


1 Department of Advanced Medical and Surgical Sciences, University of Campania Luigi Vanvitelli, I-80138 Naples, Italy
2 Department of Translational Medical Sciences, University of Campania Luigi Vanvitelli, I-80138 Naples, Italy
3 Division of Cardiology, A.O.R.N. “Sant’Anna & San Sebastiano”, I-81100 Caserta, Italy
4 Department of Precision Medicine, University of Campania Luigi Vanvitelli, I-80138 Naples, Italy
† These authors contributed equally.
Academic Editor: Peter A. McCullough
Abstract
Evidence suggests a close connection between Nonalcoholic Fatty Liver Disease (NAFLD) and increased cardiovascular (CV) risk. Several cross-sectional studies report that NAFLD is related to preclinical atherosclerotic damage, and to coronary, cerebral and peripheral vascular events. Similar results have been showed by prospective studies and also by meta-analyzes on observational studies. The pathophysiological mechanisms of NAFLD are related to insulin resistance, which causes a dysfunction in adipokine production, especially adiponectin, from adipose tissue. A proinflammatory state and an increase in oxidative stress, due to increased reacting oxygen species (ROS) formation with consequent oxidation of free fatty acids and increased de novo lipogenesis with accumulation of triglycerides, are observed. These mechanisms may have an impact on atherosclerotic plaque formation and progression, and they can lead to increased cardiovascular risk in subjects with NAFLD. This review extensively discusses and comments current and developing NAFLD therapies and their possible impact on cardiovascular outcome.
Keywords
- NAFLD
- Cardiovascular disease
- Pathophysiology
- Type 2 diabetes
- Cardiovascular risk
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver dysfunction. It affects about 25% of the population and is associated with several metabolic risk factors such as hypertension, diabetes, obesity and dyslipidemia [1, 2]. Evidence suggests a close link between Nonalcoholic Fatty Liver Disease (NAFLD) and increased cardiovascular (CV) risk. Several cross-sectional studies report hepatic steatosis as an independent risk factor for both an increased carotid intima-media thickness and diastolic cardiac dysfunction [3, 4]. Moreover, a clinical investigation conducted on a cohort of more than 2000 subjects with type 2 diabetes showed that a condition of hepatic steatosis, even after correction for the main cardio-metabolic risk factors, is associated with a higher prevalence of coronary, cerebral and peripheral vascular disease [5].
Prospective studies confirm the association between NAFLD and CV risk. A large longitudinal study with a two-year follow-up demonstrated that the presence of NAFLD at baseline assessment is an independent risk factor for the carotid disease [6]. Similarly, in the Valpolicella study conducted on diabetic subjects, after correction for cardiometabolic risk factors, NAFLD appeared as an independent risk factor for the incidence of cardio-cerebrovascular events [7].
Furthermore, a recent meta-analysis showed that subjects with NAFLD, compared to the control group without steatosis, had a 1.6 times higher risk of developing CV events, and this risk is higher for non-fatal events [8]. The same meta-analysis demonstrates that the presence of “severe NAFLD” is associated with an approximately 2.5 times higher risk of developing fatal and non-fatal CV events.
In a cohort of patients with a histological diagnosis of NAFLD, carotid intima-media thickness increased significantly and progressively along with the stage of hepatic fibrosis [9]. Furthermore, the severity of hepatic fibrosis assessed through liver biopsy has been independently associated with the worsening of both systolic and diastolic cardiac dysfunction [10].
A large retrospective study conducted on subjects with no history of CV disease undergoing to serial Computed Tomography (CT) to evaluate the Coronary Artery Calcium (CAC) score, showed that the risk of progression of CAC increased progressively from subjects without steatosis to those with steatosis and advanced fibrosis [11].
The aim of this review is to provide a comprehensive overview on the pathophysiological mechanisms underlying the development of NAFLD and its link between with CV risk. Finally, the therapeutic strategies for NAFLD treatment and their impact on CV outcome will be discussed.
NAFLD is characterized by a large spectrum of disorders, ranging from simple
steatosis to non-alcoholic steatohepatitis, which both in turn may progress into
overt cirrhosis and hepatocellular carcinoma [12, 13, 14]. NAFLD diagnosis can be
performed only in absence of other causes of chronic liver disease such as
alcohol consumption (
Recently, new non-invasive diagnostic tools, such as ultrasonography, CT, and
magnetic resonance imaging (MRI), have emerged, showing a greater safety profile
and availability, and sharing a good sensitivity and specificity compared to gold
standard [15, 18]. Ultrasonography has been recommended as the first-line
diagnostic tool to detect NAFLD, though its sensitivity decreases when less than
30% of hepatocytes are involved as well as in obese patients [15, 19]. Controlled
attenuation parameter (CAP), available on the equipment “Fibroscan
®”, represents a new and accurate technique to detect and
quantify hepatic steatosis. Moreover, transient elastography, by Fibroscan is
also able to detect liver stiffness, which is a reliable measure of liver
fibrosis [20, 21, 22]. This equipment has two M and XL probes capable of supplying
ultrasounds and an elastic wave to measure the stiffness of the liver tissue and
steatosis. XL probe allows to study obese patients who, due to their thick
panniculus adipose, can hinder the propagation of the elastic wave [23, 24].
Several clinical and imaging scores have been validated to assess liver
steatosis. The fatty liver index is calculated through serum triglycerides
levels, waist circumference, and
The primary NAFLD hallmark is fat accumulation (mostly triglycerides) in the liver. However, how this accumulation leads to a liver dysfunction is compound and incompletely understood [31, 32, 33, 34]. Of note, though triglycerides represent the major part of intrahepatic lipid in NAFLD patients, new evidence suggests that changes in liver lipid composition (including ceramides, diacylglycerol, triglyceride/diacylglycerol ratio, phospholipids and free cholesterol) may play a significant role [35, 36, 37]. Particularly, alterations of free cholesterol metabolism seem involved in the development and progression of NAFLD and consequently associated with a higher CV risk. It has been observed that alteration of microbiota could alter the absorption and elimination of free cholesterol, thus determining an increase of its serum levels. Moreover, some authors found that mitochondrial abnormalities and altered activation of acyl coenzyme A cholesterol acyl transferase, SREBP-2 maturation, altered hydroxymethylglutaryl coenzyme A reductase (HMG-CoA-R) expression and decreased phosphorylation of HMG-CoA-R, induced by genic factors, could affect synthesis and transport of free cholesterol [31, 37, 38].
Adipose tissue is also involved in hormone secretion (e.g., adipokines) [39].
Obesity represents an important cause of adipose tissue dysfunction, which plays
a relevant role in the metabolic disorders’ progression, particularly in insulin
resistance and NAFLD [40, 41]. As a result of adipose tissue dysfunction, there is
a reduction of adiponectin, an insulin-sensitizing adipokine. Adiponectin plays a
crucial role in increasing free fatty acids (FFA) oxidation and decreasing FFA
influx to the liver, gluconeogenesis, and de novo lipogenesis, thus
enhancing a protective hepatic role [42, 43, 44]. In addition, it also plays a hepatic
anti-inflammatory and antifibrotic role, by reducing proinflammatory cytokines
and the activation and proliferation of hepatic stellate cells [45, 46, 47, 48, 49]. Impaired
insulin action, through stimulation of lipogenic enzymes via sterol
receptor-binding protein 1c (SREBP-1c), results in an increase in circulating
FFA, an increased storage into the liver and increased de novo
lipogenesis [50, 51, 52, 53]. Furthermore, it seems that insulin resistance is also
involved in the triglyceride synthesis increase through the Kennedy pathway and
in fatty acids
Genetic factors associated with CV events in NAFLD are still undefined. Single-nucleotide polymorphisms (SNPs) may have a role on NAFLD-related cardiovascular disease (CVD) risk. The two most frequently SNPs in NAFLD are the patatin-like phospholipase domain-containing protein 3 (PNPLA3) and the transmembrane 6 superfamily member 2 (TM6SF2). PNPLA3 modulates the lipid droplet profile and appears to be related to tryglicerides (TG) metabolism [65]. It is shown that carriers of this mutation could increase atherosclerosis, paradoxically lowering serum TG levels [66, 67]. In a Danish cohort study, using Mendelian randomization to test a genetic variant in the gene encoding for PNPLA3, a genetically elevated hepatic fat content was not found associated to an increased risk of ischemic heart disease [68]. TM6SF2 modulates TG and cholesterol secretion in the liver through the excretion of very low-density lipoprotein (VLDL) [69]. The carriers of this mutation could raise the risk of NAFLD development because of increased TG and lipid retention in the liver, but they may exhibit some degree of cardio protection resulting in reduced serum levels of TG, low-density lipoprotein (LDL) and total cholesterol [70]. The glucokinase regulator (GCKR) is another susceptibility gene for NAFLD, which encodes liver-specific glucokinase regulatory protein (GKRP) and is involved in both hepatic de novo lipogenesis and NAFLD development [52, 71]. A recent study shown that common variants of the GCKR gene weakly could be associated with CVD risk (Odds Ratio per risk allele: 1.02, 95% confidence interval (CI): 1.0–1.04). However, the increased CVD risk might also be due to changes (increased levels) in VLDL [72].
Recent findings have proposed dysbiosis as a contributing mechanism in NAFLD development and progression. In fact, alteration in intestinal composition seems to increase intestinal tissue permeability, due to increased ethanol production. Therefore, gut-derived pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide, can reach the liver through blood flow, stimulate proinflammatory pathways and drive to liver inflammation and fibrosis [73, 74, 75, 76]. Moreover, gut microbiota increases the choline metabolism, which results in an increase in triglycerides content in the liver, due to the lack of VLDL secretion. In addition, it seems that dysbiosis is also associated to endothelial lipoprotein lipase inhibition, due to the decreased secretion of fasting-induced adipocyte factors and angiopoietin-like 4, with a consequent lack of hydrolyzation in the liver of triglycerides from VLDL particles, and increased gluconeogenesis and lipogenesis due to increased substrate [73].
Even though NAFLD is strongly associated with obesity, a substantial proportion of lean subjects without increased waist circumference can also develop NAFLD. Visceral obesity as opposed to general obesity, genetic predisposition, unhealthy dietary pattern consisting of high cholesterol and fructose intake may be associated with “lean” NAFLD [77].
Recently, numerous epidemiologic studies have documented that NAFLD is associated to increased CV morbidity and mortality both in general population and in patients with diabetes mellitus (DM), a population already burdened by chronic vascular and extravascular complications [78, 79, 80, 81, 82, 83, 84, 85].
Paik et al. [86] proved CVD as one of the major causes of death among
NAFLD patients. NAFLD and CVD share several common cardiometabolic risk factors
such as genetics, systemic inflammation, endothelial dysfunction, hepatic insulin
resistance, adipose tissue dysfunction, oxidative stress and lipid metabolism
[87, 88]. Moreover, evidence shows that NAFLD is closely interrelated with several
metabolic conditions that expose subjects to an increased risk of CV disease
[89, 90, 91, 92]. The association between NAFLD and CV events is also underlined by recent
European guidelines, which recommend CV screening in all subjects with NAFLD
[93]. Many markers of subclinical atherosclerosis, such as increased carotid
intima-media thickness, reduced flow-mediated vasodilation, arterial stiffness
and increased coronary artery calcification, are reported in patients with NAFLD
[94]. In a cohort of 755 patients, Moon et al. [95] reported a severe
and independent association between NAFLD and carotid artery inflammation, which
may be the expression of plaque vulnerability, as assessed by positron emission
tomography with 18 F-fluorodeoxyglucose. Patients with NAFLD develop
hyperglycemia, hyperinsulinemia, hyperlipidemia, and vascular endothelial cell
damage due to insulin resistance, which can also cause smooth muscle cell (SMC)
proliferation. Thus, insulin resistance counts for the development of both NAFLD
and atherosclerosis [96]. In a meta-analysis of almost 300,000 individuals,
subjects with NAFLD had a
All these pathophysiological mechanisms can result in remodeling of left ventricle, increased ventricular mass and diastolic dysfunction. In NAFLD, portal pressure is increased as a result of changes in sinusoidal morphology, reduced sinusoidal flow, and increased intrahepatic resistance, mainly to the presence of fibrosis in more advanced stages of disease [110]. NAFLD leads to an increase in body surface area, which further increases left ventricular filling pressure, cardiac output, and volume overload [116, 117]. Recently, the connection between NAFLD and increased risk of arrhythmias, mainly permanent atrial fibrillation (AF), has achieved extensive scientific interest [90]. Mantovani et al. [7] found that NAFLD doubles the risk of prevalent AF independently of other AF risk factors. In addition, several studies also shown that NAFLD is associated with an increased risk of incident AF, especially among subjects with established T2DM. Park et al. [118] observed that individuals with NAFLD and advanced fibrosis had a greater risk of prevalent AF even after adjusting for multiple CVD risk factors. The presence and severity of NAFLD is associated with a higher risk for corrected QT (QTc) interval prolongation resulting in an increased risk of both ventricular tachyarrhythmias and sudden cardiac death [90]. In a hospital-based cohort of 751 elderly patients with T2DM, NAFLD shows a three-fold greater risk of cardiac conduction defects (right bundle branch block and left anterior hemiblock, persistent first-degree atrioventricular block), so it would result in a close association between NAFLD and cardiac arrhythmias [119]. However, further research is still needed to better explain the increased risk of cardiac death observed in patients with NAFLD. Furthermore, Mantovani et al. [120] observed that ultrasound-detected NAFLD is associated with an approximately 3.5-fold higher risk of both aortic valve sclerosis and mitral annular calcification in a population of hospitalized T2DM subjects with no history of known liver disease, heart failure, moderate-to-severe heart valve disease.
Moreover, an association of excessive peri-organ adipose tissue, namely intrahepatic, epicardial/pericardial, perivascular, intramuscular, peripancreatic and perirenal fat, with cardiometabolic and CVD risk factors was observed [121].
NAFLD, especially in advanced stage of liver fibrosis and severity of disease, increases the risk of coronary atherosclerosis, cardiomyopathy and arrhythmias, which clinically result in increased CVD morbidity and mortality. Further studies are still needed to better establish whether NAFLD independently contributes to the risk of developing adverse CVD outcomes and other cardiac/arrhythmic events. These mechanisms are summarized in Fig. 1.
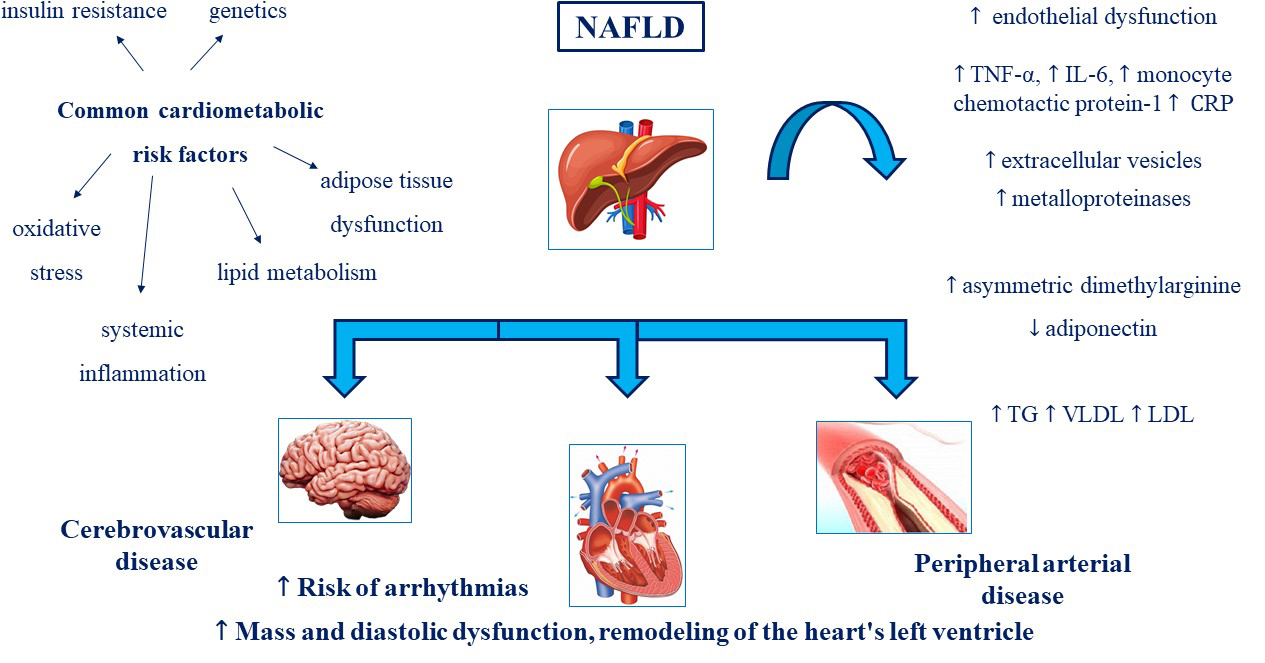 Fig. 1.
Fig. 1.NAFLD and CV risk: a pathophysiological link. NAFLD,
Non-alcoholic fatty liver disease; TNF-
Non-diabetic patients with NAFLD show a reduction in endothelium-mediated vasodilation of the brachial artery, which correlates with the extent of histologically documented liver damage, irrespective of age, sex, body mass index, and the presence of insulin resistance [122]. The implication of this association is that the treatment of NAFLD can have CV benefits and impact on CV outcomes. NAFLD is a common condition, particularly in patients who often consult the cardiologist, such as obese, diabetic and patients with metabolic syndrome. The close association with classic risk factors but especially the high prevalence of CV diseases in patients with NAFLD, mean that this disease cannot be considered the exclusive domain of the hepatologist. The possible clinical scenarios that may present to the cardiologist could be: (1) evaluation of a patient in primary prevention (based on available evidence and because it can be considered a “modifiable factor, NAFLD should be researched for optimal and complete cardiovascular risk estimation”); (2) patient with recent CV event (in these subjects the suspicion of NAFLD is less likely because, especially in the acute phase, it is possible to have an increase in transaminases related to the recent coronary event). Hence the need to treat the CV risk factors that cause NAFLD and to treat NAFLD as a CV risk factor.
If this condition of several comorbidities is observed in elderly patients, they become fragile subjects with different phenotypes. Different frailty phenotypes are differentially associated with adverse events that can worsen the prognosis [123, 124].
Currently, NAFLD has no approved drug therapy, and the objective of treatment is to prevent or reduce the liver injury. In this scenario, the target is to reduce NAFLD related risk factors and hypertriglyceridemia, diabetes, high cholesterol levels and obesity [125].
The first suggested steps are lifestyle and diet changes. It is well known that a Mediterranean diet and balanced physical activity are essential to prevent the progression of NAFLD and incidence of CVD. Through a reduction of body weight, BMI, systolic blood pressure, ALT/AST, fasting glucose, total cholesterol and low-density lipoprotein cholesterol (LDL-C) and fatty liver index and liver stiffness many authors observe a risk reduction of cirrhosis and CVD [126, 127, 128, 129, 130].
The European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO) and American Association for the Study of Liver Diseases (AASLD) practice guidelines for the management of NAFLD recommend that, in overweight/obese NAFLD patients, a 5–10% weight loss is the key goal of lifestyle interventions [17, 131]. Exercise has both direct effects on lipid metabolism and glycemic control, with an increase in VLDL clearance, promoting reduction of liver fat storage [132].
At the same time, these interventions have a direct and indirect effect on CV outcome. In fact, whereas these interventions and lifestyle modifications have shown benefit in patients with NAFLD, reducing liver involvement and consequently its CV effects by indirect impact, the direct role of lifestyle in the prevention of CV disease has been extensively established [133].
In addition to Mediterranean diet and lifestyle changes, some drugs are being evaluated for the treatment of NAFLD, and mainly those who act on CV risk factors [134].
Some authors affirm that intestinal permeability, increased in a condition of
hepatic disease, results in the release of bacterial products (e.g., endotoxins)
in mesenteric circulation, and these products could promote the progression of
liver damage [135]. Randomized clinical trials showed that by decreasing
intestinal permeability and endotoxemia, probiotics could be considered
significant treatment options [136, 137]. Compared to placebo they offered
reduction of fatty liver index, ALT/AST, total cholesterol, homeostatic model
assessment of insulin resistance (HOMA-IR),
(ALT: weighted mean difference (WMD) –23.71, 95% CI: –33.46–13.95, P
Vitamin E is a well-known anti-oxidant agent and has been proposed in this setting, although there is a lack of clinical evidence. In the PIVENS trial, after 96 weeks of treatment, Vitamin E reduces AST/ALT levels, hepatic steatosis and lobular inflammation (P = 0.001, P = 0.005 and P = 0.02 respectively) [141]. Also, in this case, no effect was demonstrated on liver stiffness or on CV risk factors.
Omega-3 fatty acid, by reducing lipogenesis, seems to determine a triglyceride serum and hepatic fat content reduction and consequently a less production of VLDL. Therefore, this drug has been proposed as a theoretically effective drug to reduce CV risk in patients with NAFLD, diabetes or hypertriglyceridemia. Results from REDUCE-IT trial have shown a beneficial effect of icosapent ethyl 4 g/d in addition to statin vs placebo, both on the primary composite end point (major adverse cardiovascular events) (hazard ratio [HR], 0.69 [95% CI, 0.59–0.80]; P = 0.000001), and on secondary CV endpoints. However, similar effects were not achieved in two recent trials, STRENGTH (NCT02104817) and EFFECT I trial (NCT02354976), respectively on the achievement of CV outcome risk reduction and on the reduction of liver fat storage compared to the control group [142, 143, 144]. Gastrointestinal effects, such as abdominal pain and flatulence, were the main reported adverse events, connected to omega-3 fatty acid assumption [142].
Statins, due to the inhibition of 3-hydroxy-3methyl-glutaryl-coenzyme A reductase (HMG-CoA reductase), determines an LDL cholesterol levels reduction, thus leading to a reduction of CV risk [145]. Also for what concerns the use of statins, it seems that these drugs have a beneficial effect on the progression of NAFLD and on the reduction of the related CV risk, despite their potential effect on CK and AST/ALT elevation [146]. However, the use of different endpoints in the different studies does not allow to confirm the efficacy of these drugs on the evolution of NAFLD. Statin and ezetimibe are well known as safe and in a population of subjects not at high CV risk, AST/ALT and LDL cholesterol were significantly reduced at the end of the follow-up [145]. In a recent work conducted on a similar population, a multidrug approach with atorvastatin and Vitamin E and C, allowed a risk reduction of disease progression after a follow up of 3.6 years [147]. Some authors have shown the same effects also in populations with NAFLD and diabetes and hypertension [148, 149]. A sub analysis of the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) study evaluated 437 patients with NAFLD (227 of whom were treated with a statin and 210 were not). The authors observed that NAFLD patients who received statins had lower incidence of CV morbidity, without significant liver-related adverse events [150]. Several studies showed that statin use in individuals with NAFLD, is associated with a considerable improvement of steatosis, inflammation, and liver fibrosis [151, 152]. An observational study of 11,593,409 Korean subjects, showed that the statins were associated with a reduced risk of NAFLD (adjusted odds ratio (OR): 0.66, 95% CI: 0.65–0.67), as well as with a reduced risk of significant liver fibrosis [152]. Interestingly, in a recent systematic review and metanalysis it was described that in the interventional studies, ALT, AST and gamma-glutamyltransferase (GGT) were reduced after statin treatment on average between 25 and 35%, while observational studies showed an effect null, suggesting the hepatic safety of statins in patients with NAFLD [153]. Moreover, genetics may play a role in the pathogenesis of NAFLD/NASH and its treatment with statins [154].
Ezetimibe, a lipid-lowering agent acting by blocking the mediator of cholesterol absorption in the intestine - namely, the Niemann-Pick C1-Like 1 (NPC1L1) protein, showed a beneficial effect on NAFLD. In fact, in a meta-analysis that considered 273 NAFLD patients with and without T2DM, it showed to significantly reduce serum liver biomarkers and hepatic steatosis [155]. It is well-known that the combination of statin and ezetimibe and their effect on LDL-C values is crucial to reduce CV risk. The use of statins, ezetimibe, or their combination strategy is a mainstay in the therapy of patients at CV risk, and their use should be even more strongly recommended in NAFLD patients in light of the liver effects [133]. Moreover, a post-hoc analysis on 14,819 patients enrolled in the IMPROVE-IT study showed that NFS identifies patients at highest risk of recurrent CV events and most likely to benefit from dual lipid-lowering therapy (ezetimibe/simvastatin) [156]. Finally, Ezetimibe is generally well tolerated and also the addition with statins does not increase the risk of side effects [133].
Inhibitors of proprotein convertase subtilisin/kexin type 9 (PCSK9-i), a new class of lipid lowering treatment, promote the activity of LDL receptors, by inhibiting their degradation. Data on PCSK9-i are poor, however, a recent observational study conducted on patients with familial hypercholesterolemia has shown as also these drugs, through their mechanism, particularly in a subgroup with low TG/HDL levels, could determine a reduction of steatosis biomarkers, triglyceride-glucose index (TyG) and hepatic steatosis index (HSI) [157]. Some authors have suggested how these drugs could be an optimal solution to prevent NAFLD and reduce CV risk, however new trials are needed to better define this hypothesis [158].
In diabetic patients with NAFLD, insulin-sensitizing drug therapy is still the preferred choice, and many patients with T2DM are already treated with these drugs. Both metformin and thiazolidinediones have been shown to be useful not only in reducing steatosis, but also in reducing inflammation and fibrosis during treatment [159, 160, 161, 162]. Nevertheless, despite beneficial effects of metformin on liver enzymes and HbA1c levels, a recent metanalysis of randomized controlled trials that involving studies with patients with biopsy-proven NAFLD, showed a small beneficial effect on liver steatosis or inflammation and no effects on the NASH resolution and liver fibrosis [163]. It should be considered that metformin, which has numerous protective extra metabolic effects, can reduce the risk for CV events and death in T2DM patients who are overweight or obese, but the EASL-EASO-EASD and AASLD practice guidelines for the management of NAFLD, to date, do not support the use of metformin for the treatment of NAFLD [17, 131, 164, 165, 166]. On the other hand, the same guidelines recommend the use of pioglitazone in patients with NASH, in fact, pioglitazone has shown to improve advanced fibrosis in NASH patients, regardless of T2DM, and in absence of significant side effects [17, 163, 167]. However, mechanisms related to this beneficial effect on the liver are still unknown.
Recently, new antidiabetic drugs have demonstrated a great efficacy on CV
outcomes [168, 169]. Moreover, some trials have evaluated if these treatments
could have a beneficial effect also on NAFLD progression. In a recent study, both
liraglutide and sitagliptin separately, in a population of subjects with diabetes
and NAFLD, have shown efficacy on the reduction of intrahepatic lipid and
visceral adipose tissue, evaluated through MRI after 24 weeks of treatment
(15.4%
Effects of SGLT2 inhibitors on NAFLD and type 2 diabetes mellitus have been
evaluated in some few studies. The sodium-glucose co-transporter 2 inhibitors
(SGLT2i) through an inhibition of SGLT2 receptors, induce urinary glucose
excretion and consequently a reduction of serum glucose levels and an increase of
insulin sensitivity. The documented adverse events are generally few and mainly
related to urinary tract infections, related to the mechanism of action. Some
animal models suggest that these effects could determine a reduction of liver
lipogenesis and a down regulation of transcription factors that play a key role
in fatty acid synthesis [179]. Dapagliflozin has shown efficacy on the reduction
of liver damage indexes and on the liver disease progression, but also a
reduction of visceral fat as compared to the control group (decrease in CAP from
314
Several specific NAFLD drugs are in development and others are being studied in phase 3 trials [185]. Obeticholic acid (OCA), which is an analogue of chenodeoxycholic acid, is a synthetic ligand agonist of the farnesoid X receptor (FXR) [186]. This class of agent showed to improve insulin resistance, modulate glucose and lipid metabolism, and have direct anti-inflammatory effects in animal NASH models. While showing efficacy on hepatic outcome, this drug appears to increase LDL-C levels, exposing patients to increased CV risk [186, 187]. On the other hand, tight glycemic control improves the CV outcome even in non-diabetic subjects [188, 189]. Moreover, it has recently been documented that a reduction of multiple risk factors significantly improves the risk of fatal and non-fatal major adverse CV events [190].
IONIS-DGAT2Rx is a 20-O-methoxyethyl chimeric antisense oligonucleotide inhibitor that mediates degradation of Diacylglycerol-O acyltransferase 2 (DGAT2) mRNA. DGAT2 is one of two enzyme isoforms involved in the final step of triglyceride synthesis. It has been shown to be effective in improving liver parameters, but its CV safety needs to be better elucidated, as some CV events (i.e., cardiac arrest, ischemic cerebral infarction, deep-vein thrombosis) have been reported [191].
NAFLD should become part of the cardiologist’s background because of its high prevalence, close association with the main CV risk factors and relationship with coronary disease that seem to indicate an autonomous role of NAFLD on the atherosclerosis progression.
In recent years some evidence from clinical trials have shown that bariatric surgery, although not considered the gold standard of treatment, could determine a beneficial effect in patients with NAFLD and liver fibrosis. Notably, it has been shown that patients observed before and after surgery, present a reduction of both steatosis and liver fibrosis, evaluated through liver biopsy. Moreover, in these patients it has been reported an improvement of biochemical parameters (e.g., AST/ALT) [192]. Some authors suggest that the beneficial effect could also improve the CV profile, however there is still no clear evidence on this point and future studies are required to better define this association [193, 194].
The recent literature has extensively documented patients with NAFLD are exposed to an increased risk of progression of liver disease, and a higher risk of extrahepatic complications, particularly CV disease. This risk seems to be conditioned by several factors among which the most relevant is the ability of NAFLD to interfere with cardiometabolic risk factors.
New drugs, evaluated in phase 3 trials for the treatment of NAFLD, can also impact metabolic risk factors such as glycemic control and lipid profile. Therefore, the possible protective effect of these drugs will be evaluated not only on liver damage but also on CV risk.
In conclusion, NAFLD seems to be the epiphenomenon of a systemic disease that can also affect the CV system, requiring a multidisciplinary diagnostic and therapeutic assessment.
FCS, RG, ACa, EV, ACe, PC designed the research study. RG, ACa, EV, ACe, FCS performed the research. ACe, LR provided help and advice on the table and figures; TS, RM, CS, EM, FG analyzed the literature data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Raffaele Galiero was supported by the Programma VALERE, University of Campania “Luigi Vanvitelli”.
This research received no external funding.
The authors declare no conflict of interest.