





 , Buyan-Ochir Orgil 2,3, Neely R. Alberson 2,3, Jeffrey A. Towbin 2,3,4, Enkhsaikhan Purevjav 2,3,*
, Buyan-Ochir Orgil 2,3, Neely R. Alberson 2,3, Jeffrey A. Towbin 2,3,4, Enkhsaikhan Purevjav 2,3,*1 College of Medicine, University of Tennessee Health Science Center, Memphis, TN 38103, USA
2 Heart Institute, Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN 38103, USA
3 Children’s Foundation Research Institute, Le Bonheur Children’s Hospital, Memphis, TN 38103, USA
4 Pediatric Cardiology, St. Jude Children's Research Hospital, Memphis, TN 38105, USA
Academic Editors: Sophie Mavrogeni and Robert C Hendel
Abstract
Restrictive cardiomyopathy (RCM), a potentially devastating heart muscle
disorder, is characterized by diastolic dysfunction due to abnormal muscle
relaxation and myocardial stiffness resulting in restrictive filling of the
ventricles. Diastolic dysfunction is often accompanied by left atrial or
bi-atrial enlargement and normal ventricular size and systolic function. RCM is
the rarest form of cardiomyopathy, accounting for 2–5% of pediatric
cardiomyopathy cases, however, survival rates have been reported to be 82%,
80%, and 68% at 1-, 2-, and 5-years after diagnosis, respectively. RCM can be
idiopathic, familial, or secondary to a systemic disorder, such as amyloidosis,
sarcoidosis, and hereditary hemochromatosis. Approximately 30% of cases are
familial RCM, and the genes that have been linked to RCM are cTnT,
cTnI, MyBP-C, MYH7, MYL2, MYL3,
DES, MYPN, TTN, BAG3, DCBLD2,
LNMA, and FLNC. Increased Ca
Keywords
- cardiomyopathy
- restrictive cardiomyopathy
- restrictive physiology
- mutation
Restrictive cardiomyopathy (RCM) is a rare form of heart muscle disease, characterized by diastolic dysfunction provoked from myocardial stiffness in the setting of normal or reduced biventricular diastolic and systolic volumes [1, 2]. Impaired diastolic function leads to left atrial or biatrial enlargement and elevated atrial pressures. RCM may eventually affect ventricular chamber size and systolic function. These patients can progress to heart failure with preserved ejection fraction (HFpEF) or heart failure with reduced ejection fraction if left untreated [1, 3]. RCM accounts for 2–5% of all diagnosed cardiomyopathies; however, it has the worst outcome among cardiomyopathies with up to 50% of deaths occurring within 2 years after diagnosis in children [4]. With a median survival of 2.2 years without heart transplantation, RCM remains a disease that is largely unexplored and with limited treatment options [5]. Advancements in molecular genetics and improved assessment by radiography and echocardiography throughout the past few decades have expanded diagnostic capabilities and understanding of pathophysiology underlying RCM.
The term cardiomyopathy was first defined by Brigden in 1957 to describe uncommon isolated and idiopathic non-coronary diseases of the myocardium with familial patterns in some families that were studied [6]. The cases of pathological asymmetric hypertrophy were first described as an obstructive cardiomyopathy by Teare in 1958 [7], and familial cases of this condition with notable arrhythmia and sudden death have subsequently been published [8]. Later, three principal clinical presentations have been used to classify cardiomyopathies: (1) congestive heart failure and cardiac enlargement with dilated and flabby myocardium, (2) cardiac constriction with stiff inelastic myocardium and associated endocardial and pericardial involvement named as constrictive cardiomyopathy, and (3) cardiac obstruction with specific asymmetrical hypertrophy of the septum (obstructive cardiomyopathy) [9]. Goodwin and Oakley distinguished the obliterative (constrictive) subtype of cardiomyopathy as a rare heart muscle disease specific to amyloid and leukemic infiltration of polyarthritis nodosa [10]. As a clinically recognizable disorder, the term “primary RCM” was introduced in 1977 by Chew et al. [11]. The authors described 11 RCM patients with endomyocardial fibrosis, who shared a common diastolic abnormality with restriction of ventricular filling and stroke output as a main feature that was not seen in other primary cardiomyopathies nor in amyloid heart disease. Along with dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM), RCM was first classified as a cardiomyopathy in 1980 by the World Health Organization (WHO) and the International Society and Federation of Cardiology (ISFC) [12]. This was the first classification of cardiac heart muscle diseases of unknown cause based on altered hemodynamics (restrictive ventricular filling) rather than pathomorphological characteristics. Later in 1996, the WHO and the Federation of Cardiology Task Force added arrhythmogenic right ventricular cardiomyopathy (ARVC) to the list of cardiomyopathies [13]. With the advent of molecular genetics and improvements in diagnostic modalities, the perspective on cardiomyopathies changed to incorporate causative genetic mutations and modifier genes to explain pathologic mechanisms. Multiple genetic mutations have been found in patients with RCM and occur primarily in genes encoding sarcomeric and cytoskeletal proteins, which are influenced by mechanosensing and mechanotransducing proteins [14]. In the time since 1996, the American Heart Association (AHA) in 2006 and the European Society of Cardiology (ESC) in 2008 updated their statements regarding the classification of cardiomyopathies [2, 15].
The difficulty in classifying cardiomyopathies lies with the overlap in morphological and physiological abnormalities, genetic mutations, and the evolving nature of the disease. The AHA consensus panel gives a broad definition of cardiomyopathies and categorizes cardiomyopathies as primary or secondary, depending on whether the disease is restricted to heart tissue or whether cardiac dysfunction results from a systemic disorder. Under the AHA classification system, primary cardiomyopathies are mostly of idiopathic or genetic origin, whereas secondary cardiomyopathies have an underlying cause. Secondary cardiomyopathies include amyloidosis, sarcoidosis, storage disorders, toxicity from drugs or chemical agents, endomyocardial fibrosis, endocrine disorders, and more. In this classification scheme, the last of the cardiomyopathies to be identified in 2006 was left ventricular noncompaction (LVNC). Under the AHA definition, RCM is based on hemodynamic dysfunction of restrictive physiology and categorized as a primary restrictive nonhypertrophied cardiomyopathy of genetic or nongenetic etiologies, whereas HCM and DCM depend on morphological abnormalities [15].
On the other hand, the ESC definition is based on both morphological and functional abnormalities that are important for diagnosis and not caused by myocardial ischemia from coronary artery disease, hypertension, valvular disease, or congenital heart disease, with no focus on the primary or secondary cause. Each classified cardiomyopathy is divided into subsets of familial or genetic cardiomyopathies and non-familial or non-genetic cardiomyopathies. Cardiomyopathies are considered familial if more than one family member presents with the same pathological phenotype caused by a pathogenic genetic variant. Both familial and non-familial cardiomyopathies are further subdivided into categories, one of which is “disease sub-type”, in which myocardial dysfunction is due to a systemic disorder, similar to the AHA secondary cardiomyopathy classification [2].
Etiologically, RCM can be idiopathic, familial (primarily autosomal dominant inheritance, but also autosomal recessive or X-linked), or secondary to a systemic disorder [16]. Secondary diseases can be characterized as infiltrative or non-infiltrative cardiomyopathies, storage disorders, fibrotic injury, or endomyocardial damage [16]. Infiltrative RCMs include amyloidosis, sarcoidosis, Gaucher’s disease, and Hurler syndrome while non-infiltrative cardiomyopathies include idiopathic RCM and scleroderma. Storage diseases that may present with RCM are Fabry’s disease, glycogen storage disorders, and hemochromatosis. The endocardium can be damaged due to endomyocardial fibrosis, hyper-eosinophilic syndromes, metastatic tumors, radiation, sickle cell disease, or drugs such as anthracyclines and other chemotherapeutic agents, ergotamine, and chloroquine [17]. In adults less than 30 years of age, RCM is most commonly linked to a wide variety of systemic disorders while the associated disorders for adults with RCM over 65 years of age are limited to amyloidosis, hemochromatosis, and radiation-induced endomyocardial damage [17].
This article will describe the epidemiology and clinical presentation of RCM, as well as the genetic basis and pathophysiology of the disease. We will also highlight family studies and animal models that have been crucial in providing insights into the pathological and pathophysiological mechanisms of RCM.
The incidence and prevalence of RCM remains unclear and variable due to differences in geographic region, sex, and ethnicity parameters. Due to its rarity and overlap in HCM, DCM, and LVNC phenotypes, a lack of epidemiological studies in RCM persists. Based on a study conducted between 1982 and 1997, RCM is the least common cardiomyopathy and accounts for approximately 2–5% of all cardiomyopathies in United States [18]. In pediatric cases, RCM accounted for 4.5% of cardiomyopathy cases (152 of 3375 children under 18 years of age) as reported by the Pediatric Cardiomyopathy Registry (PCMR) database, inclusive of 98 institutions in the United States and Canada between 1990 and 2008. Out of 152 pediatric RCM cases, 101 cases were solely RCM diagnoses as opposed to 51 cases which were a mixed RCM and HCM phenotype. Ninety-three percent of the purely RCM cases diagnosed in childhood were idiopathic in nature, 14% of the RCM patients had a family history of cardiomyopathy, 1% of cases were related to inborn errors of metabolism, and no RCM cases were secondary to a neuromuscular disorder. The mean age of pediatric diagnoses was 6.2 years [4]. A 10-year report under the National Australian Childhood Cardiomyopathy reported that 2.5% of pediatric cardiomyopathy cases between 1987 and 1996 were RCM diagnoses. Half of those patients presented with congestive heart failure (CHF). Unlike pediatric cases of DCM or HCM, which mostly present at infancy, the median age of presentation for pediatric RCM in this cohort was 3 years of age [19]. In New Delhi, India, the epidemiology of cardiomyopathy study (EPOCH) at the All India Institute of Medical Sciences reported 30 out of 190 cardiomyopathy cases (1.6%) were RCM and 63% of those RCM patients were male [20]. However, the true incidence and prevalence of RCM is unknown, as echocardiography is not a common assessment in the general population worldwide.
It is less difficult to track epidemiology for disorders underlying secondary RCM, although these systemic disorders do not equate to patients having idiopathic RCM. The incidence of cardiac amyloidosis is 3–9 cases per million people and is characterized by the extracellular deposition of amyloid fibrils in the heart. According to the most recent position statement of the ESC on cardiac amyloidosis, more than 98% of currently diagnosed cardiac amyloidosis results from fibrils composed of monoclonal immunoglobulin light chains (AL) or transthyretin (ATTR), either in its hereditary or acquired form [21]. Around 60–80% of patients with AL amyloidosis have cardiac involvement [22, 23]. AL amyloidosis is more common in male patients and at ages 55 to 60 years and death in 75% of the 393 patients was attributed to cardiac amyloidosis, including sudden death in 25% [24]. In contrast, ATTR amyloidosis is more common in patients older than 80 years [3, 25] and is prevalent among patients with heart failure, especially HFpEF [26]. However, the true prevalence of RCM in ATTR remains largely unknown [27].
Sarcoidosis, a multi-organ disease associated with secondary RCM, is characterized by inflammation and noncaseating granulomas [28]. Incidence and prevalence of sarcoidosis is high in Sweden, Norway, Denmark, Japan, and the United States [29, 30]. Scandinavian countries report an incidence of 50 to 60 cases per 100,000 per year [31]. The incidence rate of sarcoidosis was highest in black women in the United States: 46 per 100,000 per year according to the Nurses’ Health Study II and 17.8 per 100,000 per year based on the Optum health insurance medical claims database [32, 33]. Although clinical signs of cardiac involvement are present in 5% of patients with sarcoidosis, autopsy studies have demonstrated the presence of cardiac granuloma and granulomatous inflammation in up to 25% of autopsy specimens [34].
Storage diseases such as hemochromatosis (iron storage disorder), Fabry disease (lysosomal storage disorder) and Danon and Pompe diseases (glycogen storage disorders) are genetic disorders associated with diastolic dysfunction and RCM or HCM [35]. It is difficult to accurately estimate the incidence and prevalence of RCM associated with those genetic disorders [23].
Overall, the clinical presentation of RCM is nonspecific. Initially, patients are asymptomatic and do not seek medical attention unless they have a family history of cardiomyopathy or symptoms of fatigue, dyspnea, or exercise intolerance [36]. In both pediatric and adult patients with RCM, dyspnea is the most common symptom [37]. Pediatric patients with RCM usually present with fatigue, syncope, hepatomegaly, cardiomegaly on radiograph, and failure to thrive. Initial presentation of RCM in pediatric patients are typically pulmonary related, with frequent lung infections and reactive airway disease [38]. Arrhythmias or atrioventricular block increase risk for sudden cardiac death, and syncope may anticipate ischemic complications, arrhythmias, thromboembolism, and sudden death. Rivenes et al. [39] reported on 18 childhood RCM cases and identified that presentation with syncope and/or signs of ischemia had high sudden death rates of 28% with an annual mortality rate of 7% in the cohorts studied. Routine screening for ischemia in all pediatric patients with RCM should include a baseline ECG and Holter monitoring with digital ST-segment analysis as screening tools (Fig. 1, Ref. [39]).
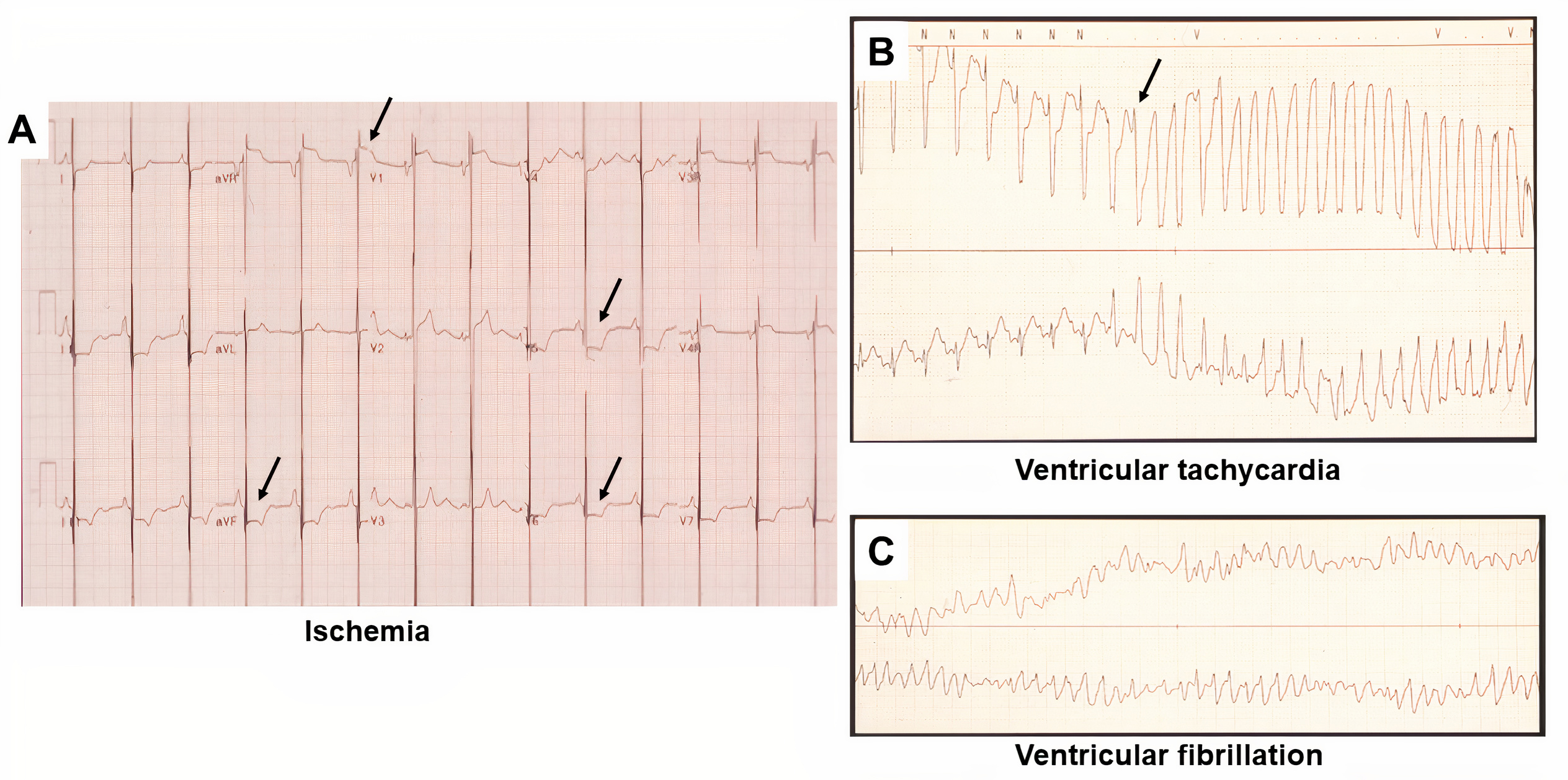 Fig. 1.
Fig. 1.ECG and Holter monitor tracing in pediatric patient with RCM and chest pain who sustained an acute myocardial infarction. (A) ECG tracing, demonstrating bi-atrial enlargement and ST-segment depression in inferior lateral and lateral precordial leads (arrows, evidence of ischemia). (B) Sinus tachycardia and ST-segment depression precede degeneration to torsade de pointes. Arrow indicates inciting “R-on-T” phenomenon. (C) Tracing obtained several seconds later, depicting classic torsade de pointes pattern [39].
Adult patients more commonly present with fatigue, dyspnea, edema, and limited exercise tolerance [16, 40, 41]. In a study cohort (1979–1996) of 94 patients with idiopathic RCM, the most common symptoms were dyspnea (71%), history of edema (46%), palpitations (33%), fatigue (32%), orthopnea (22%), and chest pain (22%). Typical physical findings were jugular venous distension (52%), systolic murmur (49%), S3 gallop (27%), and pulmonary rales (18%). Fifty-three percent of patients were categorized as NYHA class II [42]. In advanced stages of the disease, patients may develop thromboembolism, arrhythmias, heart failure, and cardiac cirrhosis [25]. RCM with HFpEF most commonly presents with heart failure symptoms such as distended jugular veins, pulsus paradoxus, pleural effusion, bilateral edema, respiratory rales, hepatomegaly, ascites, and an S3 and S4 gallop [40].
Conduction defects can be explained by the restrictive physiology of RCM, which is characterized by increased ventricular pressure caused by myocardial stiffness and impaired ventricular filling. In contrast to restrictive filling seen in other pathologies, diastolic dysfunction with abnormal relaxation in RCM is accompanied by near normal diastolic and systolic volumes, contractile function, and wall thickness [2]. Wall thickness is increased in infiltrative diseases and additional phenotypes such as HCM. In advanced stages of RCM, systolic function decreases [4]. The abnormal ventricular relaxation leads to a buildup of blood from impaired filling and atrial enlargement (Fig. 2). Bi-atrial enlargement and left ventricular stiffness may cause arrhythmias and thromboembolism from stasis of flow and resistance in pulmonary venous flow from increased atrial pressures and symptoms of heart failure. Mitral and tricuspid regurgitation can develop and consequently exacerbate atrial enlargement [17]. Fibrosis, myocardial stiffness, and the resultant restrictive remodeling are responsible for poor prognosis, but the basis of this remodeling remains to be fully understood.
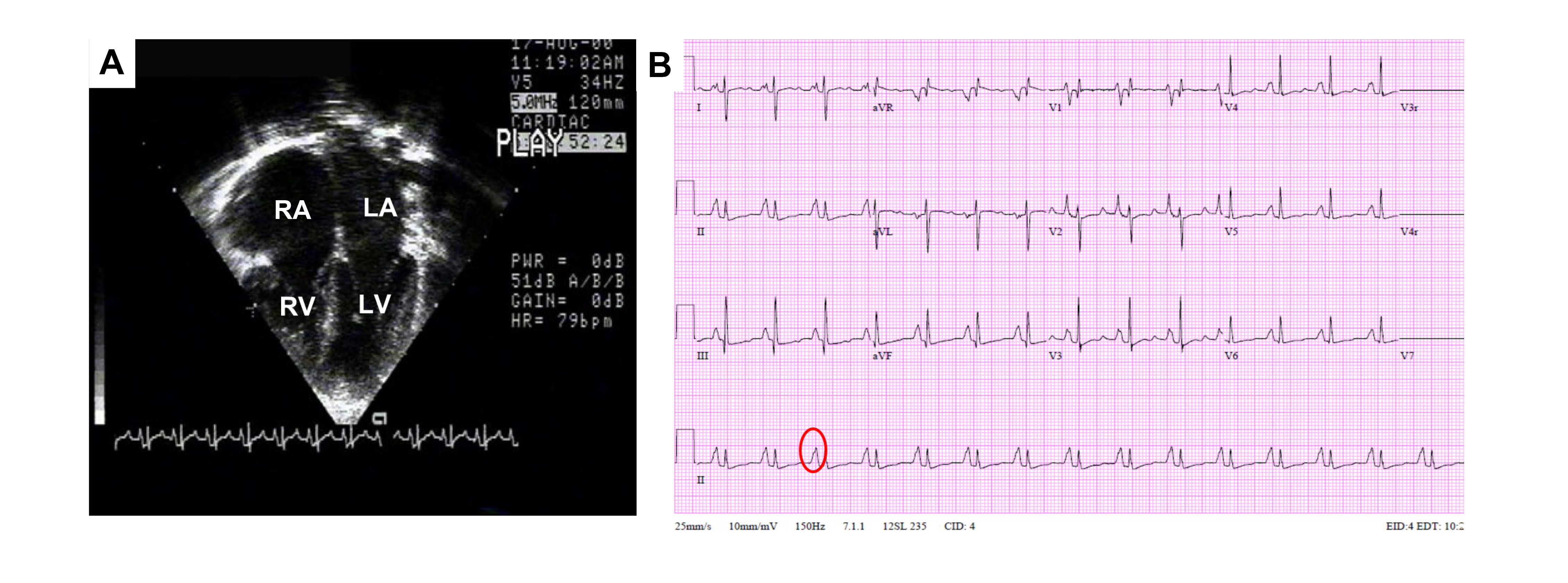 Fig. 2.
Fig. 2.Echocardiographic and ECG features of RCM. (A) Image of apical two-dimensional echocardiography in 4-chamber view. Severe right (RA) and left (LA) atrial enlargement relative to normal biventricular (RV and LV) size and thickness is captured. (B) ECG tracing, demonstrating bi-atrial enlargement (oval) and nonspecific ST-T wave changes.
Elevated ventricular filling pressures cause left and/or right atrial dilatation and symptoms of heart failure as discussed earlier. RCM must be differentiated from constrictive pericarditis (CP), HCM, and hypertensive heart disease. The differential diagnosis is established by physical examination, clinical laboratory testing, electrocardiogram, echocardiography, and cardiac magnetic resonance (CMR) with late gadolinium enhancement (LGE) [17, 40]. Myocardial stiffness eventually increases systemic filling pressures and decreases stroke volume, which in turn may affect kidney function [17]. To evaluate for heart failure and the degree of which other organs are affected, clinical labs can be sent for hematocrit, comprehensive metabolic panel, creatinine, blood urea nitrogen, urine protein, liver function panel, brain natriuretic peptide (BNP) or N-terminal pro-BNP (Nt-proBNP), and troponin T or I [40]. BNP/Nt-proBNP is markedly elevated in patients with RCM in comparison to patients with CP [43]. In primary systemic amyloidosis, measurements of Nt-proBNP can be used as a prognostic marker for advanced disease and survival rates [44]. Electrocardiogram (ECG) is often employed in the initial assessment of cardiac disorders and a 24-hour Holter monitor test should be obtained in patients with suspected cardiac sarcoidosis to document conduction and rhythm disturbances that may be missed on ECG [23]. Table 1 outlines more specific laboratory testing for systemic diseases associated with secondary RCM [40, 45].
| Disorder | Clinical laboratory test |
| Amyloidosis | 24-hour urine monoclonal protein, serum and urine immunofixation with electrophoresis, serum free light chain assay |
| Cardiac sarcoidosis | Angiotensin converting enzyme (ACE) |
| Hemochromatosis | Serum iron, total iron-binding capacity (TIBC), ferritin, liver panel, hemochromatosis gene (HFE) testing |
| Hyper-eosinophilia | Complete blood count (CBC) and differential, peripheral blood smear, anti-neutrophil cytoplasmic antibody (ANCA) |
Approximately 99% of ECG readings in RCM patients are abnormal, although only 15–30% of patients present with arrhythmias. Abnormalities are highly variable but must commonly are biphasic P wave in the precordial leads indicative of bi-atrial enlargement, elevated ST segments, Q-wave abnormalities, and notched or biphasic late peaking T waves [37, 46]. Low voltage QRS complexes indicates increased ventricular wall thickness and is a characteristic of AL amyloidosis as well as other infiltrative diseases [25]. Abnormalities in the ST segment and atrioventricular conduction and supraventricular and ventricular arrhythmias may be found in patients with RCM [40, 47]. In approximately 50–70% of patients with amyloidosis, pseudo-infarct patterns are present [48].
According to the 2016 American Society of Echocardiography (ASE) and European
Association of Cardiovascular Imaging (EACVI) guidelines, advanced myocardial
involvement with grade 3 diastolic dysfunction, associated with poor prognosis,
is detected by transmitral spectral Doppler echocardiography (Fig. 3). The
restrictive filling pattern is identified by parameters of early diastolic
velocity (increased E wave), low or absent late filling velocity (decreased A
wave), mitral inflow E/A ratio
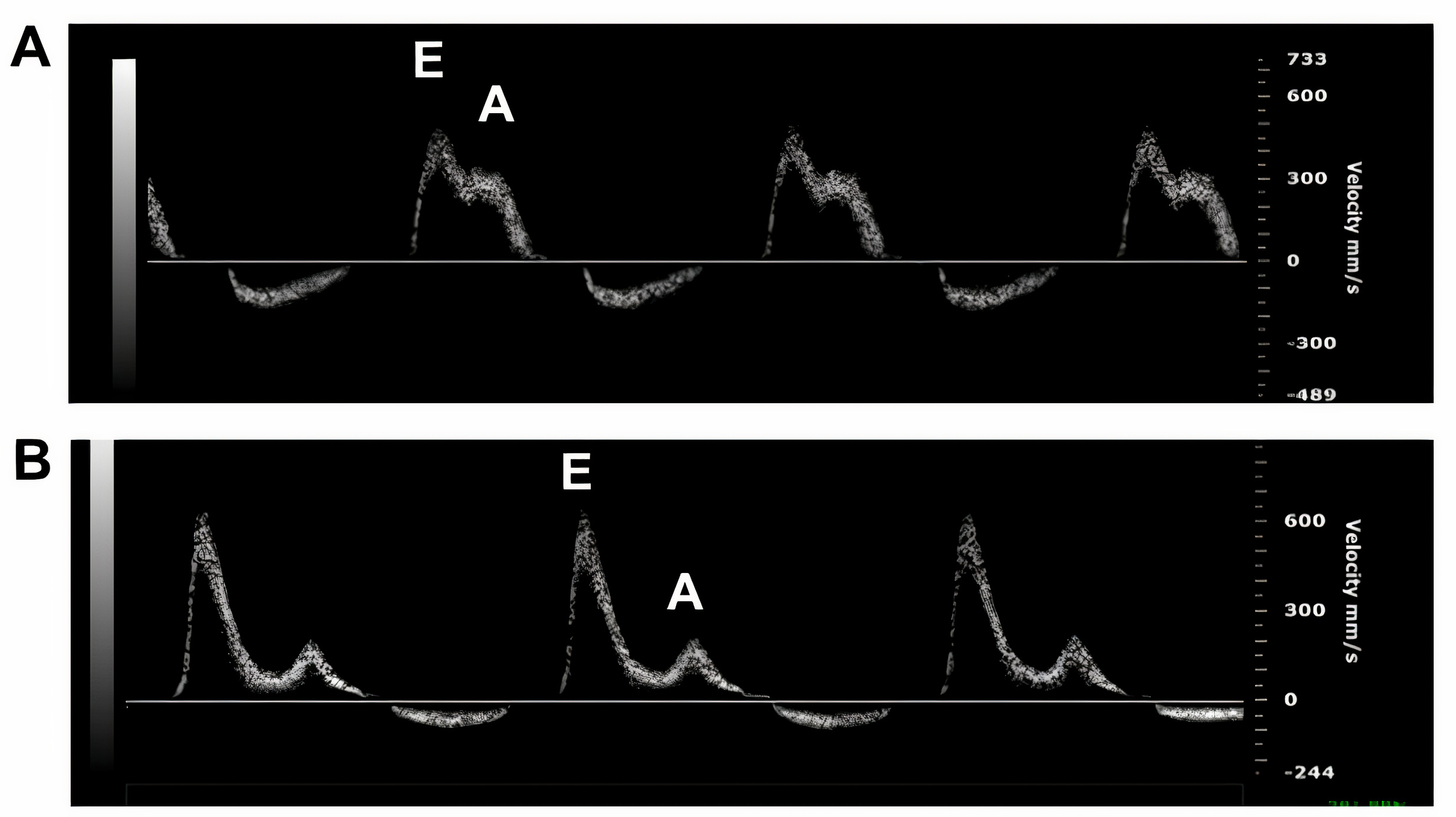 Fig. 3.
Fig. 3.
M-mode images of mitral valve movement by transthoracic
2-dimensional Doppler echocardiography showing normal diastolic function with E
Diastolic dysfunction increases atrial filling pressures, which consequently impact pulmonary venous flow with a decreased systolic to diastolic pulmonary venous flow ratio [17]. Heart failure with HFpEF is characteristic of RCM, although tissue Doppler can reveal reduced systolic waves, blunted early diastolic waves, and preserved or blunted late diastolic waves [50]. Speckle tracking echocardiography (STE), which can detect myocardial deformities, demonstrates a ratio of LV free wall strain to LV septal strain of 1, reduced longitudinal strain, and normal circumferential strain [49, 51]. Typically, chamber volumes will remain normal and there will be bi-atrial enlargement. Infiltrative types of RCM will have increased LV wall thickness. Pulmonary artery pressure may be increased in very advanced stages of disease [40].
Both RCM and CP have several similarities in echocardiography and clinical symptoms: normal to near normal ventricle size, preserved ejection fraction, dilated atria, increased filling pressure, jugular venous distension, Kussmaul sign, and diastolic sounds [25, 40]. Differentiating between RCM and CP using echocardiography coupled with the advances in cardiac CT and CMR is crucial since CP can be resolved with NSAIDs or pericardiectomy after medical therapy failure [50]. Echocardiography of a patient with RCM will display wall and valvular thickening and “sparkling” myocardium. CP may reveal pericardial thickening and a respiratory ventricular septal shift. While RCM has decreased variation in mitral and tricuspid inflow E velocity, CP has increased variation. In RCM, there may be mitral or tricuspid regurgitation as a consequence of atrial enlargement and restrictive filling [40].
CMR is beneficial in establishing a differential diagnosis among RCM, CP, and infiltrative disorders such as amyloidosis, sarcoidosis, and hemochromatosis. Imaging allows for tissue categorization, including inflammation, and provides high spatial resolution and noninvasive measurement of ventricular volumes. CMR with late gadolinium enhancement (LGE) can assist with diagnosing subtypes of RCM, particularly, amyloidosis and measuring pericardial thickness (Fig. 4). Although not applicable to the RCM diagnosis, CT scans and catheterization with coronary angiography are helpful for eliminating constrictive pericarditis and obstructive coronary artery disease, respectively, from the differential diagnosis. For RCM, chest radiographs show bi-atrial enlargement and in some cases, pulmonary congestion [52].
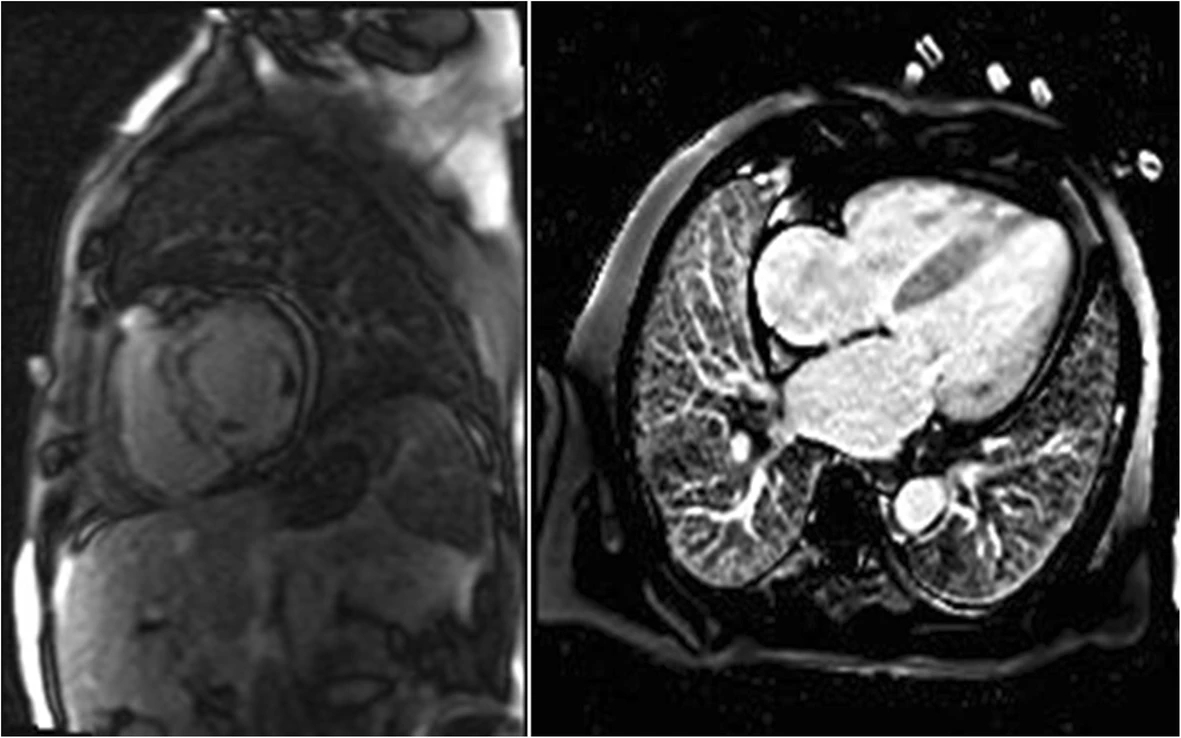 Fig. 4.
Fig. 4.Examples of cardiac magnetic resonance (CMR) in differential diagnosis between restrictive cardiomyopathy (RCM) and amyloidosis. Adopted from Kyriakou et al. 2018 [23]. Cardiac magnetic resonance (CMR) images of patients with cardiac amyloidosis. Amyloid fibril deposition pattern mainly affects subendocardial CMR imaging, leading to a shortened T1 relaxation time and a diffuse LGE of the left ventricular endocardium.
If noninvasive procedures are not decisive, endomyocardial biopsy (EMB) provides histological samples which may be helpful for infiltrative diseases like sarcoidosis [40]. EMB and a positive fat pad aspiration is indicative of cardiac amyloidosis [25]. Cardiac catheterization allows for hemodynamic measurements of filling pressures and pulmonary hypertension. This assessment is helpful in differentiating RCM diagnosis from CP [36]. Right and left ventricular pressure tracing with a dip and plateau pattern (square root sign) and elevated LV and RV diastolic pressures are characteristic of restrictive hemodynamics [16]. A comprehensively reviewed diagnostic algorithm of secondary RCM by Muchtar et al. [3] is illustrated in Fig. 5.
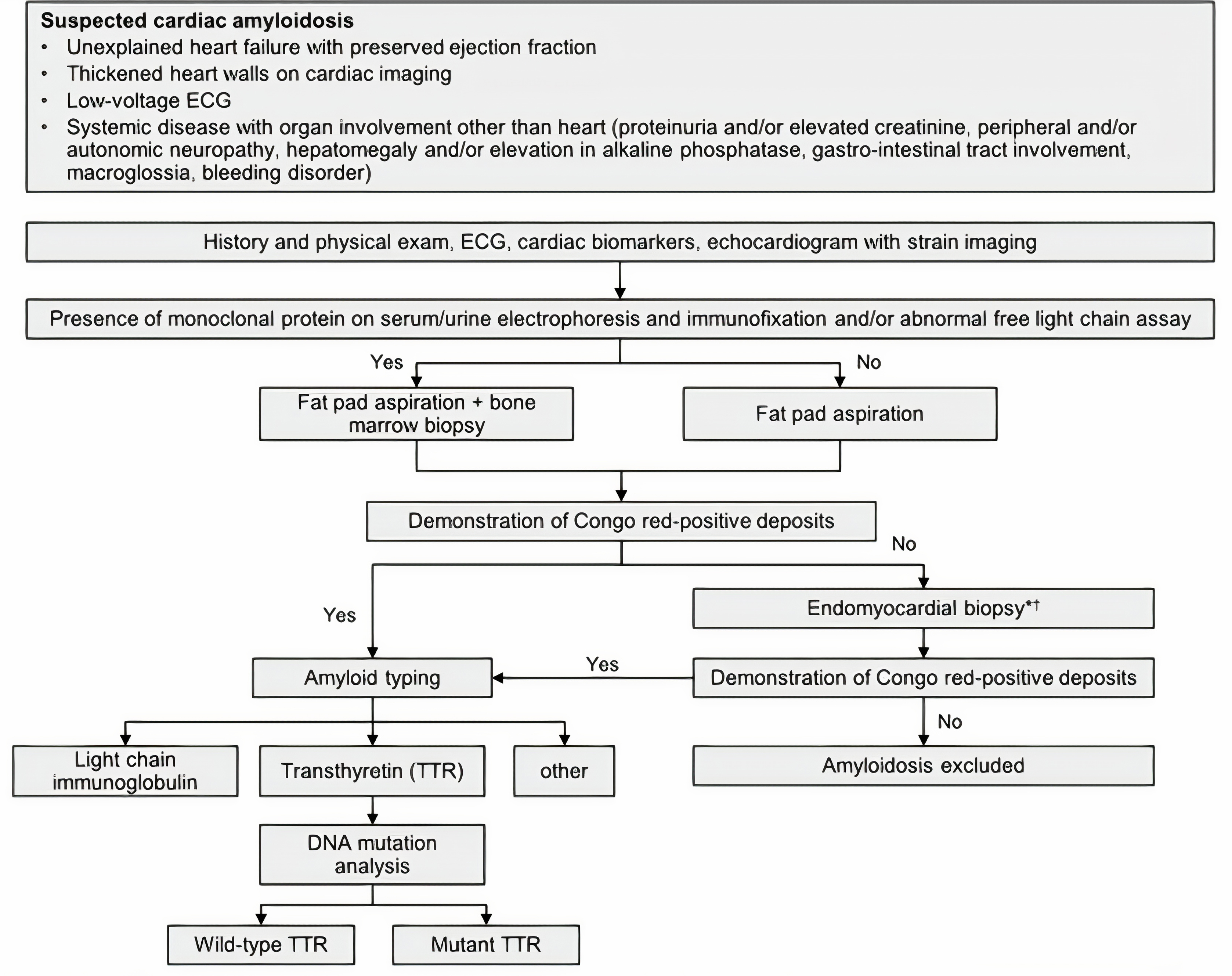 Fig. 5.
Fig. 5.Diagnostic algorithm for suspected cardiac amyloidosis. Adopted from Muchtar et al. 2012 [3]. *Alternatively other source tissue other than heart or endomyocardial biopsy (EMB) can be obtained. ᵻThe use of bone scintigraphy can replace EMB.
Genetic testing and counseling are available for patients and their relatives to determine risk of inheritance. Especially, the likelihood and screening of de novo variation(s) should be integrated into genetic counseling of pediatric cardiomyopathy cases [53]. Hereditary forms of cardiac amyloidosis commonly have an adult onset, thus genetic testing of minors is discouraged, although genetic testing is recommended for adult relatives along with genetic counseling [21]. While genetic counseling is more common for patients with HCM, genetic testing should be recommended to families of young patients with a family history of sudden cardiac death, heart failure, and implanted cardioverter defibrillators. In a multinational European study, 43% of patients with RCM were found to have one disease-causing variant [54].
There is no cure for RCM, and patients suffer from poor outcomes. Treatment should center on alleviating heart failure symptoms and treating underlying causes of RCM [23]. Without heart transplantation, survival is limited. Diuretics, with careful monitoring, can relieve volume overload and pulmonary edema or congestion [55]. Since diuretics may worsen cardiac output, patients should restrict dietary sodium [17]. Beta-blockers and calcium channel blockers may be successful in increasing filling time or treating arrhythmias, although should be administered with care since rate-slowing medication may be deleterious. Renin-angiotensin system inhibitors are also not well-tolerated; however, if heart failure advances to systolic dysfunction, angiotensin-receptor blockers and angiotensin II receptor blockers may be employed [3, 25]. Patients with enlarged atria are at increased risk of forming blood clots, and antithrombotic therapy may be administered to patients with high risk for thromboembolisms. Atrial fibrillation (AF) is a common clinical sign in cardiomyopathies, and according to a study by Mizia-Stec et al. [56], prevalence of AF is highest in RCM (52%) compared to rates in DCM, HCM, and ARVC. Stroke incidence is higher in patients with AF versus patients presenting without AF, and if not contraindicated, anticoagulants should be recommended to patients with RCM to reduce risk of thromboembolic events.
Treating the associated systemic diseases and underlying conditions of RCM is important and in need. For example, patients with both PGM1 deficiency, a congenital glycosylation disorder, and RCM were treated with oral galactose treatment. Oral galactose did not benefit heart failure symptoms but improved the myopathy and exercise tolerance [57]. Amyloidosis, one of the infiltrative disorders linked to RCM, has had recent advances in treatment strategies. Genetic and molecular mechanism-oriented therapies are emerging for treatment of amyloidosis such as orally available transthyretin stabilizers, monoclonal antibodies and gene-editing strategies that have been effective in preclinical studies and successfully advanced in human clinical trials [58]. Approved for polyneuropathy in hereditary ATTR, patisiran, vutrisiran, and inotersen are short interfering RNA (siRNA) used to silence mutant and wild type TTR genes in order to halt neuropathy impairment and improve quality of life [59, 60, 61]. In 2019, the U.S. Food and Drug Administration (FDA) approved tafamidis, a selective transthyretin stabilizer, for the treatment of transthyretin amyloid cardiomyopathy (ATTR-CM). Tafamidis is not curative but has been shown to slow the progression of disease with reduced mortality and hospitalization [62, 63]. This is particularly true in less severe cases of ATTR-CM and early diagnosis [64]. Gene-editing approaches employ innovative technologies such as transthyretin-encoding gene silencing by RNA interference (RNAi) or antisense oligonucleotides and clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 techniques [61, 65, 66].
Despite clinical management of heart failure symptoms and treatment of secondary RCM, progression to severe disease requires cardiac transplantation. Left ventricular assist device (LVAD) implantation is not commonly used in patients with RCM compared with DCM or ischemic cardiomyopathy patients [67]. A systematic review of six retrospective studies reported a higher risk of operative and short-term mortality in RCM patients with refractory heart failure undergoing LVAD implantation [68]. Prolonged LVAD implantation was pursued in two pediatric patients with pulmonary hypertension and was found to reduce pulmonary artery pressure and vascular resistance [69]. Durable mechanical circulatory support with biventricular assist device or total artificial heart has been considered for patients with amyloidosis who developed advanced heart failure [70]. Outcomes of adult RCM patients after heart transplantation have been comparable to those of non-RCM patients. However, increased mortality was observed in secondary RCM subgroup with amyloidosis and radiation/chemotherapy [71].
Pediatric patients have a waitlist mortality of around 10% at 1-year. Risk of death while on the waiting list increases with younger age, dependence on ventilator or multiple assist devices, or inotropic support. Survival rates for RCM patients 10 years after heart transplant is 63% and is comparable to non-cardiomyopathy patients and patients with other cardiomyopathies [72, 73]. Based on the PCMR database with 3375 cases of RCM between 1990–2008, 1-, 2-, and 5-year survival for RCM was 82%, 80%, and 68%, respectively [4]. Pediatric cases may result in sudden cardiac death (SCD) or complications from RCM, most commonly heart failure, arrhythmia, and ischemia-related complications. The poor clinical outcomes were due to ventricular arrhythmias, abnormal blood pressure in response to exercise, progressive elevation of pulmonary vascular resistance, and unexplained syncope [39, 74, 75]. Heart failure, lower fractional shortening z-score, and higher posterior wall thickness z-score at diagnosis are risk factors for survival without transplantation [4]. Clinical outcomes and mortality from RCM are worse in comparison to those in HCM and DCM, and survival is limited without cardiac transplantation [39, 76].
Molecular genetics has expanded the emerging potential to predict the progression of RCM and treatment strategies to prevent early mortality for inherited RCM [77]. Targeted therapies may be helpful in reversing pathogenic phenotypes or halting cardiac remodeling. For example, allele-specific silencing has been investigated in mice for the N47K mutation in the myosin light chain-2 (MYL2) gene. Injecting mice with RNAi showed the potential for a targeted therapy in RCM that partly regains normal function in diseased cells [78]. While CRISPR-Cas9 gene editing has not been studied in RCM, but it presents a potential additional therapeutic strategy and a technique to repair mutations or change gene expression in human induced pluripotent stem cell (hiPSC) [79, 80]. In 2017, Ma et al. [81] repaired a heterozygous dominant mutation of MYBC3, a mutation linked to familial HCM, in a human preimplantation embryo. MYBC3 mutations have also been seen in RCM patients; CRISPR-Cas9 gene editing studies are needed to determine its efficacy in RCM.
Due to the broad group of mechanisms of pathogenesis in RCM, such as genetic, non-genetic, or secondary to a systemic disorder, histological changes in RCM are heterogeneous. On this basis, Sen-Chowdhry et al. [82] suggested RCM as a myocardial disorder in which restrictive ventricular physiology develops an early and dominant feature in the disease course. EMB has a high diagnostic yield in infiltrative disorders. Amyloidosis, sarcoidosis, hereditary hemochromatosis, and Fabry disease are identified by Congo red stain, granulomatosis reaction, iron, and ceramide trihexoside, respectively [25]. Additional techniques such as immunohistochemistry, Western blotting, mass spectrometry, amino acid sequencing, and electron microscopy are useful for amyloid fibril protein identification for an early diagnosis to prevent rapid disease deterioration [83, 84].
Autopsy studies of idiopathic RCM patients reveal ischemic damage preceding SCD [16]. In a cohort of seven patients diagnosed with RCM between 1974 and 1993, EMB revealed interstitial fibrosis [85]. A study by Kawano et al. [86] describes electron microscopy findings of a 65-year-old Japanese woman with a myosin heavy chain 7 (MYH7) causative mutation for RCM. Results from EMB of the left ventricle revealed focal myocyte necrosis, myofibrillar disarrangement, and scattered electron-dense material, some associated with Z-bands. Myofibrils without Z-bands were also identified, and tubular structures and pseudo-inclusion bodies were imaged in the nucleus [86]. Histological samples from a patient with a Q529X mutation in myopalladin (MYPN) revealed disorganized cardiomyocytes with bizarrely shaped nuclei, myofibrillar disarrangement, and hypertrophy. Side-branching and side-to-side junctions were observed in cardiomyocytes [87]. In biopsies from RCM patients with mutations in sarcomeric proteins, electron-dense material and Z-band disruption are the most common histopathological finding [86].
Approximately 30% of RCM cases have a family history of RCM considered as a primary disease of genetic origin, but only a few genes, such as cardiac troponin T (cTnT) and troponin I (cTnI), myosin-binding protein C (MYBPC), MYH7, myosin light chain-2 and 3 (MYL2, MYL3), desmin (DES), MYPN, titin (TTN), Bcl2-associated athanogene 3 (BAG3), discoidin, CUB and LCCL domain containing 2 (DCBLD2), lamin A/C (LMNA), and filamin C (FLNC) have been associated with familial RCM [87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105] (Table 2, Ref. [87, 89, 97, 98, 102, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126]).
| Gene | Protein | Human mutation | Animal mutation models | Pathogenesis |
| BAG3 | Bcl2-associated athanogene 3 | P209L | P209L overexpression [102] | Autophagy, Z-disc disruption, protein aggregates, and increased cardiac fibroblasts. |
| DCBLD2 | Discodin, CUB, LCCL domain-containing protein 2 | W27* | Dcbld2 KO [106] | Impaired normal vascular development via altered VEGF signaling. |
| DES | Desmin | R406W | R405W | Desmin aggregates and disruption of intercalated discs. |
| KI [107] | ||||
| FLNC | Filamin C | S1624L | A1186V | Myotube defects, decreased sarcomere localization, protein aggregates, Z-disc disruption, autophagy. |
| I2160F [97] | A1183L | |||
| V2297M [108] | zebrafish overexpression | |||
| A1186V | ||||
| A1183L [109] | ||||
| P2298L [110] | ||||
| T2563C | ||||
| T2710X [110] | ||||
| LMNA | Lamin A/C | 835delG [104] | N/A | Not known. Likely protein truncation or loss of protein via nonsense‐mediated mRNA decay. |
| MYBPC-3 | Myosin-binding protein C | Ala1105Glyfs*85 [105] | N/A | Not known. Likely protein truncation or loss of protein via nonsense‐mediated mRNA decay. |
| MYH7 | β-myosin heavy chain | G768R [111] | N/A | Not known. |
| P838L [112] | ||||
| MYL2 | Myosin light chain 2 | N47K [113] | N47K Tg | Silencing of the 47K allele reduced cardiac fibrosis, LV mass, hypertrophic biomarkers, and increased survival. |
| N47K AAV9-RNAi [78] | ||||
| MYL3 | Myosin light chain 3 | E143K [114] | E143K [115] | Hypercontractility, increased binding affinity to actin, and slow dissociation of acto-myosin complex. |
| MYPN | Myopalladin | Q529X [87] | Q526X KI [116] | Reduced levels of CARP, Mek1/2, Erk1/2, Smad2 and Akt. |
| TNNI3 | Cardiac troponin I | L144Q [117] | Increased Ca | |
| R145W | R145W [118, 119] | |||
| R170W | ||||
| A171T | ||||
| K178E [120] | K178E [121, 120, 122] | |||
| D190G | ||||
| D190H | ||||
| R192H | R193H [123] | |||
| R192C [124] | ||||
| TNNT2 | Troponin T type 2 | E136K [89] | Drosophila melanogaster E87K | Increased resting tension and reduced sarcomere length. |
| I79N [125] | Rat E138K | |||
| I79N Tg [126] | ||||
| TTN | Titin | Y7621C [98] | N/A | Myocyte degeneration, Z-disc disruption, and fibrosis. |
| Comment: All mutations are studied in mouse models unless indicated otherwise. | ||||
The genetic mutations associated with inherited RCM are primarily in structural proteins of the sarcomere or proteins associated with the sarcomere [77]. The sarcomere is the contractile unit of skeletal and cardiac muscle with thin, thick, and elastic filaments anchored by the M-line and Z-disc [127]. The thick filaments are made up of myosin and myosin-binding protein C, a regulatory protein at the C region cross-bridging zone, and thin filaments are comprised of actin and regulatory proteins troponin and tropomyosin. At low calcium concentrations, troponin and tropomyosin bind and inhibit the myosin ATPase and force generation. Calcium release from the sarcoplasmic reticulum causes the sarcomere to contract from thick and thin filaments sliding past one another [128]. Contractile function relies on a delicate balance between the filaments and troponin complexes to control the state of the sarcomere involved in homeostasis of cardiac function [129]. Most of the RCM cases with genetic variants involved directly or indirectly in sarcomere function have been reported to cause childhood RCM or cases that rapidly deteriorate due to conduction defects and cardiac restrictive dysfunction necessitating heart transplantation [97, 98, 103, 104, 124, 130].
Several mutations found from family studies have been used in animal studies of RCM as shown in Table 2. Animal models with analogous human mutations in myosin, cTnI, MYPN, and DES yield a RCM phenotype of diastolic dysfunction [131]. Cells from patients with cardiomyopathies can be taken to develop hiPSC lines for cell studies on cardiomyocytes to test therapeutics and detect cardiotoxicity [79]. Using CRISPR-Cas9 gene editing, researchers can create isogenic variants of hiPSC lines to identify cardiotoxic mutations and investigate a pathogenic mutation’s effect on physiology, contractility, and morphology [132]. In addition, single short guide RNAs through CRISPR-Cas9 editing has been shown to disrupt genes Myh6, Sav1, and Tbx20 in postnatal mice [133]. With the hope of developing a human RCM therapy, the adeno-associated virus (AAV) - RNAi approach showed the feasibility, efficacy, and safety of preclinical therapeutics in MYL2 N47K mutation transgenic mice [78].
Some of the examples of clinical or histopathological changes in RCM patients
with genetic mutations listed in Table 2 may reveal features of HCM,
suggesting that both diseases may share similar pathogenic and genetic mechanisms
[134]. While HCM and RCM are phenotypically different, both are considered
disorders of diastolic dysfunction. Vio et al. [35] suggested that
familial RCM cases may represent a part of the phenotypic spectrum of HCM rather
than a different genetic cardiomyopathy. Differential diagnosis between primary
RCM and HCM can be difficult when ventricular hypertrophy is mild. Kubo
et al. [135] distinguished the “restrictive phenotype” as a severe
diastolic dysfunction with restrictive filling that is in the clinical spectrum
of HCM with no or minimal left ventricular hypertrophy of
Beyond genetic mutations of genes expressed in cardiac myocytes, cardiac fibroblasts and the extracellular matrix (ECM) may be involved with the pathogenesis of RCM [138]. Natural feline RCM is characterized by severe and extensive fibrous thickening of the endocardium and chamber deformity [139]. Despite these animal models and cell lines elucidating the pathogenesis and pathophysiology as well genetic therapies, primary RCM remains one of the least studied and understood cardiomyopathies. The following sections will outline the genetic variants linked to RCM and the role of paracrine factors secreted by cardiac fibroblasts.
Troponin I (cTnI) inhibits sarcomere contraction by binding to the Ca
Transgenic R145W mice have also been studied as a model for RCM. Papillary
fibers in mutant mice demonstrate increased Ca
Certain amino acid substitutions can have varying effects depending on the charge differences. Removing or substituting charged residues on tropomyosin-binding sites impairs normal function of cTnT and relaxation [147]. Equivalent E136K mutations were studied in Drosophila melanogaster (E87K) and in rats (E138K). Mutant E87K Drosophila demonstrate increased resting tension and slight reduction in sarcomere length. The rat E138K mutation showed similar results with increased resting tension, reduced sarcomere lengths, greater contractility, and prolonged relaxation periods [147].
Myosin is a protein complex comprised of two heavy chains -
Allele-specific silencing, using AAV-RNAi (AAV9-M7.8L), was found to reduce mutant allele expression, hypertrophic biomarkers, and intramyocardial fibrosis in mice with the human RCM-analogous mutation N47K. In single cell studies, cardiac myocytes treated with RNAi were shown to partially restore normal contraction, relaxation, and calcium reuptake [78]. Although this therapeutic strategy has only been conducted in animal studies, RNAi therapies may be an effective strategy to reverse pathogenic phenotypes.
Myosin heavy chain, along with myosin binding protein C, are the genes most affected by HCM. The RCM mutation G768R in exon 21 in the MYH7 was described in a 15-month-old infant presenting with a heart murmur, gallop, and diastolic dysfunction [95].
Titin, a giant filament protein, is frequently mutated in cardiomyopathies and
phosphorylation of titin affects passive tension, Ca
Filamins A, B, and C cross-link actin rods at the Z-disc of sarcomeres, and FLNC is primarily expressed in cardiac and skeletal muscle, which explains the pleiotropic effects of its genetic mutations in cardiomyopathies and myopathies [151, 152]. Mutations S1624L, V2297M, and I2160F have been associated with RCM, and it is hypothesized that loss-of-function of the gene contributes to pathogenesis [108]. Histology of heart tissue of patients with S1624L and I2160F revealed eosinophilic cytoplasmic inclusions and filament accumulation, suggestive of protein aggregation [97]. The mutation V2297M was identified as the pathogenic mutation in a pedigree with phenotypes of RCM, atrial fibrillation, ventricular tachycardia, and congenital heart defects. Histology of the proband’s heart revealed interstitial fibrosis, myocyte hypertrophy, and mild atrial amyloid deposition, and the pathology report of the proband’s sister, also diagnosed with an RCM phenotype, identified severe interstitial fibrosis and myocyte hypertrophy. In bi-allelic CRISPR-Cas9 insertions of V2297M in human embryonic stem cells, contractile function was diminished with no changes in FLNC expression [108]. Recently, the P2298L, T2563C and T2710X variants have been reported in pediatric RCM [110].
Mutations in LMNA, an intermediate protein meshwork lining the nucleoplasmic face of the inner nuclear membrane, are mostly associated with DCM, metabolic diseases, conduction disorders, and neuromuscular disorders [153, 154]. The heterozygous 835delG mutation was shown to be associated with RCM in a 53-year-old man with exertional dyspnea who received a cardiac transplant two years after diagnosis. His sister presented with the same mutation; however, she was asymptomatic [155].
Myosin-binding protein C is a myosin-associated protein found in the cross-bridge-bearing zone (C region) of A-bands in striated muscle [156]. Mutations in MYBPC are common in HCM and less common in RCM. The frameshift mutation Ala1105Glyfs*85 was identified as the causative mutation of RCM in a 74-year-old Korean male with exertional dyspnea, palpitations, and pitting edema in the lower extremities. The patient’s two children were diagnosed with HCM, speaking to the pleiotropic nature of this mutation and probable impact of modifier genes and environmental factors [105].
Myopalladin, a 145-kDa protein with five immunoglobulin domains, is encoded by
MYPN gene located on chromosome 10q21.3 [157]. The protein interacts
with cardiac ankyrin repeat protein (CARP/ANKRD1) to regulate Z-disc
stretch-signaling to the nucleus of cardiomyocytes. In family studies,
MYPN heterozygous mutations have been associated with HCM, DCM, and RCM
[87]. Using one such human Q529X mutation, heterozygote knock-in mice and
homozygote mice carried the corresponding murine MYPN mutation (Q526X)
in exon 10 as a model for RCM [158]. At 12 weeks of age, heterozygote
Mypn
Gu et al. [160] applied a systems genetics approach to investigate the regulation of MYPN for discovery its gene network involved in RCM. Gene expression values were measured in the heart of a large family of BXD recombinant inbred (RI) mice derived from C57BL/6J and DBA/2J, and expression quantitative trait locus (eQTL) mapping and gene enrichment analysis results were validated on proteomic levels in Mypn knock-in mice. Two novel modifier genes, Syne1 and Myom1, have been confirmed at the protein level that could be involved in RCM development.
BAG3 is an anti-apoptotic protein that also plays a role in mediating autophagy
and targeting aggregated proteins for degradation [161]. Mutation P209L causes
severe pediatric RCM, muscular dystrophy, and respiratory distress. The mutation
is located at the heat shock protein (HSP) 8 binding site; however, it does not
affect binding of HSP8 - instead, it promotes aggregation [102]. Interestingly,
murine models have yielded various phenotypes depending on the study. The murine
equivalent mutation, P215L, did not lead to cardiomyopathy in knock-in mice for
up to 16 months. Physiology and morphology of mutant hearts revealed no changes
from wildtype hearts, and measurements of ubiquitinated proteins, Hsps, and
autophagy were unaffected [162]. On the other hand, cardiomyocyte-specific
Desmin is a muscle-specific intermediate filament that integrates the sarcomere, Z-discs, nuclear membrane, and intercalated discs acting as a structural component as well as a regulator of the sarcomeric architecture [164]. A mutation in DES, R406W, was identified in a young male patient as the causative gene of progressive RCM. Mutated desmin filaments can integrate with wildtype filaments; however, under mechanical stress, aggregates will form. The R406W mutation did not allow desmin filaments to attach to intercalated discs and affected organization of spectrin proteins, N-cadherin, and connexin-43. Disruption in the intercalated discs may produce conduction abnormalities and arrhythmias, a common clinical manifestation of RCM [165]. The R406W mutation was recapitulated in knock-in heterozygous and homozygous mice with an analogous murine mutation, Des R405W. Histology demonstrated similar pathology with desmin aggregates and misalignment of myofibrils. Homozygous mutant mice died within the first three months of life from complications in gut smooth muscle [107].
A DCBLD2 gene variant has recently been identified in a 5-year-old patient with RCM, developmental delay, and dysmorphic features. The DCBLD2 gene encodes a type-I transmembrane receptor involved with intercellular signaling, cell growth regulation, and wound healing. Heterozygous V429L variant in ACVRL1 and homozygous nonsense mutation W27* in DCBLD2 were identified in the patient, although the DCBLD2 was determined as the causative mutation. Angiogenesis was reduced in knockout DCBLD2 mice [166].
Impaired ventricular filling and fibrosis, hallmarks of RCM, was evaluated by
Tsuru et al. [139] to determine how paracrine factors secreted by
cardiac fibroblasts may impact cardiomyocyte dysfunction, causing the pathogenic
phenotype. Under mechanical stress and ischemic damage, quiescent cardiac
fibroblasts differentiate into myofibroblasts and secrete ECM proteins and
cytokines. Cardiac fibroblasts are the most common cells found in the heart and
play a significant role in ischemic damage and pathogenic hemodynamics.
Myofibroblasts participate in scar formation and play a role in ischemic damage,
expressing
RCM is pleiotropic and, in some family studies, the genotype-phenotype
correlation can be ambiguous due to the complex genetic network and influence
from environmental factors and modifier genes. Family studies, human cell lines,
animal models, and systems genetics analysis are important tools for determining
disease-causing variants of RCM and elucidating the pathogenic mechanism(s).
Cardiomyopathies overlap in causative mutations and associated genes. Hence,
there is a pressing need for genetic testing in family members of patients with
cardiomyopathy. Whole exome sequencing and next generation sequencing are
important techniques for identifying gene variants. Proteins encoded by
BAG3, DES, and FLNC exhibit histopathological
protein aggregation, and sarcomeric proteins cTnI and titin affect Ca
MC—Data curation, Investigation, Project administration, Writing - review and editing; BOO, NRA—Data curation, Investigation, Writing - review & editing; JAT—Funding acquisition, Resources, Writing - review & editing; EP—Conceptualization, Methodology, Writing - original draft, Writing - review & editing. All authors have read and agreed to the published version of the manuscript.
Not applicable.
Thanks to all the peer reviewers for their opinions and suggestions.
The study was supported in part by National Institutes of Health [R01HL53392 and R01HL116906 (Towbin) and R01HL151438 (Multiple PI: Towbin, Purevjav, Lu)].
The authors declare no conflict of interest.