



 , Nick G Bellenger 1,2, Andrew J Ludman 1,2, Angela C Shore 1,3, W David Strain 1,3
, Nick G Bellenger 1,2, Andrew J Ludman 1,2, Angela C Shore 1,3, W David Strain 1,31 Diabetes and Vascular Medicine Research Centre, Institute of Biomedical and Clinical Science and University of Exeter, College of Medicine and Health, Royal Devon & Exeter Hospital, EX2 5AX Exeter, UK
2 Department of Cardiology, Royal Devon & Exeter Hospital, EX2 5AX Exeter, UK
3 NIHR Exeter Clinical Research Facility, Royal Devon & Exeter NHS Foundation Trust and University of Exeter, College of Medicine and Health, RILD building, Royal Devon & Exeter Hospital, EX2 5DW Exeter, UK
Academic Editors: Filippos Triposkiadis, Massimo Volpe, Grigorios Korosoglou and Matteo Cameli
Abstract
Acute myocardial infarction (AMI) is a major cause of morbidity and mortality worldwide. Timely reperfusion with primary percutaneous coronary intervention (PPCI) remains the gold standard in patients presenting with ST-segment elevation myocardial infarction (STEMI), limiting infarct size, preserving left ventricular ejection fraction (LVEF), and improving clinical outcomes. Despite this, a significant proportion of STEMI patients develop post-infarct heart failure. We review the current understanding and up-to-date evidence base for therapeutic intervention of ischaemia-reperfusion injury (IRI), a combination of myocardial ischaemia secondary to acute coronary occlusion and reperfusion injury leading to further myocardial injury and cell death. Multiple treatment modalities have been shown to be cardioprotective and reduce IRI in experimental animal models. Recent phase II/III randomised controlled trials (RCT) have assessed multiple cardioprotective strategies ranging from ischaemic conditioning, therapeutic hypothermia and hyperoxaemia to pharmacological therapies. While several therapies have been shown to reduce infarct size in animal models or proof-of-concept studies, many larger scale trial results have proven inconsistent and disappointing. Hard clinical outcomes remain elusive. We discuss potential reasons for the difficulties in translation to clinical practice.
Keywords
- Acute myocardial infarction
- Coronary microvascular dysfunction
- Ischaemia reperfusion injury
- Cardioprotection
- Ischaemic conditioning
Ischaemic heart disease (IHD) is the leading cause of death worldwide with around 1.8 million deaths per year in Europe alone, accounting for 20% of all deaths [1]. Primary percutaneous coronary intervention (PPCI) is the gold standard reperfusion strategy for patients presenting with an acute ST-segment elevation myocardial infarction (STEMI) within 12 h of symptom onset. The end goal is to ensure adequate blood supply to ischaemic but viable myocardium in order to reduce infarct size, preserve left ventricular (LV) function, and reduce mortality [2, 3].
Up to 95% of occluded coronary vessels are re-opened in this manner [4]. However, despite prompt reperfusion and optimal medical therapy, up to 22% of individuals will experience a prolonged or new hospitalisation for heart failure within a year, with up to 7% mortality at 1 year [5]. One of the main determinants of mortality, heart failure, and arrhythmia after acute myocardial infarction is infarct size [6, 7, 8, 9]. A meta-analysis by Stone et al. [10] of 1889 patients assessed after STEMI by cardiac magnetic resonance imaging (CMRI) and 743 by single-photon emission computerised tomography (SPECT) found an independent association between infarct size within 1 month, and subsequent mortality and hospitalisation for heart failure at 1 year.
While timely reperfusion with PPCI is essential to salvage myocardium, the process of restoring coronary blood flow can induce myocardial injury and death of cells that were only reversibly injured during the preceding ischaemic episode [11, 12]. The ongoing damage to myocardial tissue and coronary microvasculature that occurs after blood supply is restored is a complex multifactorial process that is estimated to account for up to 50% of the final infarct size [11]. It is therefore the summative effects of both myocardial ischaemia and lethal reperfusion injury that contribute to the final infarct size and clinical outcomes.
There is no doubt regarding the evidence and efficacy for PPCI in reducing infarct size. However, cardioprotective strategies for prevention of lethal reperfusion injury and coronary microvascular dysfunction (CMD) remain limited. This mini review aims to cover the basic pathophysiology of ischaemia-reperfusion injury (IRI) and summarise the attempts made to translate cardioprotective strategies into the clinical setting.
Acute coronary artery occlusion in ST-segment elevation myocardial infarction results in myocardial ischaemia downstream of the blockage. This initiates a cascade of abnormalities affecting the function, metabolism, and structure of myocytes, ultimately leading to necrosis and cell death [13]. Reimer and Jennings illustrated this in 1977 after work on myocardial ischaemia in the dog model, describing a “wavefront phenomenon”, where irreversible injury of ischaemic myocardium occurs first in the subendocardium before extending to the subepicardial layers [14, 15]. The resulting infarct size is determined by several key factors and is summarised in Fig. 1.
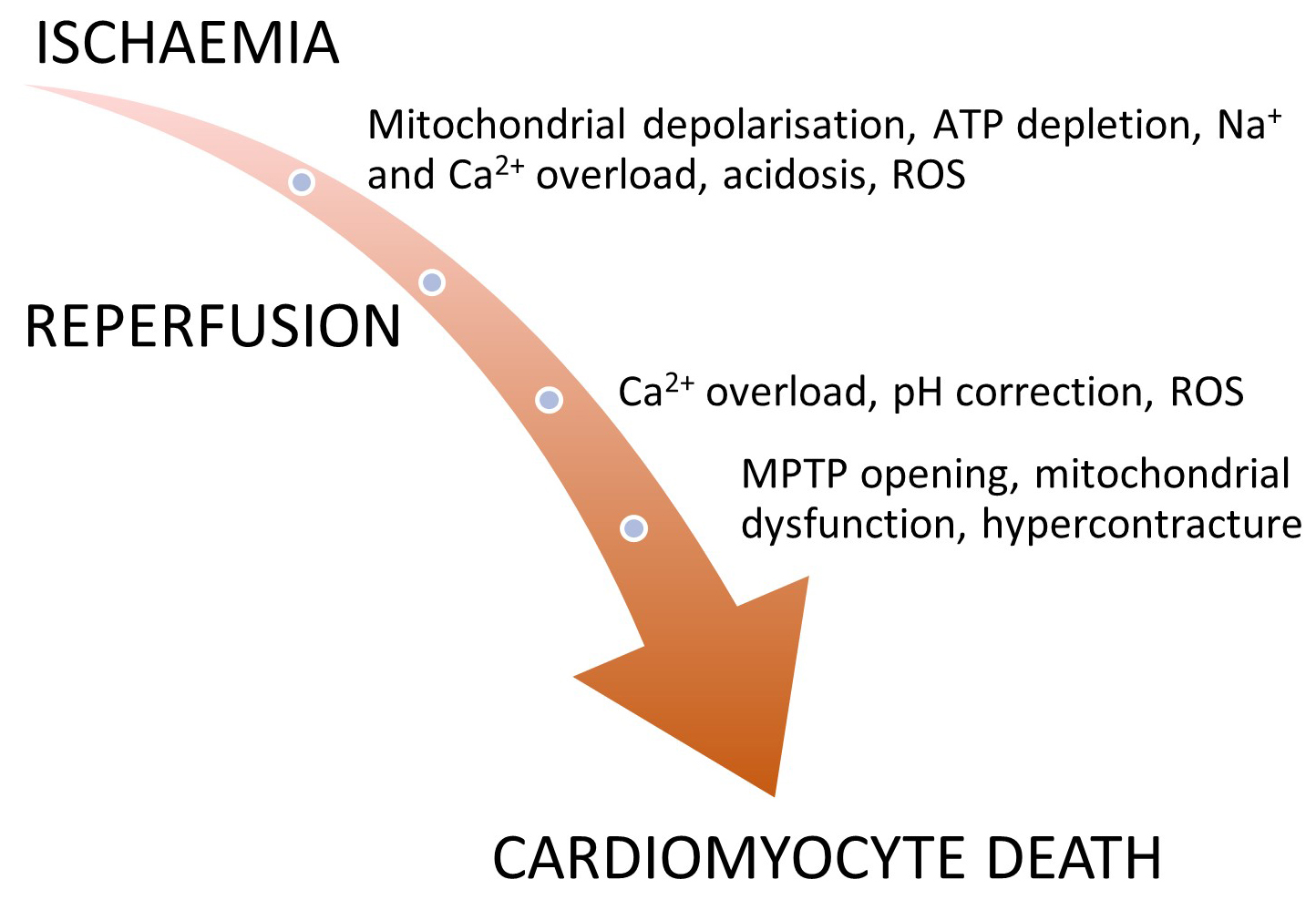 Fig. 1.
Fig. 1.Proposed mechanisms of cardiomyocyte death during ischaemia-reperfusion. A diagrammatic overview of the processes involved during both the ischaemic, and reperfusion phases with oxidative stress leading to production of reactive oxygen species (ROS), mitochondrial calcium overload, and effects on the mitochondrial permeability transition pore (MPTP) resulting in cardiomyocyte death.
The myocardial tissue in the vascular territory distal to the occlusion is at definite risk of ischaemic death if not reperfused in a timely fashion and is known as the myocardial area at risk [13]. In humans, the proportion of AAR that is irreversibly injured ranges from 0% (aborted infarction) up to 88% (infarction), and is dependent on the infarct artery, reperfusion time, collateral flow, reperfusion injury, and microvascular dysfunction [13, 16]. Myocardial salvage describes the proportion of the AAR that survives, and is an important determinant of final infarct size.
Assessment of AAR can be difficult. In humans, a commonly used technique is single-photon emission tomography but this requires radioisotope to be injected during occlusion, and before reperfusion [13]. Given the logistical issues, this technique has limited clinical applicability. More recently, cardiac magnetic resonance (CMR) has been used to assess AAR. T2-weighted imaging enhances oedema, which is present in ischaemic myocardium [17, 18, 19]. However, T2 imaging can be prone to artifact and requires long breath-holds, with insufficient image quality in up to 62% of patients [20, 21, 22]. More recently, contrast-enhanced cine (CE-SSFP) was compared to T2-weighted imaging in 166 participants involved in a substudy of the DANAMI-3 trial (Complete revascularisation versus treatment of the culprit lesion only in patients with ST-segment elevation myocardial infarction and multivessel disease). This showed good internal consistency in assessment of the AAR along with good inter-observer agreement of analysis of contrast enhanced cine with the authors suggesting that CE-cine can replace T2-weighted imaging for a more valid assessment of myocardial AAR [23].
Cardiomyocyte death from ischaemia commences if this period is greater than 20
minutes, arising from the subendocardium, and progressing into the subepicardium
[14]. Despite this, some 30–50% of myocardium in the AAR is still viable after
4–6 h of symptom onset [24], with evidence from thrombolysis data that
reperfusion can limit infarct size and reduce mortality in patients presenting
with STEMI up to 12 h post coronary occlusion [25, 26, 27, 28]. It is from this data that
consensus guidelines, such as those from the European Society of Cardiology (ESC)
recommend reperfusion therapy in all patients with symptoms of ischaemia and
ST-segment elevation of
Collateral vessels connect one epicardial vascular territory to another, providing an alternative source of blood supply to myocardium, should one epicardial artery occlude [31, 32]. The phase 1 thrombolysis in myocardial infarction (TIMI) trial demonstrated reduced creatine kinase (CK) enzyme release and improved LV ejection fraction at discharge in patients presenting with acute myocardial infarction (AMI) and failed reperfusion in the presence of coronary collaterals [33]. Furthermore, the presence of well-developed vs poorly grown collateral vessels has been shown to significantly reduce major adverse cardiac events (MACE), and even cardiac mortality at 10 years [34, 35].
While reperfusion is necessary for salvaging ischaemic myocardium, the process also leads to reversible injury by way of myocardial stunning [36], CMD, and death of cardiomyocytes that were viable at the end of the index ischaemic event (lethal reperfusion injury).
The most severe end of this spectrum is the phenomenon of “no-reflow”, where there is inadequate myocardial perfusion despite re-establishing infarct related artery patency, with no angiographic evidence of mechanical obstruction. No-reflow affects between 11% and 41% of patients undergoing PPCI and significantly diminishes the beneficial impact of reperfusion therapy, results in poor clinical and functional outcomes, and is an independent predictor of increased infarct size and 5-year mortality [37, 38, 39, 40, 41, 42].
The pathophysiology of no-reflow is complex and multifactorial. Mechanisms implicated include:
(1) Pre-existing microvascular dysfunction. This may be structural or functional and impairs coronary flow reserve, increasing susceptibility to ischaemic injury. Risk factors include age, abnormal insulin resistance and lipid metabolism and chronic inflammatory disease. In addition, pre-existing microvascular dysfunction is an independent predictor of adverse cardiac events [30, 31].
(2) Distal micro-thromboembolism. Thrombus debris migrate distally during
balloon dilatation/stenting. It has been shown that myocardial perfusion falls
when embolic microspheres block
(3) Ischaemic injury. Endothelial cell damage and necrosis causes loss of vascular integrity, extravascular accumulation of fluid and blood cells, altered production of nitric oxide, and cell swelling [46, 47].
(4) Lethal reperfusion injury. This is discussed in more detail below, but major contributory factors include infiltration of neutrophils and platelets, oxidative stress, and mitochondrial dysfunction. This can lead to intra-myocardial haemorrhage, a predictor of infarct size and poor clinical outcomes [11, 48].
The timeframe for lethal reperfusion injury (RI) is difficult to accurately quantify. Clearly, a significant amount of damage takes place within the first few minutes of reperfusion, which has been demonstrated in both rat hearts and in humans [49, 50, 51]. However, it is difficult to distinguish between ischaemic cell death or lethal RI (or a combination of both) as the cause of apoptosis and late cell death [12]. Several important processes have been identified in experimental studies that act in concert leading to lethal RI.
(1) Oxidative stress. Myocardial hypoxia leads to dysfunction of the electron transport chain in the mitochondria, with decreased adenosine triphosphate production (ATP) and anaerobic metabolism. One of the subsequent consequences is decreased anti-oxidative intracellular agents [52]. During the first few minutes of reperfusion, reactive oxygen species (ROS) are generated by the xanthine oxidase and nicotinamide adenine dinucleotide phosphate oxidase systems, mitochondrial electron transport chain, and uncoupling of the nitric oxide synthase (NOS) system [52, 53, 54] . ROS induce opening of the mitochondrial permeability transition pore (MPTP), act as a neutrophil chemoattractant, and lead to dysfunction of the sarcoplasmic reticulum. They also induce cell damage and ultimately cause cell death via autophagy (damaged components in the cytoplasm that are degraded in membrane vesicles), necrosis (unlike apoptosis, is passive and unregulated), and apoptosis (programmed cell death activated under hypoxic stress and in response to ROS) [52, 55, 56].
(2) Intracellular and mitochondrial calcium overload. This starts during acute ischaemia and is exacerbated during reperfusion by ROS, disruption of the plasma membrane, and opening of the MPTP [57].
(3) Rapid restoration of physiological pH. During reperfusion, intracellular myocardial pH rapidly normalises from less than 7.0 during acute ischaemia. This contributes to myocyte death by permitting MPTP opening and hypercontracture [58].
(4) Mitochondrial permeability transition pore. Many proposed mechanisms of ischaemia-reperfusion injury (IRI) converge on MPTP, a mitochondrial membrane channel. Rat heart models show that opening leads to membrane depolarisation and uncoupling of oxidative phosphorylation, leading to ATP depletion and cell death [59, 60]. During IRI, MPTP remains closed during ischaemia, before opening at reperfusion in response to mitochondrial calcium and phosphate overload, ROS, and rapid pH correction [61].
Blood contains large numbers of EVs that originate from a variety of cells, including platelets, erythrocytes, leucocytes, and endothelium [62, 63, 64]. These small vesicles aid intercellular communication during IRI by transferring their contents (lipids, amino-acids, proteins, mRNAs, miRNAs) [65]. There are different types of EVs.
In the past few years, there has been increasing interest in Exosomes, a lipid vesicle 50–150 nm in diameter that is secreted by all cells [63]. The exact mechanism of cardioprotection in IRI has yet to be fully unravelled but they have been shown to reduce scar size when injected into the infarct border, and improve cardiac function after AMI in mice [66]. Some exosomes contain short RNA, that have been shown to reduce infarct size 48 h after reperfusion in rats following 45 min of coronary artery occlusion [67].
Other EVs include microvesicles (MVs) which are involved in intercellular communication. Patients with heart failure have been found to have increasing circulating MVs, and they may also play a negative role in IRI, with MVs released from endothelial cells after ischaemic-reperfusion acting as pro-apoptotic and pro-oxidative to cardiomyocytes [68]. More recently, EVs taken from patients presenting with acute coronary syndrome before PCI have been shown to protect against IRI, both in-vitro and in the rat heart [69].
Prompt reperfusion in patients presenting with STEMI with PPCI remains the gold standard and improves outcomes. However, both experimental animal models and studies in patients with STEMI suggest that up to 50% of the final infarct size is a result of lethal reperfusion injury [11], making it an attractive therapeutic target (Fig. 2). Over the past 30 years, multiple cardioprotective strategies aimed at reducing myocardial IRI have been proposed, and although pre-clinical studies have often been promising, this has not translated into clinical efficacy [70]. Therefore, there remains an urgent need to discover novel therapies that can be given peri-procedurally to reduce infarct size and improve mortality and morbidity in reperfused STEMI patients. Numerous cardioprotective strategies have been proposed and tested, broadly divided into 3 treatment modalities (Fig. 3).
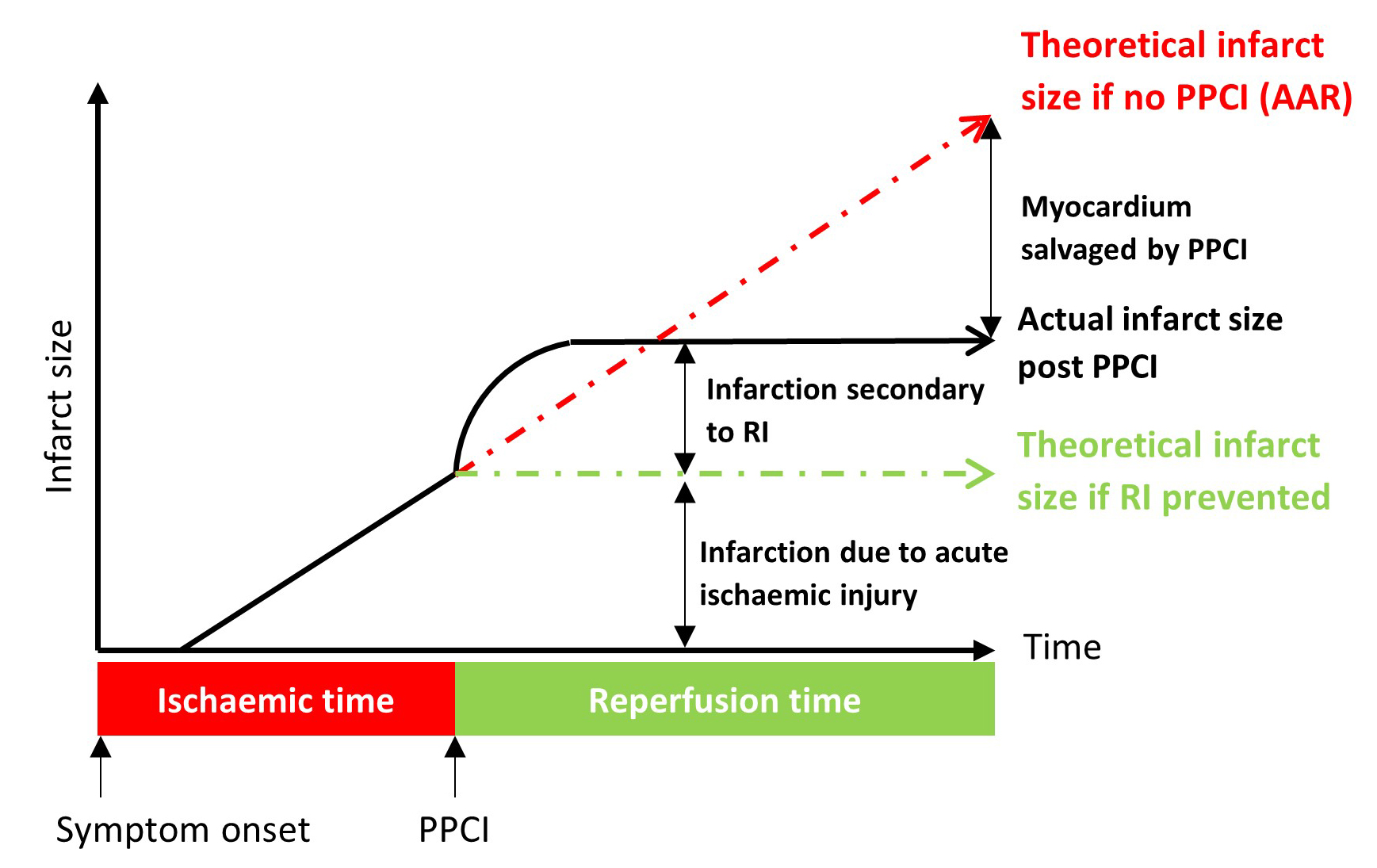 Fig. 2.
Fig. 2.This highlights the individual components that make up final myocardial infarct size (arbitrary value) post STEMI up to 24 h (black solid line). The dotted red line represents the theoretical infarct size if no PPCI (AAR – area at risk). The dotted green line represents the theoretical infarct size if RI is prevented. Figure modified from J Clinical Res [57].
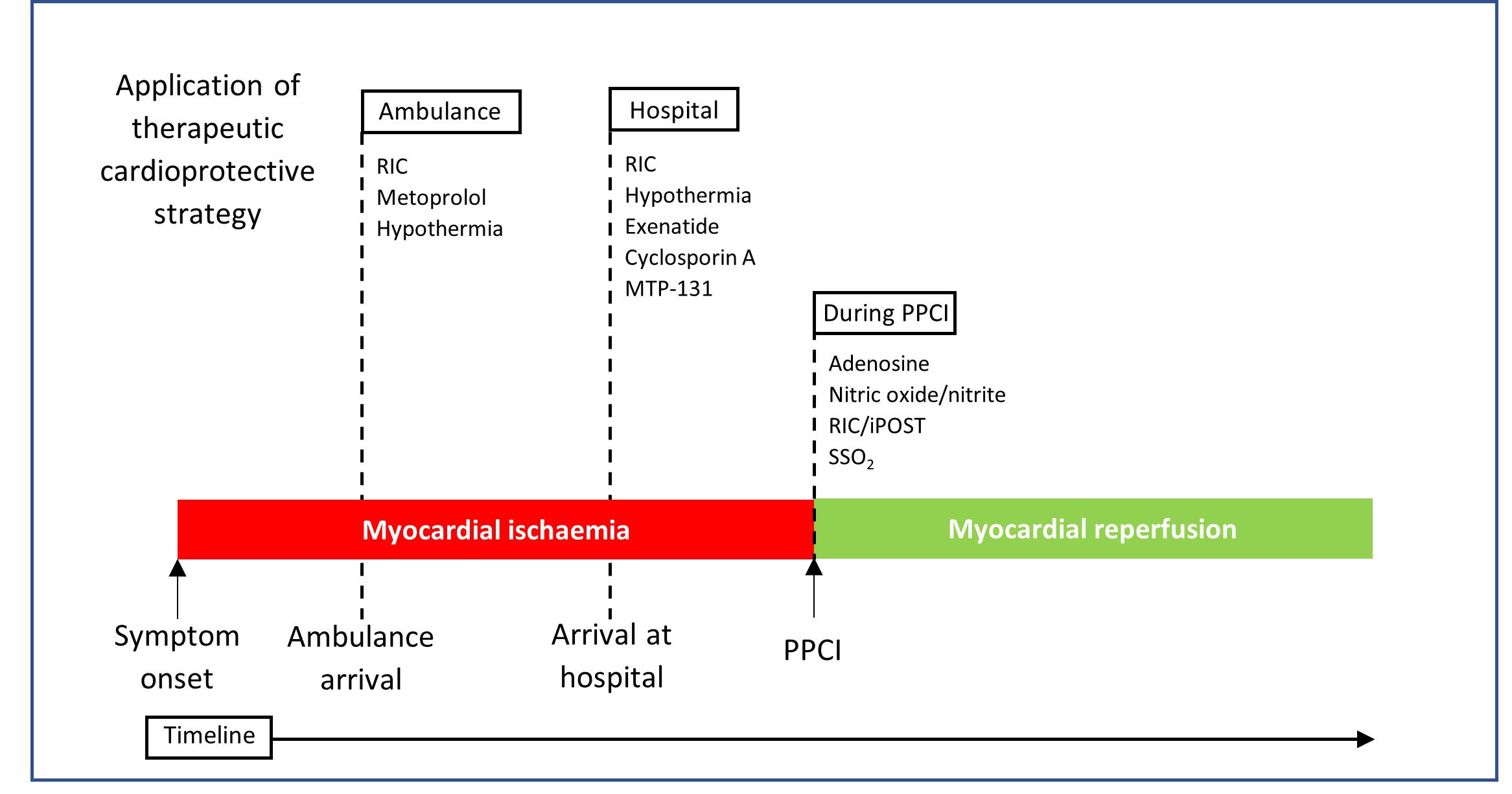 Fig. 3.
Fig. 3.Diagrammatic representation of the various timeframes of
cardioprotective treatments designed to reduce MI size in STEMI
patients. Treatment strategies may be attempted during the ischaemic
phase, that is, prior to reperfusion with PPCI or thrombolysis, such as remote
ischaemic conditioning (RIC), hypothermia, and some pharmacological therapies.
They may also be given during myocardial reperfusion e.g., ischaemic
post-conditioning (iPOST) and super saturated oxygen (SSO
This involves brief cycles of ischaemia and reperfusion applied to an organ or tissue that may be pre, during, or post reperfusion; local or remote. The mechanisms are incompletely understood, but may involve changes to intracellular pH, effects on nitric oxide synthases (NOS) and their subsequent generation of reactive oxygen and nitrogen species, along with increased protein kinase G and reperfusion injury salvage kinase [71].
Ischaemic post-conditioning (iPOST): alternating angioplasty balloon inflation/deflations immediately after opening the culprit artery has been shown to be protective in multiple Phase II trials [72, 73]. However, the largest randomised control trial (RCT) to date (DANAMI-3, 2017) of 1234 patients presenting within 12 h of a STEMI with a thrombolysis in myocardial infarction (TIMI) flow of 0–1 (absence or faint antegrade coronary flow beyond occlusion) showed that routine iPOST during PPCI failed to reduce the composite outcome of all cause death and hospitalisation for heart failure [74]. Limitations to iPOST include treatment being administered after reperfusion, thereby suggesting that reperfusion injury occurs before any potential benefit in this study protocol. The authors suggested a modified protocol involving post-conditioning immediately after reperfusion (e.g., within 15 seconds) [74].
Remote ischaemic conditioning (RIC): serial inflations and deflations with a pneumatic cuff on the arm or thigh to induce brief cycles of ischaemia and reperfusion showed promise in increasing myocardial salvage and reducing infarct size by 20–30% in animal (dog) and small clinical human trials [75, 76, 77, 78]. Despite these encouraging results, the largest RCT to date (CONDI-2/ERIC-PPCI, 2019) of 5401 patients presenting with STEMI failed to show improved clinical outcomes (cardiac death or hospitalisation for heart failure) at 12 months [79]. Several potential reasons for this have been proposed: (1) animal models used for IRI do not adequately represent a typical STEMI patient, (2) limb RIC protocol not sufficiently optimised for maximal cardioprotection, and (3) presence of co-morbidities that may confound results [80].
Multiple therapeutic agents have been shown to improve myocardial infarct size in animal/pre-clinical models. Here, we focus on those that have progressed to clinical trials.
Exenatide: this glucagon-like peptide-1 (GLP-1) receptor agonist has been shown in both small animal (rat) models and small clinical trials to significantly reduce myocardial infarct size, and increase myocardial salvage index [81, 82]. The exact mechanism of cardio-protection is unclear but evidence suggests multiple modes of action, involving activation of phosphoinositide 3 kinase/cyclic adenosine monophosphate/cyclic guanosine monophosphate, inhibition of apoptotic factors, and attenuating the oxidative stress injury [82, 83]. Further studies are required to assess for improved clinical outcomes.
Nitric oxide and nitrite: Two recent clinical studies have failed to demonstrate
significant reduction in MI size with either intravenous, or intracoronary routes
in STEMI patients treated with PPCI [84, 85]. However, the latter did
significantly reduce MACE at 1 year (p = 0.04) and infarct size in
patients with STEMI and TIMI flow
Cyclosporin A: This inhibitor of MPTP showed promise in several small clinical trials, reducing CK AUC by 40%, and infarct size by between 20–28% as assessed on CMR [86, 87]. The Cyclosporine and Prognosis in AMI patients trial (CIRCUS, 2015), the largest to date involving 970 patients in patients with anterior STEMI showed no clinical benefit nor difference in CK, Troponin I release, and adverse left ventricular remodelling at 1 year when compared to placebo [88].
MTP-131: This peptide targets mitochondria, inhibiting cardiolipin, thereby
reducing production of ROS. Once again, small animal (rat) models reported
reduced MI size [89, 90], but in the Phase II 117 patient EMBRACE STEMI trial, no
significant reduction in MI size was seen in a carefully selected population of
patients presenting with an anterior STEMI, TIMI 0, and symptom onset time
Adenosine: There have been a total of 13 RCTs (4273 STEMI patients) assessing both intracoronary adenosine (8 RCTs) and intravenous adenosine (5 RCTs). A recent meta-analysis in 2015 found that patients who were administered intracoronary adenosine showed a significantly lower incidence of heart failure (risk ratio 0.44 [95% confidence interval 0.25–0.78], p = 0.005) and coronary re-flow [92]. There was no difference in non-fatal MI or all-cause mortality.
Metoprolol: IV metoprolol administered after 90 mins of coronary occlusion prior to reperfusion reduces MI size and improves LV systolic function in the porcine heart [93]. Results from clinical trials have been mixed. The proof-of-concept study (The Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) trial) in 270 anterior STEMI patients given metoprolol as early as possible showed reduced MI size, preserved LV systolic function, and lowered hospital readmissions for heart failure [94].
However, the more recent phase III trial (the Early Intravenous Beta-Blockers in
Patients with ST-Segment Elevation Myocardial Infarction Before PPCI (EARLY-BAMI)
trial) in 683 patients in a non-restricted STEMI population showed effect on
infarct size or LV function [95]. This difference may be in part due to the more
select population in the METOCARD-CNIC trial (symptom onset
Sodium thiosulphate (STS): this metabolite of hydrogen sulphide (H
Therapeutic hyperoxaemia/super-saturated oxygen (SSO
Therapeutic hypothermia: this has been consistently shown to reduce MI in animal models, and human pre-clinical studies [110]. A meta-analysis in 2016 included 6 RCTs, with a total of 819 patients found no significant reduction in all-cause mortality or heart failure [111]. More recently, both the Out-of-hospital initiation of hypothermia in STEMI trial and Cooling As an Adjunctive Therapy to Percutaneous Intervention in Patients with AMI EU (COOL AMI EU) showed no significant reduction in MI size, with the latter showing a significantly increased risk of paroxysmal atrial fibrillation and cardiogenic shock in the hypothermia group [112, 113].
The accumulation of evidence described has shown numerous cardioprotective strategies that are effective at reducing infarct size in animal models. However, they have yet to translate into improvements in hard outcomes in clinical trials. In fact, many treatment modalities have shown early promise in small phase I/II trials but have fallen at the last hurdle when tested in large RCTs (in particular, iPOST and RIC). Many of the pharmacological therapies require larger studies to provide more definitive answers.
There are several possible reasons for these conflicting and often disappointing results.
Often, pre-clinical studies are performed on small animals, such as mice, rats,
and rabbits. Some potential treatments have not been studied in large animals.
Those that have usually include healthy, young animals. This contrasts with the
typical demographic of the STEMI patient who is older, with multiple
co-morbidities, an on multiple medications that may interfere with the
cardioprotective intervention, such as statins and platelet P2Y
The inclusion criteria should only include patients that have been shown to
benefit most from adjunctive therapy to PPCI. This includes: a large AAR such as
anterior STEMI, occluded culprit artery on presentation (TIMI
Without good pre-clinical trials and pilot data, it may be difficult to establish effective treatment dosage or interventional protocol resulting in failure to show positive results in some studies. For example, the limb RIC protocol in the CONDI-2/ERIC-PPCI trial was not optimised for maximal cardioprotection (duration of limb ischaemia, and reperfusion cycles) which the authors note as one of the potential reasons why the trial failed to show a positive outcome [80].
The concentration of drug may vary significantly when given intravenous vs intracoronary. An example of this can be seen with the use of adenosine where intracoronary adenosine was associated with clinical improvement compared to intravenous injection [92].
There is evidence that intervention that is present at time of reperfusion can
reduce MI size [118]. This may be difficult to apply in STEMI given the early
administration required. Furthermore, treatment may not reach ischaemic
myocardium in those with a completely occluded artery. Timing of intervention may
be more complex than simply as soon as possible, as demonstrated with the use of
IV metoprolol in the pig model, where no effect on infarct size was found if the
coronary artery was occluded for too short (
The pathophysiology of ischaemic RI is complex and multifactorial. Routinely used animal models may not adequately simulate this but are rather one element of it. It may be that a multi-pronged approach is required targeting the different players involved in IRI, or a therapy that acts on multiple pathways.
Clinical trials should be undertaken based on solid pre-clinical data, including
those performed on large animals, with large RCTs only performed if Phase II
trials have shown reduction in MI size. Furthermore, the trial design is crucial,
and should ideally include patients with large AAR (e.g., anterior STEMI), TIMI
AMI, acute myocardial infarction; IHD, ischaemic heart disease; IRI, ischaemia-reperfusion injury; MPTP, mitochondrial permeability transition pore; MVO, microvascular obstruction; STEMI, ST-segment elevation myocardial infarction; iPOST, ischaemic post-conditioning; RIC, remote ischaemic conditioning; PPC, primary percutaneous coronary intervention; ROS, reactive oxygen species; AAR, area at risk.
The first draft of the manuscript was written by JH with supervision and contribution by WDS. NGB and AJL provided specialist expertise and advice regarding manuscript content and contributed to the final manuscript. WDS and ACS supervised the project. All authors read and approved the final manuscript.
Not applicable.
We would like to express our gratitude to all those who helped us during the writing of this manuscript and for all the peer reviewers for their expert opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.