (~3%), that lead
to its overproduction [11, 12, 13, 14, 15]. In addition, the APOE gene represents
another FH-causative gene, where single p.(Leu167del) mutation might occur in 1%
to 2% of patients with FH phenotype and result in LDL-R downregulation [16]. In
addition, there are also some other, sporadically occurring gene variants, e.g.,
within the genes encoding for signal-transducing adaptor protein family 1
(STAP1), patatin-like phospholipase-domain-containing family
(PNPLA5) or some rare mutations, related to severe, recessive
hypercholesterolemia, including LDLR adapter protein 1 (LDLRAP1),
lysosomal acid lipase (LIPA) or ATP- binding cassette subfamily G member
5 (ABCG5) [11, 12, 17, 18]. Interestingly, some of these genes might also
cause distinctive non-FH syndromes such as sitosterolemia (ABCG5),
dysbetalipoproteinemia (APOE) or cholesteryl ester storage disease
(LIPA) [19]. Despite a huge genetic heterogeneity (
Typically, the likelihood for FH can be estimated primarily on the basis of the clinical phenotype. To date, there are several diagnostic algorithms available [29] (e.g., the Dutch Lipid Clinic Network (DLCN) Criteria, the Simon Broome (SB) system, the Make Early Diagnosis to Prevent Early Deaths (MEDPED) system, as well as the American Heart Association Agenda for FH criteria), which can be applied to diagnose FH, although DLCN, SB and MEDPED remain the most commonly used scores so far. All of these scores focus on LDL-C concentration, most of them also include personal and/or family history of premature coronary artery disease or dyslipidemia. Additionally, physical signs, all reflecting extravasal cholesterol deposits such as arcus cornea or bilateral xanthomas (within the Achilles tendons or within extensor tendon of the hand) might also be included (see Table 1 for the comparison between main existing algorithms).
| Dutch Lipid Clinic Network | Simon Broome Register Group’s | MEDPED | |
| Family history of hypercholesterolemia | I° relative with LDL-C |
I° or II° relative with TC |
Relative with confirmed FH diagnosis (I°/II°/III°) |
| Children ( |
|||
| Elevated LDL-C (untreated) | Relative I°/II°/III°/general population | ||
| 250–329 mg/dL (6.5–8.4 mmol/L) (5 points) | |||
| 190–249 mg/dL (5.0–6.4 mmol/L) (3 points) | 20–29 y: 240/250/260/290 mg/dL / 6.2/6.5/6.7/7.5 mmol/L | ||
| 155–189 mg/dL (4.0–4.9 mmol/L) (1 point) | 30–39 y: 270/280/290/340 mg/dL / 7.0/7.2/7.5/8.8 mmol/L | ||
| Family history of premature coronary artery disease | I° relative with known premature coronary and/or vascular disease (♂ |
I° relative with MI ( |
- |
| Family history of tendon xanthomas | I° relative with tendinous xanthomata and/or arcus cornealis (2 points) | I° relative with xanthomas (B) | - |
| Personal history | - Patients with premature coronary artery disease (♂ |
- | - |
| - Patients with premature cerebral or peripheral vascular disease (♂ |
|||
| Physical examination | Tendinous xanthomata (6 points) | Xanthomas in the proband (B) | - |
| Arcus cornealis |
|||
| Genetic analysis | Mutation in the LDLR, APOB or PCSK9 gene (8 points) | Mutation in the LDLR, APOB or PCSK9 gene (C) | - |
| Diagnosis | Unlikely FH: |
Possible FH: A + D or A and E | FH is diagnosed if LDL-C exceed the cut point |
| Possible FH: 3–5 | Definitive FH: A + B or C | ||
| Probable FH: 6–8 | |||
| Definite FH: |
FH, familial hypercholesterolemia; MEDPED, “Make Early Diagnosis to Prevent Early Death”; LDL-C, low density lipoprotein cholesterol; pctl, percentile; TC, total cholesterol; y, year; MI, myocardial infarction; LDLR, low-density lipoprotein receptor; APOB, apolipoprotein B; PCSK9, proprotein convertase subtilisin/kexin 9.
Subsequent genetic testing with confirmation of a pathogenic mutation in the FH
causative gene would provide diagnostic certainty, although this is not essential
for the diagnosis itself. Nonetheless, identification of a positive mutation is
important for the initiation of cascade screening to detect FH in other family
members as well as for the early initiation of lipid-lowering treatment (LLT) and
might be implicated in the choice of treatment in FH. Moreover, genetic
confirmation of FH is extremely helpful in identifying subjects with the highest
risk for ASCVD, since the presence of a “classic” FH mutation in subjects with
LDL-C levels
Nonetheless, optimal screening strategies to identify index patients on the
population level have not yet been determined. The commonly used diagnostic tools
for the severe hypercholesterolemic phenotype in the clinical setting rely on
already manifested clinical symptoms/diseases, thereby having only limited
utility in primary care for the early (asymptomatic) FH case-finding.
Furthermore, xanthomas and corneal arcus can be detected only in
In line with all of the above mentioned findings are the results of several
studies, showing significant variability in the accuracy of clinical algorithms
in those with genetically confirmed FH [33, 34]. One extreme example represents
the analysis by Mohammadnia et al. [33], who demonstrated that the
sensitivity of currently available scores for the clinical FH diagnosis in
subjects, positive for FH gene mutations is only modest, being 9% for DLCN
On the other hand, recent data from the large-scale population-based studies
using next-generation DNA sequencing have found that on a molecular-genetic level
FH is more complex than previously assumed. Importantly, the majority of
individuals who meet clinical FH criteria do not possess a causative gene defect
within the main, “classical” FH genes [30, 35, 36, 37, 38, 39], although the prevalence of
identified mutations might vary significantly depending of the applied clinical
criteria or the clinical setting (from the general population to the tertiary
care lipid clinics). For instance, data from one large study, including
~20,000 individuals from the general population have demonstrated
that in subjects with LDL-C
Yet, the prevalence of causative FH mutation among patients with suspected FH,
referred to the tertiary care lipid clinics, seems to be higher. Genetic
confirmation might be found among up to two-thirds of such patients and even
~90% in those with untreated LDL-C levels
Thus, a substantial number of patients with clinical FH phenotype (both very high LDL-C levels and positive family history) but without monogenic mutation would suggest polygenic causes of FH, where small but cumulative effects of several LDL-C raising alleles can cause the LDL-C increase up to the same range as that caused by the three primary FH-causing genes [12, 17, 18]. Indeed, up to 100 polymorphic loci might contribute to polygenic susceptibility to elevated LDL-C [12, 41, 42]. In addition, presence of negative genetic test results might also imply presence of causal mutations within the still unidentified genes, so called genetically undefined hypercholesterolemia [43].
Interestingly, some substantial differences between monogenic and polygenic FH
might exist with regard to the clinical presentation, cardiovascular risk and
responsiveness to therapy (for comprehensive review please see Ref. [43, 44]). For
instance, it could be shown that subjects with monogenic FH not only have
statistically higher LDL-C concentration (typically by ~
Taken together, there is not only a clear mismatch between clinical and genetic
diagnosis of FH, but also significant differences in its genetic background
(mono- versus polygenic), that might significantly complicate the recognition of
FH in daily practice. Nonetheless, a simple combination of untreated LDL-C
Another challenge in the diagnosis of FH is the LDL-C measurement. Conventional assays for LDL-C determination quantify a composite of atherogenic cholesterol, which is attributable not only to LDL-C, but also to lipoprotein(a)-cholesterol (Lp(a)-C) due to their overlapping densities. Lp(a) represents a genetically determined highly atherogenic LDL-like particle, which has been considered a novel risk factor for ASCVD and aortic stenosis [49]. Early studies have shown that subjects with diagnosed FH had higher levels of Lp(a) [50, 51], compared to non-affected individuals, thereby assuming that FH might lead to an increase in Lp(a). Indeed, elevated Lp(a) is present in 30–50% of FH patients [52]. However, the role of Lp(a) in FH seems to be much more complex. Recent data suggest that increased Lp(a) levels might, at least in part, mimic the clinical diagnosis of FH, probably due to the Lp(a)-C component within “overall” LDL-C quantification [53, 54, 55, 56]. For instance, among 46,200 individuals from the Copenhagen General Population Study, about 25% of individuals with clinical FH were diagnosed because of high Lp(a) levels [54]. A series of studies, conducted within the last 2–3 years provided very consistent results, showing that the Lp(a)-C content in LDL-C in subjects with suspected HeFH can lead to reclassification of clinical FH status [54, 55, 56, 57, 58]. For instance, Hedegaard et al. [58] recalculated the DLCN scores after adjusting for the contribution of Lp(a) to LDL-C values and found that 16.6% of patients fell into a lower DLCN category. Furthermore, two other studies showed that up to 10 patients with clinical suspicion of FH could be down-classified to the category “unlikely FH” using Lp(a)-corrected LDL-C [55, 56], thereby avoiding unnecessary genetic analysis for FH.
Unfortunately, how LDL-C should be corrected for its Lp(a)-C content is not
entirely clear, especially taking into account a possible variation of Lp(a)-C
relative to its mass, which might vary from 6% to 60% [59, 60, 61]. Most
importantly, by applying the “wrong” correction one could also miss mutation
positive subjects. Nonetheless, taking into account the fact that all diagnostic
FH algorithms rely on plasma LDL-C level, an interrelationship between Lp(a)-C
and “true” LDL-C should not be underestimated, especially in those with LDL-C
levels that are borderline consistent with HeFH. Although several issues still
have to be clarified on the role of Lp(a) in FH, it is clear, having both FH and
high Lp(a) values
Being mostly asymptomatic, lifelong exposure to elevated LDL-C, if untreated,
leads to premature development and accelerated progression of ASCVD. Thus, early
introduction of therapeutic interventions is essential for improved prognosis of
patients with FH. Currently, the European Society of Cardiology/European
Atherosclerosis Society (ESC/EAS) 2019 guidelines recommend at least 50% LDL-C
reduction and LDL-C target
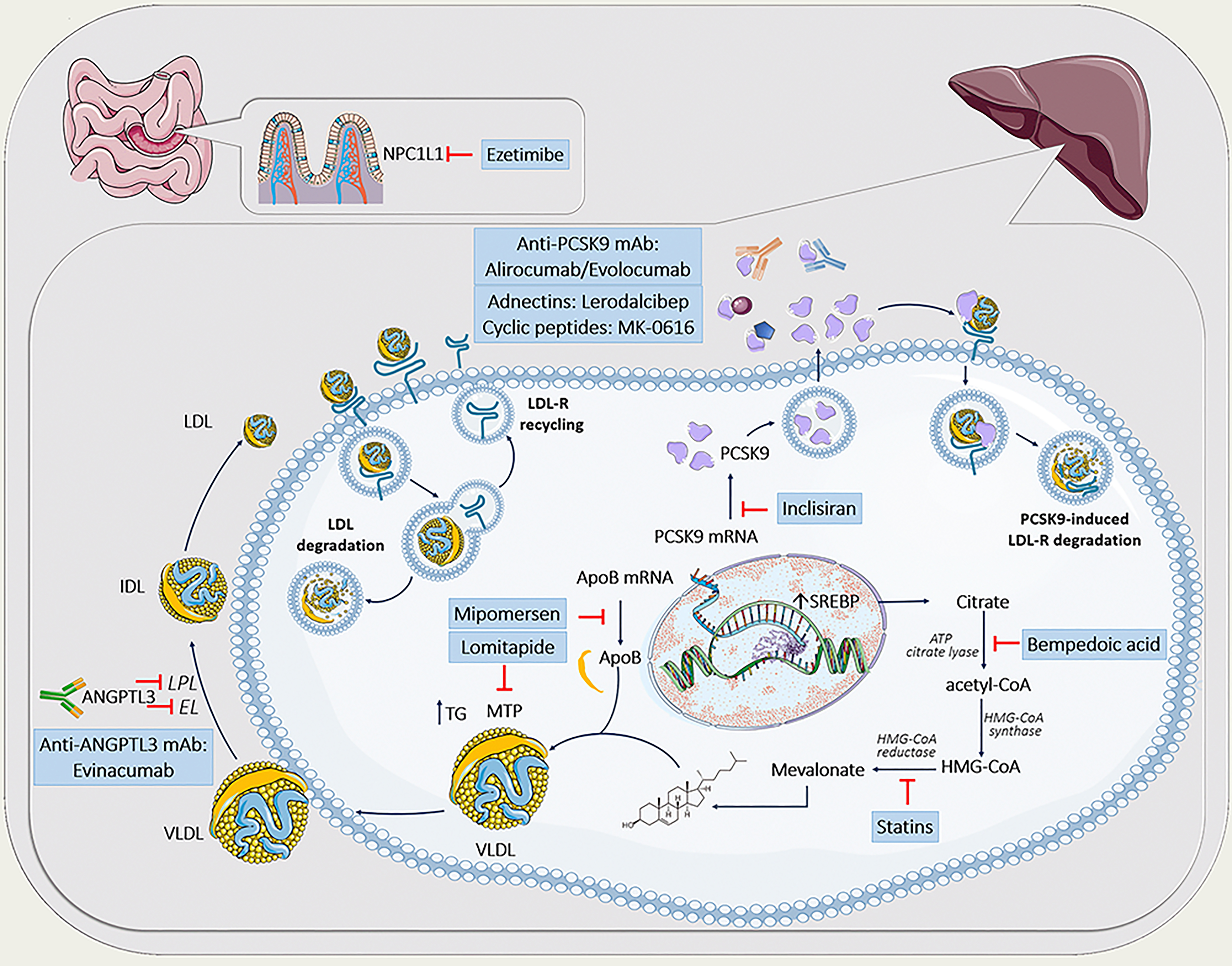 Fig. 1.
Fig. 1.Lipid-lowering agents in the treatment of familial hypercholesterolemia. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. ANGPTL3, angiopoietin-like protein 3; apoB, apolipoprotein B; ATP, adenosine triphosphate; CETP, cholesteryl ester transfer protein; EL, endothelial lipase; HDL, high density lipoprotein; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LDL-R, low-density lipoprotein receptor; LPL, lipoprotein lipase; mAb, monoclonal antibodies; MTP, microsomal triglyceride transfer protein; NPC1L1, Niemann-Pick C1-like 1 protein; PCSK9, proprotein convertase subtilisin kexin type 9; SREBP, sterol regulatory element-binding protein; mRNA, messenger RNA; TG, triglycerides; VLDL, very-low-density lipoprotein.
For years, statins (alone or in combination) represent a cost-effective first-line therapy in subjects with FH, particularly in heterozygous patients [63, 64]. In general, high-potency statins are capable of lowering LDL-C by 50% to 60% as monotherapy and even by 65 to 70% if combined, e.g., with ezetimibe, a Niemann-Pick C1-like protein inhibitor [65, 66]. Both compounds, although acting differentially (statins by decreasing cholesterol production via selective inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase; ezetimibe by blocking cholesterol uptake from the jejunum) result in a compensatory increase in LDL-R and subsequently enhanced LDL-C clearance. So, in subjects with FH, having a dysfunctional LDL-R, LDL-C lowering effects of this standard LLT might be only modest [63]. In addition, the presence of increased Lp(a) might also influence the LDL-C lowering ability of statins, particularly in those with smaller apo(a) isoforms, either by decreasing an apparent response to LDL-C lowering or even increasing the LDL-C concentration [67, 68]. Nonetheless, statins (alone or in combination with ezetimibe) demonstrated a significant reduction of future ASCVD events even in subjects with LDL-R defective forms [69, 70, 71, 72, 73].
More recently, bempedoic acid (BA), another inhibitor of intracellular cholesterol biosynthesis, has been introduced in the clinical setting. It acts as an inhibitor of adenosine triphosphate (ATP) citrate lyase, a hepatic enzyme that works upstream of HMG-CoA reductase with subsequent upregulation of LDL-R activity, similar to statins [74]. Pooled analysis of 112 patients with a clinical phenotype of HeFH, participating in phase 3 trials (CLEAR Harmony and CLEAR wisdom) showed a mean LDL-C reduction of 22.3% by BA, applied as an adjunct or alternatively to currently existing LLT [75]. However, whether BA would also reduce LDL-C in HoFH has not been investigated so far. But, based on the mechanism of action of BA, which is similar to statins, it is possible that patients with residual LDL-R activity will respond to it as well.
The development of PCSK9 inhibitors has provided an additional therapeutic tool to control LDL-C in FH patients with residual LDL-receptor activity. In 2003, a novel gain-of-function mutation within the PCSK9 gene, contributing to a phenotype with markedly elevated LDL-C levels and premature ASCVD, had been identified in patients with severe hypercholesterolemia [15]. PCSK9 decreases recycling and increases degradation of the LDL-R (Fig. 1). To date, there are only two approved modalities to inhibit PCSK9 activity. Alirocumab and evolocumab are fully human monoclonal antibodies (mAbs) targeted against PCSK9, whereas inclisiran represents a first-in-class cholesterol-lowering small interfering ribonucleic acid (siRNA), targeting PCSK9 messenger RNA (mRNA) in hepatocytes. In contrast to anti-PCSK9 mAbs, inclisiran inactivates PCSK9 by inhibition of its hepatic synthesis [76].
So far, there are several trials that have assessed the efficacy of anti-PCSK9 mAbs in HeFH, including ODYSSEY FH I/II, ODYSSEY HIGH FH, RUTHERFORD-2, as well as HAUSER-RCT, all reporting meaningful LDL-C reductions by alirocumab or evolocumab between 45 and 65% [77, 78, 79, 80, 81]. However, in HoFH and LDL-R-negative mutations, anti-PCSK9 mAbs would probably only be mildly effective or even fail to lower LDL-C [82, 83, 84]. So, current guidelines also recommend the use of PCSK9 inhibitors to treat homozygous FH patients except those with confirmed negative/negative LDLR mutations.
Inclisiran might also be a promising option to treat FH patients, showing a mean
LDL-C reduction of 40% in HeFH subjects within the ORION-9 trial [85]. Also, in
the ORION-2 trial, an open-label pilot study in 5 HoFH patients receiving
high-intensity statin plus ezetimibe, inclisiran exhibited similar LDL-C
lowering, compared to those, observed for anti PCSK9-mAb, although with a longer
effect duration [86]. A phase 3 study of inclisiran in HoFH (NCT03851705),
including 56 patients with HoFH and LDL-C
There are also several emerging PCSK9 inhibitors such as Lerodalcibep (recombinant fusion protein, consisting of a PCSK9-binding domain (adnectin)) or MK-0616 (synthetic cyclic peptide, being a first orally bioavailable PCSK9 inhibitor) which are currently being tested in FH patients (Lerodalcibep: NCT04034485 for HoFH and NCT04797104 for HeFH; MK-0616: NCT05261126).
Despite a large armamentarium of potent lipid-lowering medication, including
statins, ezetimibe, BA and PCSK9 inhibitors, which demonstrate a cumulative
ability to lower LDL-C
Being a cellular protein, responsible for the transport of neutral lipids between membrane vesicles, microsomal triglyceride transfer protein (MTP) plays a pivotal role in apoB secretion [90]. Lomitapide, the first MTP inhibitor, exclusively used for patients with HoFH with or without lipid apheresis, reduce LDL-C concentration by 40–50% primarily via decreased VLDL and apoB secretion [91].
Another inhibitor of apoB/VLDL secretion is Mipomersen, an antisense oligonucleotide (ASO) targeted to the APOB mRNA [90], having a potential to lower LDL-C by ~25% in patients with HoFH [92, 93]. It is approved for HoFH patients in the U.S. but EMA refused mipomersen marketing authorization.
Unfortunately, both therapeutic compounds have strong gastrointestinal side effects and might significantly increase hepatic fat deposition, leading to hepatosteatosis, thereby limiting their use in patients with FH [90, 93].
Angiopoietin-like 3 protein (ANGPTL3) is an endogenous inhibitor of endothelial and lipoprotein lipase, the latter representing a key enzyme involved in the removal of triglycerides rich lipoproteins from the circulation [94, 95]. The discovery of ANGPTL3 as a potential treatment target came from Genome-wide association study (GWAS), where subjects with a loss-of-function mutation within the ANGPTL3 gene demonstrated a 41% lower risk of ASCVD due to life-long low levels of both LDL-C and triglycerides [96]. Although the role of ANGPTL3 in lowering LDL-C is still not completely understood one might suggest, that ANGPTL3 inhibition enhances fractional catabolic rate of large VLDL thereby reducing LDL-C through faster clearance of their remnants by non-LDL-R-mediated pathways [97].
Evinacumab, the first available ANGPTL3 inhibitor is a human monoclonal antibody for ANGPTL3, which has been approved for the treatment of patients with HoFH [98]. Approximately 50% LDL-C reduction under evinacumab therapy has been demonstrated in HoFH subjects and those with refractory hypercholesterolemia [99, 100]. More importantly, even in patients with null/null variants in the LDL-R a 43% reduction in LDL-C has been seen, indicating LDL-R independent pathway of lipid lowering.
Finally, a first siRNA targeting ANGPTL3 mRNA is also under development (ARO-ANG3), demonstrating an approximately 40% LDL-C reduction in a phase I trial [101]. ARO-ANG3 is currently being tested in phase 2 trials in patients with HoFH (NCT05217667) or mixed dyslipidemia (NCT04832971).
Since FH patients might have dysfunctional high density lipoproteins (HDL), resulting in defective reverse cholesterol transport (RCT) and subsequent increase in cholesteryl ester transfer protein (CETP) in the circulation [102], inhibition of CETP, a hydrophobic glycoprotein that promotes the transfer of cholesteryl ester and triglyceride between all lipoproteins, might represent another possible target in FH. Although initial studies on CETP inhibitors were rather disappointing [103], the data on the newest CETP inhibitor obicetrapib seems to be more promising, achieving reductions in LDL-C up to 50% [104]. Currently, obicetrapib has been tested within the phase III study (BROOKLYN) (NCT05425745) in patients with HeFH on top of maximum tolerated lipid-modifying therapies. More interestingly, obicetrapib might also lower Lp(a) level by approximately 50%. However, the pathophysiological mechanism responsible for such profound Lp(a) lowering is still poorly understood.
Taken together, our therapeutic armamentarium to combat FH increased significantly during the last years allowing us to prevent/reduce future cardiovascular events more successfully [105]. However, despite such significant improvement in the pharmacologic intervention, initiation of lipoprotein apheresis (LA) in addition to existing drug therapy is foundational for a still substantial proportion of FH patients (HoFH or with increased Lp(a) level) and might represent the only way to attain the guideline-recommended LDL-C targets [89]. On the other hand, there is clear evidence that novel FH therapeutics might significantly reduce the need for LA [89].
Familial hypercholesterolemia remains vastly underdiagnosed and as a consequence, undertreated, resulting in a missed opportunity to delay or even prevent clinical manifestations of atherosclerosis. Early detection of FH, including wide-spread pediatric screening programs, as well as increased medical community awareness of FH should become a priority worldwide to improve the low diagnostic rates of FH. A further major challenge in FH represents its definition, since recent research has reshaped our understanding of the pathogenesis of FH, indicating that FH is not an exclusively monogenic disorder. Finally, significant treatment gaps are still existing, demanding not only novel therapeutics, but also their broad accessibility. Although current efforts in FH management are still hampered, integrated implementation of strategies worldwide to identify, diagnose and successfully treat FH patients would undoubtedly lead to a significant reduction of FH burden.
NA and WK equally concepted the present review and researched, collected and interpreted the relevant data. NA wrote the first draft of the manuscript and WK critically revised the manuscript for important intellectual content. NA designed the figure. Both authors read and approved the final manuscript and have participated sufficiently in the work, as well as agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Not applicable.
Not applicable.
Financial support from the Open Access Publication Fund of UKE (University Medical Center Hamburg-Eppendorf) and DFG (German Research Foundation).
N.A. has no conflicts of interest. W.K. reports receiving consulting fees and lecture fees from AstraZeneca, Novartis, and Amgen; consulting fees from Pfizer, the Medicines Company, DalCor Pharmaceuticals, Kowa, Corvidia Therapeutics, Genentech, Esperion, Novo Nordisk, OMEICOS, New Amsterdam Pharma, TenSixteen Bio, Daiichi Sankyo; lecture fees from Berlin-Chemie, Bristol-Myers Squibb, Amgen, AstraZeneca, Novartis, and Sanofi; and grant support and provision of reagents from Singulex, Abbott and Roche Diagnostics, and Dr. Beckmann Pharma. W.K. has been a member of the executive steering committees of ORION, JUPITER, CANTOS, SPIRE, GLAGOV, and COLCOT.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.