

 , Le-Feng Wang 1,*,†
, Le-Feng Wang 1,*,†1 Heart Center and Beijing Key Laboratory of Hypertension, Beijing Chaoyang Hospital, Capital Medical University, 100020 Beijing, P. R. China
2 Miyun teaching hospital, Capital Medical University, 101500 Beijing, P. R. China
†These authors contributed equally.
Academic Editor: Peter A. McCullough
Abstract
Cardiovascular
diseases (CVD), especially acute myocardial infarction, are the leading cause of
death, morbidity and disability across the world, affecting millions of people
each year. Atherosclerosis (AS) is the major cause of CVD, and is a chronic
inflammation involving different cell types and various molecular mechanisms.
Ca
Keywords
- TRPC5
- Atherosclerosis
- Cardiovascular disease
- Review
CVD, especially coronary heart disease
(CHD), is associated with high morbidity and mortality worldwide. Presently
mortality and incidence rates among the elderly are increasing [1].
Although significant progress in the diagnosis and treatment of
CVD has been made in recent years, the incidence rate and mortality rate remain
high. Therefore, it is of high clinical significance that new therapeutic targets
of CVD are explored. The intracellular Ca
As the firstly encoded TRP gene family
discovered in mammal, the canonical transient receptor potential channels (TRPCs)
are the most leading non-voltage-gated, Ca
TRPC5 expression has been found in a lot of cell types with inheriting
mechanosensitive Ca
Atherosclerosis (AS) is one of the leading causes of coronary artery disease. EC injury is an essential basis of AS, whilst vascular SMCs (VSMCs), which play a critical role in maintaining blood vessel homeostasis, are also affected in this disease. Therefore the dysfunction of ECs and SMCs play essential roles in the pathogenesis of AS (Fig. 1). In response to injury, VSMCs proliferate, migrate, and release cytokines that can promote and increase inflammation. Interestingly the TRPC family, most commonly TRPC1, TRPC4 and TRPC5, are abundantly expressed in vascular SMCs. Fahy and colleagues (2008) confirmed that sphingosine 1-phosphate (S1P) can activate TRPC5, which is an endogenous signaling phospholipid participating in the migration of VSMCs. Therefore, activation of the TRPC5 channel can significantly promote the proliferation and migration of VSMCs [22]. Immunohistochemical techniques to detect the expression of TRPC5 in ECs and SMCs from murine carotid arteries have shown that blocking TRPC5 led to a great decrease in endothelium-dependent vasoconstriction [23].
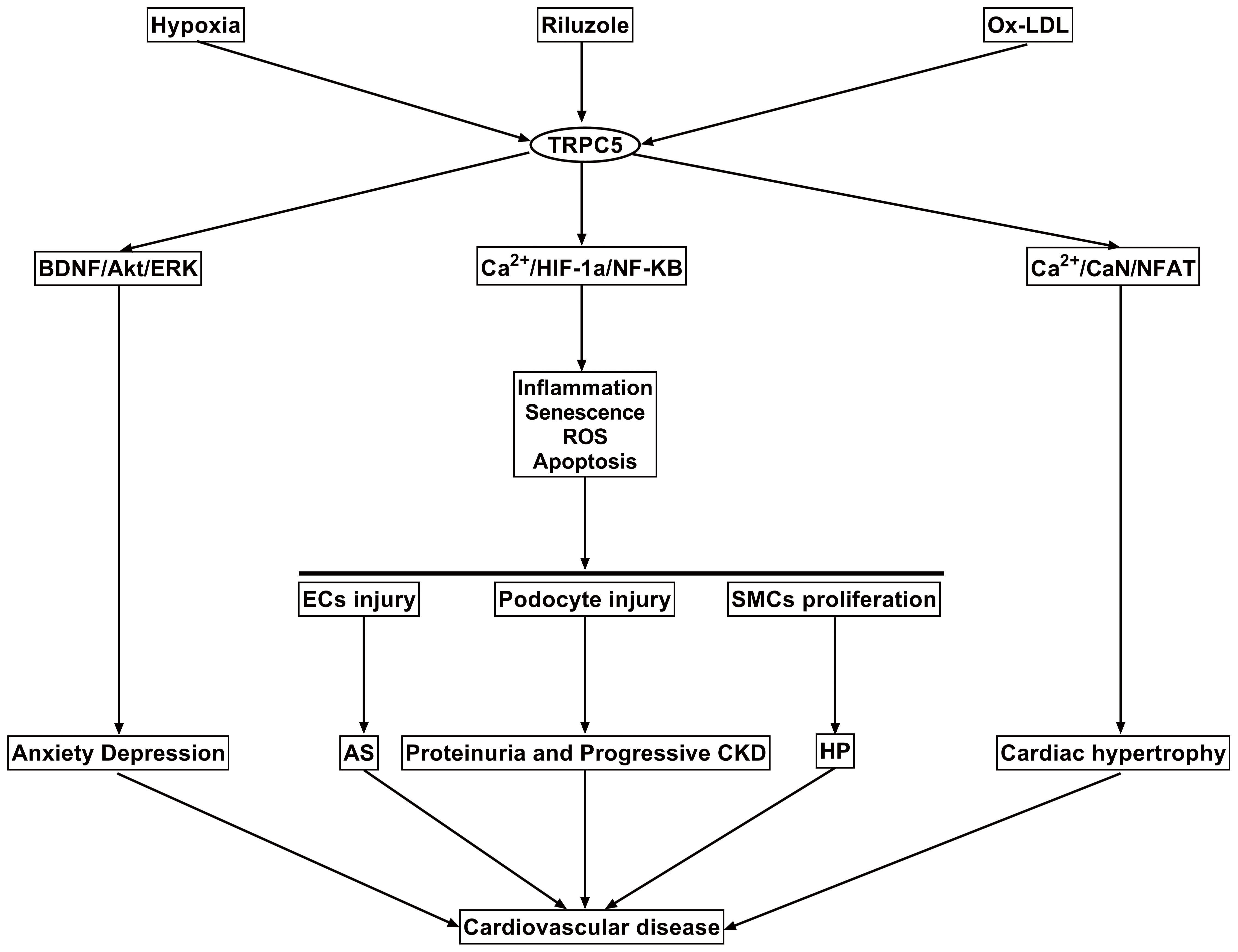 Fig. 1.
Fig. 1.The mechanisms of TRPC5 in the cardiovascular and related
systems are outlined. First, under the stimulation of hypoxia, ox-LDL, or its
activator, TRPC5 can cause endothelial cells damage and dysfunction, smooth
muscle cell proliferation, atherosclerosis and high blood pressure by increasing
inflammatory response, ROS, senescence and apoptosis. TRPC5 can also cause renal
filtration barrier damage by injuring podocyte through the above mechanisms.
Furthermore, TRPC5 can enhance the transcription of cardiac hypertrophy gene by
regulating Ca
Inflammation plays vital roles throughout the entire AS processes ranging from
foam cell accumulation, fatty streak organization and fibrous plaque formation,
through to acute plaque fissuring, rupture, and thrombosis. Numerous evidence
sources define AS as a complicated and systemic pathology where hyperlipidaemia
acts as an important element. The existence of inflammation is necessary for
plaque evolution and destabilization and exerts a crucial effect on the
pathogenesis and progression of coronary artery disease. Therefore it is of
interest that TRPC5 is upregulated in inflammatory diseases. TRPC5 expression has
been positively correlated with eosinophils, interleukin- 6 (IL-6), and
phosphorylation levels of nuclear factor kappa-B (NF-
Dyslipidemia is an independent risk factor for CVD. TRPC5 can promote cholestasis and dyslipidemia [26], while hypercholesterolemia can also positively activate TRPC5 and impair endothelial cell healing [27, 28]. TRPC5 channel protein has the characteristics of a lipid subtype receptor, which can recognize lysophosphatidylcholine as the main component of oxidation low lipoprotein (ox-LDL) and is activated by S1P in ox-LDL. The functioning mechanism of Statins, a commonly used lipid-lowering drug, is enabling delayed progression of AS by downregulating the TRPC5 protein expression. TRPC5 plays a vital role in the process of atherosclerosis mediated by hyperlipidemia, therefore blocking TRPC5 can delay the process of atherosclerosis.
Endothelial cell apoptosis is another critical step in the pathogenesis of AS
[29]. Phospholipid scramblase 1 (PLSCR1) critically participates in
phosphatidylserine (PS) externalization, a vital process in cell apoptosis. TRPC5
can mediate apoptosis by interacting with PLSCR1 and hypoxia-inducible
factor-1
Most
patients with acute myocardial infarction receive routine treatment with
pharmacological reperfusion therapy and/or widening of the vessel with
angioplasty. Nevertheless, critical injuries happen when the vulnerable
myocardial tissue is reperfuses by oxygen-rich blood notably via mass
manufacturing of mitochondrial reactive oxygen species. This situation is called
ischemia/reperfusion (I/R) syndrome [33].
Apoptosis and tissue damage caused by ischemia-reperfusion are the leading causes
of death in patients with fatal diseases such as myocardial infarction and
stroke. According to the study, the growth in cytosolic Ca
Therapeutic angiogenesis promoting blood
flow is essential while curing ischemia-related diseases [37, 38, 39], therefore TRPC5
may also have the potential to treat ischemic diseases. By activating
HIF-1
In early stage of heart failure (HF),
changes in the expression and activity of Ca
Recently, a lot of researches have paid
attention to the relationships between initial hypertension and TRPCs [50].
Members of the TRPC subfamily are involved in primary hypertension due to the
Ca
As a polymodal channel enriched in neuronal cells, TRPC5 also localizes to the aortic baroreceptor termini, sensory neuronal termini for blood pressure detection [12]. In addition, TRPC5 can be directly activated by membrane stretch [21], which is independent of phospholipase C. In TRPC5 knockout mice, the pressure-induced action potential firings in the afferent nerve and the baroreflex-mediated heart rate reduction were attenuated. Telemetric measurements of blood pressure demonstrate that these TRPC5 knockout mice also display severe daily blood pressure fluctuations [60]. This suggests that TRPC5 channels stand for a core pressure transducer in the baroreceptors and exert a primary role in keeping blood pressure stable.
Therefore, in relation to the aspect of prevention and treatment of hypertension, could blocking TRPC5 reduce blood pressure and offset the blood pressure fluctuation caused by it? We look forward to further support from future relevant research.
Intracellular calcium ion (Ca
Additionally, oxidizing agents can directly activate TRPC5 channels through cysteine modification [69]. This implies that TRPC5 may be a useful target for the prevention of AF. But more researches on TRCP5 in arrhythmias are needed in the future.
According to various researches, CVD is closely associated with mental health disorders. As an independent risk factor of CVD, emotional disorders play vital roles in the occurrence and development of CVD. The effective control of depression in patients with CVD, for example, has a potential auxiliary effect on the prevention of various types of CVD, such as coronary heart disease and heart failure. TRPC5 is highly expressed in the hippocampus and amygdala, which can regulate anxiety. Within the nervous system, one of the most studied fields relating to TRPC5 is its anti-anxiety and anti-depression effects. The evidence for this effect is obtained from studies using transgenic mouse models and pharmacological modulators. For example, M084, a TRPC5 inhibitor, plays a rapid anti-depressant and anti-anxiety role by increasing the expression of brain-derived neurotrophic factor (BDNF) mRNA and protein in the prefrontal cortex, as well as increasing the phosphorylation level of protein kinase B (Akt) and extracellular regulated protein kinases (ERK) [70]. Following TRPC5 molecule blocker treatment in experimental mice, behavioural evaluation showed that the inhibitor had the potential for anti-anxiety and anti-depression effects [17, 71] (Fig. 1). This suggests that TRPC5 has potential for not only treating nervous system diseases, but also as an additional treatment for CVD.
Renal barrier dysfunction is a risk factor for CVD and kidney disease. In the treatment of coronary heart disease, drugs and devices inevitably produce different degrees of renal damage. Acute renal injury caused by the use of contrast medium in interventional surgery, i.e. contrast medium nephropathy, is a common clinical focus in both heart and kidney disease. TRPC5 is also involved in kidney disease [72]. Podocytes are special cells that form the filtration membrane of kidney. TRPC5 channels are expressed in podocytes and are involved in the regulation of cell migration and actin remodeling stimulated by angiotensin [73] (Fig. 1). Schaldecker and colleagues confirmed that TRPC5 is the main pathway for lipopolysaccharide (LPS) induced proteinuria. Application of TRPC5 inhibitors can block LPS induced proteinuria, and protect renal podocytes from LPS induced microfilament remodeling [74]. Yiming Zhou (2017) applied a TRPC5 inhibitor to treat rats with glomerulonephritis, and achieved the same results. AC1903, a small molecule inhibitor of TRPC5 channel, inhibited severe proteinuria in focal segmental glomerular sclerosis (FSGS) transgenic rats and prevented podocyte loss. AC1903 also had a therapeutic effect on hypertensive proteinuria in rats. These trials suggest that TRPC5 inhibitors may provide significant value during the treatment of hypertensive renal injury [75]. GFB-8438, another more recent TRPC5 inhibitor, significantly reduced urinary total protein and albumin concentrations and protected podocytes from protamine sulphate (PS) injury [76]. Hypertension acts as an ordinary complication among hemodialysis patients during erythropoietin (EPO) treatment. EPO increased the stability of TRPC5 mRNA and the concentrations of TRPC5 channel protein. Additionally, TRPC5 gene knockout can reduce the generation of reactive oxygen species in endothelial cells induced by EPO [32]. This indicated that upregulated functional TRPC5 gene may act as one of the causes of EPO-induced hypertension among patients with chronic kidney disease. Therefore, inhibition of TRPC5 may protect renal filtration barrier from acute reversible injury and has a great potential in the field of heart and kidney treatment.
In contrast to other studies, the protective role of blocking TRPC5 in progressive kidney disease was not supported in transgenic mice overexpressing either WT TRPC5 or a dominant-negative TRPC5 mutant [77]. No differences in LPS-induced kidney damage were observed among the different groups, and there is no effect of the treatment with the inhibitor ML204 on proteinuria in LPS-challenged animals. And treatment with the TRPC5 activator (-)EA exerted no bad influence on proteinuria in mice. Nevertheless, due to the poor stability of (-)EA in plasma [78], it is still controversial as to whether the plasma concentration of (-)EA can activate the TRPC5 channel and lead to renal injury. In addition, as van der Wijst and Bindels [79] mentioned, the dosage and administration scheme of ML204 used in the above studies are different, which may lead to differential results. In order to discriminate and conclude potential actions, the roles of TRPC5 in renal diseases shall be elucidated by further studies.
Over the last 20 years, research into TRPC5 has been limited to relatively basic experiments and clinical translational research is proceeding very slowly. This may be due to the following reasons: Firstly, TRPC5 channels are widely distributed around the human body, and inhibition of TRPC5 may lead to various complications. Since all TRPC channels, including TRPC5, are commonly distributed throughout the body and can also regulate a variety of cellular functions, the adverse effects of nonselective inhibition, such as significantly increased bleeding time [80] and cognitive impairment [81], could become a major obstacle to the use of TRPC blockers. Therefore, due to safety reasons, clinical translation of TRPC5 inhibitors have progressed slowly. Secondly, current research on TRPC5 have mainly focused on cell lines, and gene knockout or overexpression models in vivo. There is a gap between these studies and those needed in humans. Prior to clinical drug development, it is necessary to demonstrate how TRPC5 is altered in human biopsy samples. Thirdly, from the perspective of the current clinical development history of drugs targeting ion channels, such as Bolapamil, lidocaine and diazepam, early research results were restricted because of the shortage of specificity and significant off-target effects. This also restricted their clinical applicability [82]. Consequently researchers need to develop drugs that are more specific and have fewer off-target effects. Fourthly, the specific roles and mechanisms of TRPC5 in heart disease are still unclear, and further researches shall be made to illustrate the molecular mechanisms (including key regulatory sites and signaling pathways) of TRPC5 channels involved in cardiovascular disease. This will help to design drugs specifically targeting the TRPC5 channels. Finally, TRPC5 channels are abundantly expressed in the brain and exert a significant effect on signal transmission within the nervous system. Therefore, TRPC5 inhibitors that cannot pass the blood-brain barrier should be developed, thus ensuring they can be safely used in clinical practice.
Translational research needs to be combined with research in clinical and pharmaceutical settings in addition to other fields. Therefore, we believe that the future research direction of TRPC5 is as follows: (1) Search for highly specific inhibitors to reduce the incidence of side effects. (2) Validate the results in more animal models, preferably those most similar to humans. (3) Clarify the roles and molecular mechanisms of TRPC5 in cardiovascular diseases. (4) Regulate molecules in the upstream and downstream signaling pathways of the TRPC5 channel.
Currently, several small molecular compounds have been used in basic experiments, such as HC-070 [17], ML204 [83], AC1903 [75], in addition to others. Laboratory studies of these smaller molecular compounds may provide valuable insights into the roles of TRPC5 within both cells and animal disease models. This is beneficial for the development of new drugs targeting TRPC5. Also, to conveniently detect the expression of TRPC5 under different conditions, researchers have synthesized a TRPC5 PET radiotracer, [11C]HC608, to quantify TRPC5 expression changes in the brain [84]. Unfortunately, to dtae, there have been no similar studies in the heart. These approaches would benefit our understanding of the roles of TRPC5 in the heart, especially if PET-CT and tracers were used for quantification of TRPC5 expression in cardiovascular diseases.
We have focused on the critical effect of TRPC5 in multi-organ areas, especially
within the cardiovascular and AS fields. By regulating intracellular Ca
| Role | Reference | Object | Outcome | Effects of blocking TRPC5 |
| inflammation | [24] | patient | TRPC5 expression was positively correlated with eosinophils, IL-6, and phosphorylation levels of NF-κB. | anti-inflammatory |
| VSMC | [22] | animal cell | TRPC5 mediates VSMC migration. | anti-VSMC migration |
| dyslipidemia | [25] | animal | TRPC5 contributes to the development of cholestasis and dyslipidemia. | regulate blood lipids |
| apoptosis | [30, 31] | animal cell | TRPC5-PLSCR1 is a signaling complex mediating PS externalization and apoptosis, decreased TRPC5 protein may resist cell apoptosis injured by CoCl |
anti-apoptosis |
| oxidative stress | [32] | patients cell | Knockdown of TRPC5 alleviated EPO-induced reactive oxygen species generation in endothelial cells. | Antioxidant stress |
| I/R injury | [30, 36] | animal cell | Reduced I/R-induced adverse cardiac remodeling was associated with decreased TRPC5 expression. | Reduce I/R injury |
| angiogenesis | [40, 41, 42, 43] | animal cell | TRPC5 mediated HIF-1 |
Reduce angiogenesis, which is detrimental to ischemic disease |
| cardiac hypertrophy | [36] | animal cell | Reduced I/R-induced adverse cardiac remodeling was associated with decreased TRPC5 expression. | Anti-cardiac hypertrophy |
| blood pressure | [60] | animal | TRPC5 knockout mice display severe daily blood pressure fluctuation, suggest TRPC5 channels stand for a core pressure transducer in the baroreceptors and exert a significant effect on keeping blood pressure stable. | Display severe daily blood pressure fluctuation |
| cardiac rhythm | [63] | patient | TRPC5 gene expressions in NVAF patients had a marked increasing. | May be beneficial to the treatment of NVAF |
| emotion | [70, 71] | animal | TRPC5 |
anti-anxiety, anti-depression |
| kidney | [74, 75] | animal | TRPC5 mediates filtration barrier injury. genetic deletion or pharmacologic inhibition of TRPC5 protected mice from albuminuria and podocyte loss. | Protect kidney podocyte, reduce albuminuria |
| EPO = erythropoietin; HIF-1 | ||||
Du Sheng-li and Jia Zeng-qin conducted literature search and wrote the paper. Wang le-feng and Zhong Jiu-chang reviewed and edited the manuscript.
This work was supported by the Digestive Medical Coordinated Development Center of Beijing Hospitals Authority, No.XXZ0607.
The author declares no conflicts of interests.