


1 Department of Cardiovascular Medicine, The First People's Hospital of Changzhou, The Third Affiliated Hospital of Soochow University, 213000 Changzhou, Jiangsu, China
†These authors contributed equally.
Abstract
Atherosclerotic cardiovascular disease is currently the leading cause of death worldwide. Its pathophysiological basis includes endothelial dysfunction, macrophage activation, vascular smooth muscle cell (VSMC) proliferation, lipid metabolism, platelet aggregation, and changes in the gut microbiota. Salidroside has beneficial effects on atherosclerosis through multiple pathways. In this review, we present studies on the regulatory effect of salidroside on atherosclerosis. Furthermore, we report the protective effects of salidroside against atherosclerosis by ameliorating endothelial dysfunction, suppressing macrophage activation and polarization, inhibiting VSMC proliferation, adjusting lipid metabolism, attenuating platelet aggregation, and modulating the gut microbiota. This review provides further understanding of the molecular mechanism of salidroside and new ideas for atherosclerosis management.
Keywords
- salidroside
- atherosclerosis
- endothelial dysfunction
- cell targets
- lipid metabolism
- gut microbiota
With the aging of the population, the morbidity and mortality of cardiovascular disease (CVD) remain high. In China, the number of deaths due to CVD was nearly 3.97 million, and the prevalence of CVD was estimated to be 93.8 million in 2016 [1]. The common pathological basis of CVD is atherosclerosis. The complex pathophysiologic process of atherosclerosis includes dyslipidemia, oxidative stress, endothelial dysfunction, thrombocyte aggregation [2, 3], foam cell formation and accumulation [4], and vascular smooth muscle cell (VSMC) migration and proliferation [5]. Oxidative modification and subendothelial retention of low-density lipoprotein cholesterol (LDL-C) represent the initial events in atherogenesis [6]. Oxidized low-density lipoprotein (Ox-LDL) enters the intima-media of the vascular wall and contributes to atherosclerotic plaque formation and progression by inducing endothelial cell (EC) activation and dysfunction, macrophage foam cell formation, and vascular smooth muscle cell (VSMC) migration and proliferation [7]. At present, statins [8] and antiplatelet therapy [9] are widely used to prevent atherosclerosis-related complications, and the effect of these therapies has pros and cons. For example, statins may lead to hepatotoxicity and skeletal muscle toxicity [10], and antiplatelet therapy inevitably leads to a risk of hemorrhage [11]. In recent years, interest in the role of herbal plants in treating CVD has grown. Chinese herbal medicine has long been used for the treatment of atherosclerotic complications [12]. Many studies suggest that salidroside, which has low toxicity and few side effects, possesses a wide range of biological properties, such as inhibiting inflammation, regulating dyslipidemia, improving endothelial function [13, 14, 15], suppressing macrophage phenotypic switching, decreasing the proliferation of VSMCs and impairing the activation of platelets. Thus, salidroside may be a valuable and promising drug candidate for the treatment of CVDs, but it is not in widespread use in clinical practice. In particular, salidroside can influence the gut microbiota; however, the mechanism that drives this phenomenon remains unclear. In this review, we provide an overview of the molecular mechanism by which salidroside attenuates atherosclerosis. The underlying mechanisms of salidroside in protecting against atherosclerosis as shown in Fig. 1.
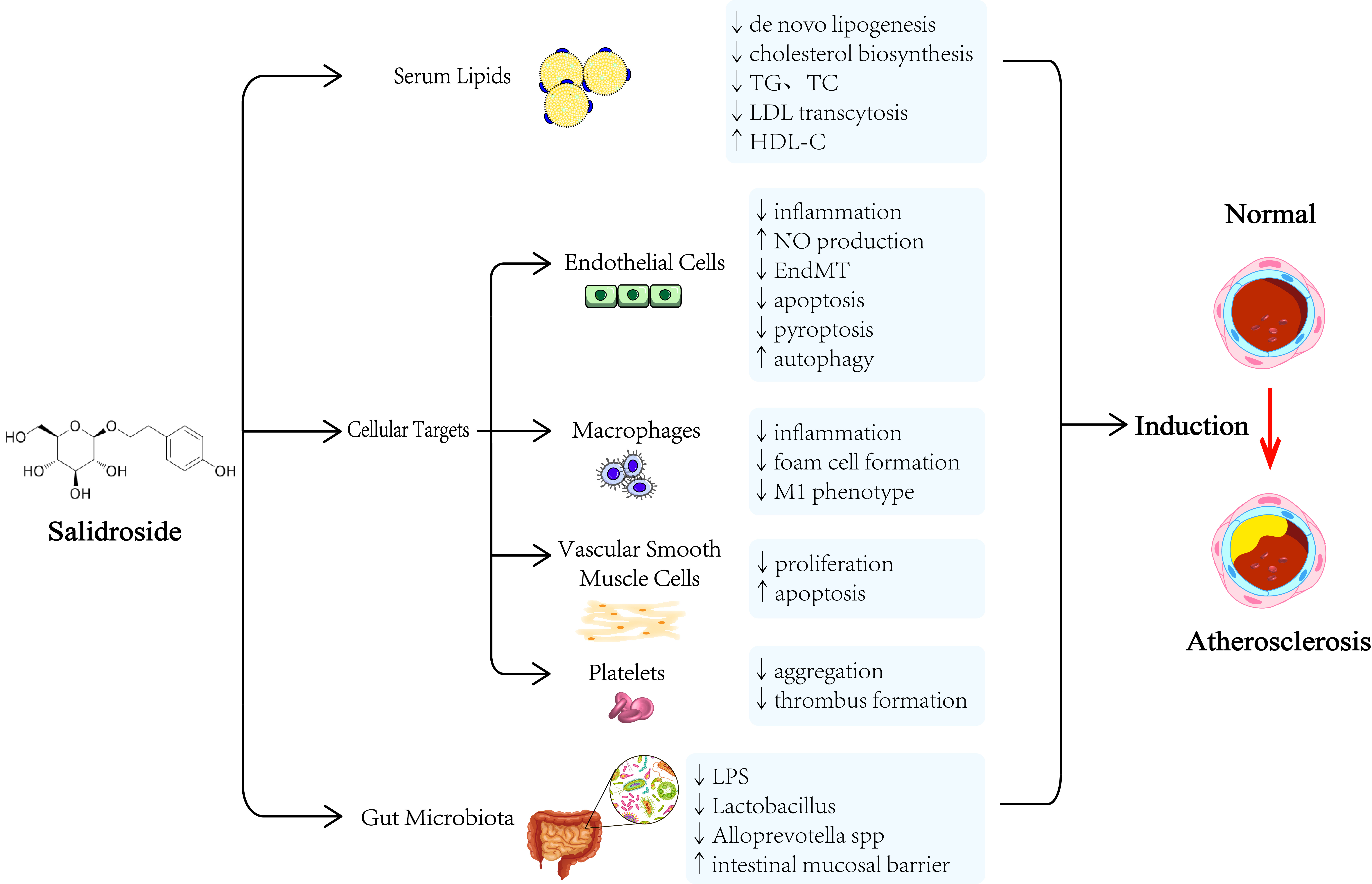 Fig. 1.
Fig. 1.The underlying mechanisms of salidroside in protecting against atherosclerosis. TC, total cholesterol; TG, triglyceride (TG); LDL, low-density lipoprotein; NO, nitric oxide; HDL-C, high-density lipoprotein cholesterol; EndMT, endothelial-mesenchymal transition; LPS, lipopolysaccharide.
Considerable evidence has suggested that dysfunction of the vascular endothelium plays a significant role in atherosclerosis development and progression. First, excessive reactive oxygen species (ROS) [16] and malondialdehyde (MDA) [17] can increase oxidative stress, which is linked to endothelial dysfunction and atherogenesis. Second, due to decreases in nitric oxide (NO) and endothelial nitric oxide synthase (eNOS) levels, endothelium-dependent vasodilation is impaired, which confers a risk of atherogenesis [18]. Third, endothelial-mesenchymal transition (EndMT), a process in which ECs acquire myofibroblast-like properties, is one of the main mechanisms of atherogenesis that increases endothelial dysfunction [19]. Furthermore, increasing levels of apoptosis and autophagy in ECs can also influence the development of atherosclerosis. The activation of apoptosis [20] and pyroptosis [21] in ECs can increase the levels of inflammatory factors, such as ROS and caspase-1, and lead to vessel wall inflammation, which may be involved in atherogenesis. On the other hand, endothelial autophagy may prolong the survival of ECs by inhibiting endothelial apoptosis and has antiatherogenic effects [22]. Excessive autophagy in ECs can promote atherosclerotic plaque destabilization, which leads to accelerated atherogenesis [23]. Overall, multiple pathways work together to confer a high risk of endothelial dysfunction and atherosclerosis. Therefore, improving endothelial function is critical in the treatment and prognosis of atherosclerosis. Endothelial activity regulated by salidroside are summarized in Table 1.
| Cell activity | Inflammatory factors or receptors | Possible targeting pathways by SAL | Effect |
|---|---|---|---|
| Endothelial oxidative stress | ROS | cAMP/PKA/RhoA | Downregulate |
| IL-1 |
AMPK/NF-κB/NLRP3 | Downregulate | |
| ROS, MDA | AMPK/SIRT1 | Downregulate | |
| SOD, CAT | Nrf2 | Upregulate | |
| Endothelium-dependent contraction | NO | AMPK/PI3K/Akt/eNOS | Upregulate |
| Nox2, ROS | — | Downregulate | |
| Endothelial-mesenchymal transition | NO | KLF4/eNOS | Downregulate |
| Endothelial apoptosis | Bcl-xL | miR-133a | Upregulate |
| Caspase-3 | — | Downregulate | |
| Endothelial pyroptosis | Caspase-1, IL-1 |
— | Downregulate |
| Endothelial autophagy | LC3-II/ LC3-I | AMPK-mTOR | Upregulate |
| — | SIRT1-FoxO1 | Upregulate | |
| ROS, Reactive oxygen species; cAMP, Cyclic adenosine monophosphate; PKA, Protein
kinase A; RhoA, Ras homolog gene family member A; IL-1 | |||
A major cause of endothelial dysfunction is oxidative stress. Salidroside plays
a crucial role in downregulating endothelial oxidative stress not only by
decreasing the level of proinflammatory factors but also by increasing the level
of anti-inflammatory factors. First, ROS have been shown to enhance oxidative
stress in ECs [24], which plays an important role in the signaling pathways
associated with endothelial dysfunction [25]. Li et al. [26] found that
the endothelial barrier in intermittent hypoxia (IH)-induced human coronary vein
endothelial cell was damaged by ROS, and salidroside (10
Endothelium-dependent contraction (EDC) is also associated with endothelial
dysfunction. EDC leads to vasospasm, which may exacerbate ischemia and hypoxia at
the beginning of atherogenesis [31]. It is well documented that increased eNOS
phosphorylation and expression can enhance NO production to improve endothelial
function [32]. Xing et al. [33] administered different salidroside (1
EndMT can be exacerbated by inflammation, hypoxia, and oxidative stress in the
endothelium through the activation of TGF-
Various forms of endothelial death, such as apoptosis [42], pyroptosis [43], and
autophagy [44], can influence the development and progression of atherosclerosis.
First, salidroside upregulates the expression of B-cell lymphoma-extra large
(Bcl-xL), an antiapoptotic protein, and inhibits Ox-LDL-induced EC apoptosis
[45]. Zhang et al. [46] used human coronary artery endothelial cell
(HCAECs) to analyze the effect of salidroside (100
Overall, salidroside can improve endothelial function in many ways, such as through anti-inflammatory effects, increasing the production of NO, inhibiting EndMT, and regulating EC death. These findings support the clinical importance of salidroside.
Macrophages play a critical role in the initiation and progression of
atherosclerosis. In the early stage of atherosclerosis, macrophages can be
recruited to the lesioned arterial wall by proinflammatory cytokines [53].
Macrophage activation is an essential event in early atherosclerosis. Wang and
colleagues [54] found that salidroside (50, 100 or 50
Finally, studies have shown that different macrophage phenotypes play different
roles in atherosclerosis. It is widely known that M1 macrophages play a
proinflammatory role in atherosclerosis, while M2 macrophages play a preventive
role [57, 58, 59]. Li et al. [60] discovered that salidroside
(25~100
VSMCs are one of the main cell types in the blood vessel wall. Increased VSMC proliferation can induce pathological intimal thickening, which can induce the progression of atherosclerosis [64]. Some studies have shown that VSMCs switch from a contractile to synthetic phenotype, and these cell possess highly proliferative and migratory capacities, which may impair plaque stability [64, 65]. In addition, atherosclerotic plaque stability is negatively associated with increased VSMC apoptosis [66]. Whether salidroside can inhibit the switching of VSMCs is still unclear and needs further examination. The studies which have focused on the beneficial effects of salidroside on inhibiting VSMC proliferation and apoptosis are as follows.
Zhuang and other researchers [67] investigated the protective effect of
salidroside (0.3 and 0.5 mM, 24 h) on VSMCs under high glucose stimulation. The
results showed that salidroside could decrease the proliferation of VSMCs not
only by downregulating the activation of NADPH and reducing the level of ROS but
also by inhibiting mitochondrial fission through the downregulation of
dynamin-related protein (Drp1) and mitofusin 2 (Mfn2). Moreover, salidroside (100
Platelet activation leads to adhesion, aggregation, and thrombosis, playing a significant role in atherosclerosis [71]. Recently, antiplatelet therapies, such as aspirin, clopidogrel, and ticagrelor, have been shown to play a significant role in reducing clinical atherothrombotic events among high-risk patients [72]. Salidroside, which is a botanical medicine, has also been demonstrated to produce beneficial effects on inhibiting platelets.
Wei et al. [73] demonstrated that salidroside (5, 10 and 20
An aberrant lipid profile, including increased total cholesterol (TC), triglyceride (TG), and LDL-C and decreased high-density lipoprotein cholesterol (HDL-C), is associated with an increased risk of atherosclerosis [74]. Therefore, lipid lowering is regarded as the key treatment in the primary and secondary prevention of atherosclerosis. Currently, statins, and proprotein convertase subtilisin/Kexin type 9 (PCSK9) inhibitors, and icosapent ethyl (IPE), which are essential lipid-lowering therapies, play vital roles in controlling atherosclerosis [75]. The studies which have shown that salidroside may also lower lipid levels are as follows.
First, some animal studies have suggested that salidroside
(100 mg
Recent research has highlighted the significant role of the gut microbiota in
CVD [84], especially in atherosclerosis. On the one hand, some studies have shown
that gut dysbiosis plays an important role in atherosclerosis [85]. On the other
hand, increasing intestinal permeability and disruption of the intestinal barrier
can lead to bacterial translocation, which may release LPS, trimethylamine (TMA)
and trimethylamine-N-oxide (TMAO) into the circulation. These gut
microbiota-derived products can not only induce systemic inflammation but are
also connected with atherosclerosis [86]. In other words, dysregulation of the
gut microbiota leads to low-grade chronic inflammation, which can accelerate
atherosclerotic progression [87]. Zhu et al. [52] collected fecal
samples from 218 individuals with atherosclerotic cardiovascular disease (ASCVD)
and compared the composition of the gut microbiota with the samples from healthy
controls. They discovered that ASCVD patients had a higher level of Streptococcus
and Escherichia [88]. Moreover, scholars from Japan reported that Lactobacillales
was increased in CAD patients, while Bacteroidetes was decreased [89]. In
addition, changes in the gut microbiota and gut permeability can increase IL-6,
TNF-
Several studies have reported the protective effect of salidroside on the gut
microbiome. First, Xie and other scholars [92] observed that salidroside
(50 mg
This review provides a modern scientific perspective to further understanding the molecular mechanism of salidroside attenuating atherosclerosis and supply new ideas for atherosclerosis management.
Based on the present studies, salidroside affects atherosclerosis through
multiple signaling pathways and related mechanisms. Salidroside protects against
atherosclerosis through multiple targets and multiple pathways. (1) Salidroside
ameliorates endothelial dysfunction through anti-inflammatory effects, increasing
the production of NO, inhibiting EndMT, and regulating the death of ECs. (2)
Salidroside suppresses macrophage activation by inhibiting the
MAPK/NF-
In conclusion, these findings suggest that salidroside may be a promising drug for preventing and treating atherosclerosis. At present, the anti-atherosclerotic signaling pathways and targets of salidroside are not comprehensively understood, and few animal studies have been conducted. Besides, its clinical application has progressed slowly and some details remain unknown, and the best optimum dose is not determined. Some studies were restricted to a single model, and toxicity issues were not included. Therefore, more studies, especially clinical trials, are needed to further confirm the therapeutic effects and molecular mechanisms of salidroside.
SFF and DBT are the primary author who performed the literature review and manuscript preparation. FJ is the senior author who assisted with literature review, edits, and revisions of text. All authors have read and agreed to the published version of the manuscript.
Not applicable.
Not applicable.
This work was Funding from Major Technology Projects of Changzhou Health Commission (ZD202212).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.