


 , Norma Serrano-Garcia 1,†, Ricardo Pérez-Rubio 1,2, Javier Pérez-Villavicencio 1,3, Héctor Romo-Parra 1,4, Ángel Lee 5, Moisés Rubio-Osornio 6,*
, Norma Serrano-Garcia 1,†, Ricardo Pérez-Rubio 1,2, Javier Pérez-Villavicencio 1,3, Héctor Romo-Parra 1,4, Ángel Lee 5, Moisés Rubio-Osornio 6,*
1 Department of Neurophysiology, National Institute of Neurology and Neurosurgery, 14269 Mexico City, Mexico
2 Mexican Faculty of Medicine, La Salle University, 14000 Mexico City, Mexico
3 Department of Electrical Engineering, Basic Sciences and Engineering Division Metropolitan Autonomous University, Iztapalapa Campus, 09340 Mexico City, Mexico
4 Department of Psychology, Ibero-American University, Santa Fe Campus, 01376 Mexico City, Mexico
5 National Institute of Public Health, Cuernavaca, 62100 Morelos, Mexico
6 Department of Neurochemistry, National Institute of Neurology and Neurosurgery, 14269 Mexico City, Mexico
†These authors contributed equally.
Abstract
Antiseizure medications (ASMs) have traditionally been characterized by their modulation of neuronal ion channels and synaptic processes; however, accumulating evidence indicates that numerous ASMs also directly modulate mitochondrial function. Specifically, several ASMs interact with ion channels located in both the inner and outer mitochondrial membranes, including the voltage-dependent anion channel (VDAC), the mitochondrial calcium uniporter (MCU), the mitochondrial Na+/Ca2+ exchanger (NCLX), the mitochondrial permeability transition pore (mPTP), and mitochondrial ATP-sensitive potassium channels (mitoKATP). Modulation of these channels regulates critical processes in epilepsy, including Ca2+ homeostasis, ATP synthesis, redox equilibrium, and susceptibility to neuronal apoptosis. Phenytoin and carbamazepine reduce voltage-dependent anion channel isoform 1 (VDAC1)-associated mitochondrial permeability by modulating the Bcl-2-associated X protein (Bax)/B-cell lymphoma 2 protein (Bcl-2) ratio; ethosuximide limits mitochondrial Ca2+ overload through modulation of the MCU complex; valproic acid stabilizes NCLX function and prevents mPTP opening via antioxidant mechanisms; levetiracetam contributes to preserving intracellular Ca2+ handling; and mitoKATP activators, including diazoxide and retigabine, promote mitochondrial membrane potential stability and reduce seizure-induced reactive oxygen species (ROS) generation. The mitochondrial effects vary according to epilepsy subtype, contributing to the attenuation of hippocampal apoptosis in temporal lobe epilepsy and thalamocortical network modulation in generalized epilepsies. In this narrative review we examine the experimental and molecular evidence demonstrating how ASMs modulate mitochondrial ion channels and how these interactions contribute to their anticonvulsant mechanisms, thereby broadening the understanding of mitochondria as key functional components in antiseizure pharmacology.
Keywords
- mitochondria
- anticonvulsants
- voltage-dependent anion channels
- ion channels
- mitochondrial membrane transport proteins
- epilepsy
- reactive oxygen species
- calcium signaling
Epilepsy encompasses a diverse array of neurological conditions characterized by a chronic propensity to generate epileptic seizures arising from aberrant, excessive, and hypersynchronous neural activity [1,2]. Seizures arise from a neurobiological imbalance between excitatory and inhibitory mechanisms, including disruptions in glutamatergic and GABAergic transmission, variations in intrinsic neuronal excitability, and reconfigurations of neuronal networks [3,4,5].
Despite the development of numerous antiseizure medications (ASMs), approximately one-third of patients continue to suffer from drug-resistant epilepsy, highlighting significant limitations in current therapeutic approaches and pointing to the potential contribution of incompletely understood cellular mechanisms [6,7]. ASMs have traditionally been classified according to their ability to modulate voltage-gated neuronal ion channels, specifically sodium, calcium, and potassium channels as well as their effects on inhibitory or excitatory synaptic neurotransmission [8,9].
These mechanisms partially explain the reduction of neuronal hyperexcitability; however, they do not fully account for the neuroprotective benefits reported with various pharmacological agents or the clinical heterogeneity observed across patients and epilepsy subtypes [10,11]. More recently, a new paradigm has emerged acknowledging the mitochondria as a pivotal regulator of neuronal excitability [12,13]. Beyond its well-established role in energy production, the mitochondria plays a critical function in intracellular calcium regulation, formation of reactive oxygen species (ROS), redox signaling, and the initiation of apoptotic pathways [14,15]. These processes are closely associated with the pathophysiology of epileptic seizures [16].
The study of mitochondrial ion channels therefore represents both a conceptual and therapeutic priority, as these systems govern the interplay between cellular metabolism and brain electrical activity [17,18]. Understanding how ASMs interact with these channels extends the traditional view of their mechanisms of action and helps explain the anticonvulsant and neuroprotective effects that were previously attributed solely to synaptic mechanisms [19,20].
This narrative review synthesizes the molecular and experimental evidence supporting the role of mitochondrial ion channels in epilepsy and elucidates how various ASMs modulate these structures, thereby contributing to their anticonvulsant efficacy. Representative preclinical, mechanistic, and pharmacological studies were selected on the basis of their relevance to the interaction between ASMs and mitochondrial ion channels, without applying formal systematic search or risk-of-bias appraisal protocols.
The present review focuses primarily on classical ASMs (phenytoin, carbamazepine, ethosuximide, valproate, levetiracetam) and on selected mitochondrial-channel modulators (diazoxide, retigabine), as these agents possess the most extensively characterized preclinical evidence regarding interactions with specific mitochondrial ion channels; mechanistic data on most newer-generation ASMs remain comparatively scarce, an issue revisited in Section 5.
Neuronal excitability is closely linked to cellular metabolic status [21,22]. Mitochondria serves as a central nexus connecting electrical activity with energy supply and intracellular ionic regulation, allowing neurons to maintain elevated activity levels without compromising cell viability [23,24].
The relationship between metabolism and neural excitability depends on the mitochondria’s ability to modulate energy production according to synaptic requirements [12,25]. Intense neural activity increases adenosine triphosphate (ATP) consumption, which activates oxidative phosphorylation supporting the activity of ion pumps like Na+/K+-ATPase and calcium-transporting ATPase (Ca2+-ATPase), crucial for reestablishing electrochemical gradients following depolarization [26,27]. During epileptiform episodes, energy demand increases sharply [28]. Mitochondria augment ATP synthesis through calcium-dependent activation of the Krebs cycle enzymes [29,30]. Excessive or sustained stimulation, however, overwhelms bioenergetic capacity, leading to metabolic dysfunction and promoting further neuronal discharges [16,31].
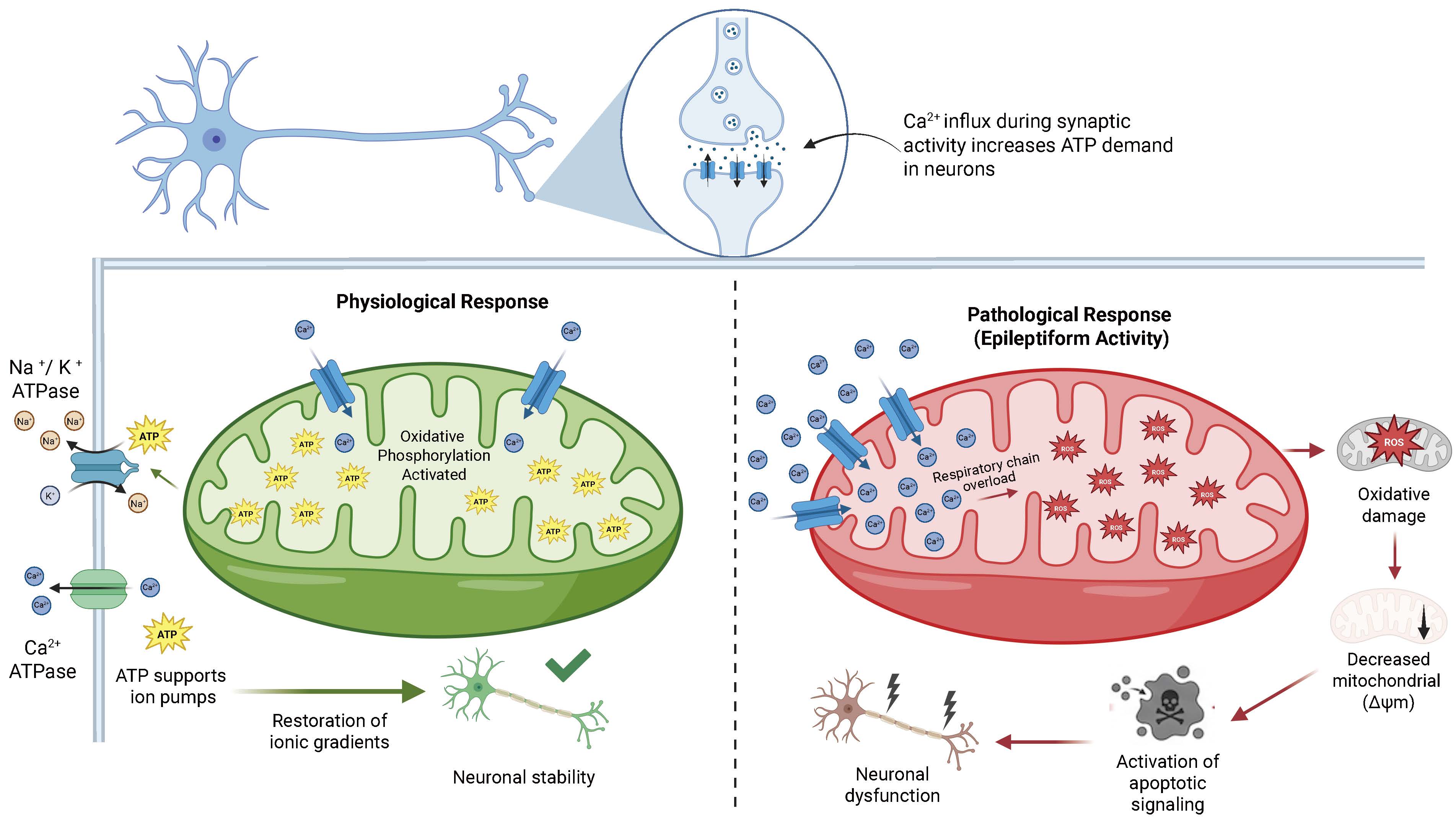
Epileptic seizures markedly increase ROS generation because of respiratory chain overload [32,33]. The excess of free radicals induces oxidative damage to lipids, proteins, and mitochondrial deoxyribonucleic acid (DNA), impairing energy efficiency and establishing a detrimental cycle between oxidative stress and neuronal excitability [34,35]. Mitochondrial Ca2+ homeostasis is a fundamental component of this regulatory mechanism [36,37]. Mitochondria buffer synaptically generated calcium to prevent excessive cytosolic accumulation [38,39]. In hyperexcitable neurons, sustained mitochondrial Ca2+ accumulation induces metabolic alterations, dissipates membrane potential, and triggers apoptotic pathways [40,41]. The dual role of mitochondria as both supporters of physiological neuronal activity and contributors to seizure-related injury is illustrated in Fig. 1, which contrasts the adaptive bioenergetic responses observed under normal conditions with the cascade of mitochondrial dysfunction including ROS overproduction, membrane potential collapse, and apoptotic signaling that characterizes epileptiform activity.
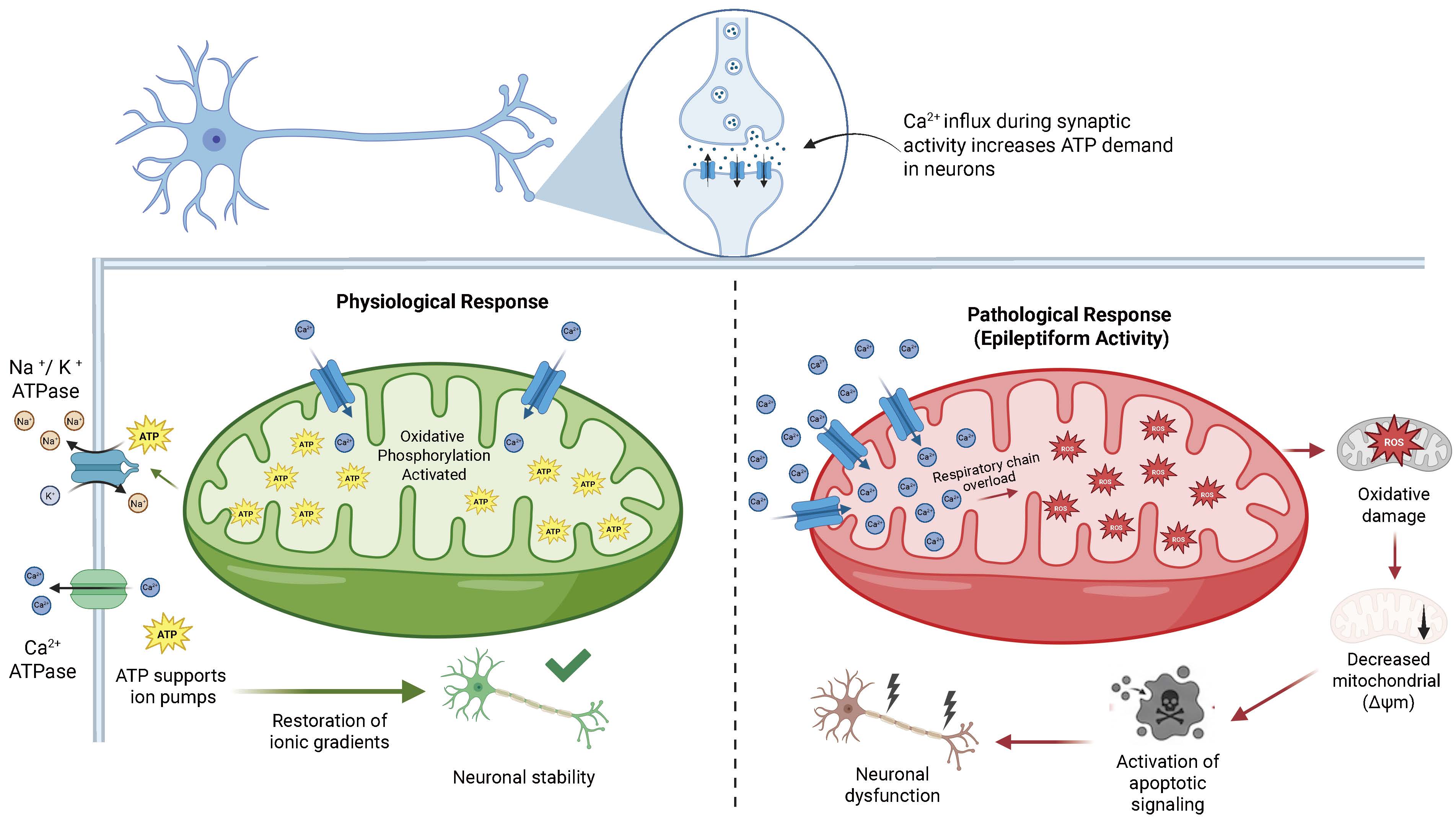 Fig. 1.
Fig. 1.Mitochondrial regulation of neuronal excitability under physiological and epileptiform conditions. Under physiological conditions (left), activity-dependent Ca2+ influx increases metabolic demand and stimulates mitochondrial oxidative phosphorylation, enhancing adenosine triphosphate (ATP) production to support ion transport mechanisms including Na+/K+-ATPase and Ca2+-ATPase that restore electrochemical gradients and maintain neuronal stability. Under pathological conditions such as epileptiform activity (right), excessive mitochondrial Ca2+ accumulation leads to respiratory chain overload and increased generation of reactive oxygen species (ROS). These alterations promote oxidative damage, mitochondrial membrane potential dissipation (loss of mitochondrial membrane potential ΔΨm), and activation of apoptotic signaling cascades, ultimately resulting in neuronal dysfunction and injury. Created in BioRender. Pérez-Rubio, R. (2026) https://BioRender.com/x23h795. ATP, adenosine triphosphate; ΔΨm, mitochondrial membrane potential.
Mitochondrial ion channels are specialized protein complexes that govern the exchange of metabolites and ions between the mitochondria and the cytoplasm, thus regulating cellular bioenergetics and neuronal survival. Biochemical, electrophysiological, and structural studies have established that these channels regulate ATP synthesis, calcium homeostasis, and apoptotic signaling, thereby positioning the mitochondria as a central hub for metabolic integration and the determination of cellular fate [17,36,42,43]. The voltage-dependent anion channel (VDAC), situated in the outer mitochondrial membrane, functions as the principal conduit for metabolic exchange between the cytosol and the intermembrane space [44,45]. Early studies using lipid bilayer reconstitution demonstrated that VDAC facilitates the transport of nucleotides such as ATP and adenosine diphosphate (ADP), as well as essential respiratory metabolites [46,47]. Subsequent studies demonstrated that VDAC serves as a platform for interactions with apoptosis-regulating proteins including Bcl-2 associated X protein (Bax), Bcl-2 homologous antagonist/killer (Bak), and B-cell lymphoma 2 protein (Bcl-2) thereby influencing outer membrane permeabilization and cytochrome c release [48,49]. These interactions directly link mitochondrial metabolic status to the activation of intrinsic apoptotic pathways, particularly in neurons exposed to oxidative stress or excitotoxicity [50,51].
The mitochondrial calcium uniporter (MCU) complex resides in the inner mitochondrial membrane and serves as the principal conduit for Ca2+ influx into the mitochondrial matrix. Its molecular identification through genetic and proteomic analyses confirmed its critical role in coupling cytosolic calcium signaling to mitochondrial energy metabolism [52,53,54]. MCU activity is precisely regulated by EF-hand Ca2+-sensing proteins, including mitochondrial calcium uptake protein 1 (MICU1) and mitochondrial calcium uptake protein 2 (MICU2), which establish a Ca2+-dependent activation threshold that prevents mitochondrial overload under basal conditions while permitting rapid Ca2+ uptake during physiological calcium transients [55,56,57]. This regulatory mechanism is especially important in neurons, where calcium-dependent signaling governs synaptic plasticity and ATP synthesis [58,59].
The mitochondrial sodium-calcium exchanger (NCLX) serves as the primary transporter mediating calcium efflux from mitochondria. Functional studies indicate that NCLX maintains mitochondrial calcium homeostasis by extruding Ca2+ accumulated during intense neuronal activity, thereby preventing respiratory chain dysfunction and excessive ROS production [60,61]. Experimental models have shown that NCLX suppression or genetic ablation leads to mitochondrial calcium overload, loss of membrane potential, and neuronal death, underscoring its neuroprotective role [62,63]. The mitochondrial permeability transition pore (mPTP) is a multiprotein complex whose opening is regulated by the cellular redox state, calcium levels, and metabolic conditions [64]. Seminal studies have shown that sustained mPTP opening results in dissipation of the mitochondrial membrane potential, osmotic swelling, and the release of pro-apoptotic factors, including cytochrome c and apoptosis-inducing factors (AIF) [65,66]. Recent studies have identified ATP synthase and cyclophilin D (CypD) as critical regulatory elements of the pore, associating its activation with neurodegenerative mechanisms and excitotoxic injury [67,68,69,70]. Mitochondrial ATP-sensitive potassium channels (mitoKATP) are involved in regulating mitochondrial membrane potential and facilitating metabolic adaptability under cellular stress. Pharmacological studies and ischemia models have demonstrated that mitoKATP activation induces mild mitochondrial depolarization, which limits excessive ROS production and attenuates mPTP opening, thereby promoting cellular preconditioning and enhancing neuronal survival [71,72,73]. This protective effect has been extensively documented in neurodegenerative disease and brain ischemia models, where mitoKATP activation sustains mitochondrial function during energy-stress conditions. Accumulated data indicate that mitochondrial ion channels regulate metabolite and ion flux while also integrating metabolic, redox, and apoptotic signals, serving as crucial components in neuronal homeostasis and the pathogenesis of neurodegenerative disorders. The structural diversity and functional specialization of these channels are summarized in Table 1 (Ref. [46,48,50,52,53,56,58,60,61,62,64,65,67,69,71,72,73,74,75]), which provides a comparative overview of each channel’s subcellular localization, transported ions or molecules, physiological roles, and specific relevance to epilepsy-related pathology.
| Channel/pore | Mitochondrial location | Transported ions/molecules | Physiological role | Relevance in epilepsy | Key references |
|---|---|---|---|---|---|
| VDAC (1/2) | Outer membrane | Ca2+, ATP, ADP, metabolites | Metabolite exchange between cytosol and mitochondria and regulation of outer membrane permeability | Regulation of outer mitochondrial membrane permeability and apoptotic signaling pathways | [46,48,50,74] |
| MCU complex | Inner membrane | Ca2+ (influx) | Mediates mitochondrial Ca2+ uptake linking cytosolic Ca2+ signaling with mitochondrial metabolism | Excessive Ca2+ uptake contributes to mitochondrial dysfunction during seizures | [52,53,56,58] |
| NCLX | Inner membrane | Na+ (influx) and Ca2+ (efflux) | Maintains mitochondrial Ca2+ homeostasis through Ca2+ extrusion | Prevents Ca2+ overload and subsequent neuronal degeneration | [60,61,62] |
| mPTP | Inner membrane complex | Non-selective solutes | Regulates mitochondrial membrane permeability under metabolic stress | Persistent opening induces mitochondrial depolarization and apoptotic pathways | [64,65,67,69] |
| mitoKATP | Inner membrane | K+ (influx) | Modulates mitochondrial membrane potential and limits excessive ROS generation | Activation reduces ROS generation and enhances neuronal survival | [71,72,73,75] |
Summary of the principal mitochondrial ion channels, including their subcellular localization, transported ions or molecules, physiological roles in mitochondrial bioenergetics and calcium homeostasis, and their relevance in neuronal excitability and epilepsy. Abbreviations: VDAC, voltage-dependent anion channel; MCU, mitochondrial calcium uniporter; NCLX, mitochondrial Na+/Ca2+ exchanger; mPTP, mitochondrial permeability transition pore; mitoKATP, mitochondrial ATP-sensitive potassium channel; ADP, adenosine diphosphate.
Numerous experimental studies have demonstrated that epileptic seizures are associated with profound changes in mitochondrial channel activity. A prominent finding is mitochondrial Ca2+ overload driven by excessive calcium influx during prolonged neural activity. This condition impairs oxidative metabolism and facilitates the activation of cell death pathways. Moreover, the dissipation of mitochondrial membrane potential is recognized as an early indicator of bioenergetic compromise. This change reduces ATP production and impairs the ability of neurons to restore ionic equilibrium following seizures [76].
Apoptotic activation mediated by VDAC and mPTP represents another critical pathway. Mitochondrial outer membrane permeabilization promotes cytochrome c release and caspase activation, thereby contributing to neurodegeneration, particularly in hippocampal regions. Simultaneously, persistently elevated ROS levels induce cumulative oxidative damage that exacerbates bioenergetic failure and promotes seizure recurrence, creating a pathogenic cycle between metabolic stress and neuronal excitability [77]. Beyond these acute alterations, growing preclinical evidence implicates mitochondrial channels in epileptogenesis. This term refers to the latent process by which an initial brain insult, such as status epilepticus, traumatic brain injury, or prolonged febrile seizures, gradually transforms a normal neuronal network into a chronically hyperexcitable circuit [16,28]. Longitudinal studies in chemoconvulsant models have shown that mitochondrial oxidative damage and Complex I inactivation persist beyond the acute phase. These alterations emerge during the seizure-free latent period and continue into chronic epilepsy [78,79]. Within this window, sustained Ca2+ overload, mPTP sensitization, and VDAC1-mediated apoptotic signaling contribute to selective hippocampal neuronal loss and to the synaptic reorganization that underlies hippocampal sclerosis [80,81]. Direct human evidence remains limited, and most currently available ASMs lack demonstrated antiepileptogenic activity. This unmet need positions mitochondrial channels as candidate targets for disease-modifying intervention rather than purely symptomatic treatment [34,82].
A genetic dimension underlies many of these mechanisms. Most mitochondrial channels discussed in this review, including VDAC, MCU, NCLX, mitoKATP, and the principal regulators of the mPTP, are encoded by nuclear DNA. By contrast, mitochondrial DNA (mtDNA), encodes only thirteen respiratory chain subunits, two ribosomal RNA (rRNA), and twenty-two transfer RNA (tRNA). Pathogenic mtDNA variants therefore rarely mutate these channels directly. Instead, they impair channel function indirectly by compromising oxidative phosphorylation, depolarizing the inner mitochondrial membrane, and increasing matrix Ca2+ and ROS levels. These changes sensitize the mPTP, alter VDAC gating, and disturb Ca2+ handling through mitoKATP, MCU, and NCLX [16,28]. This convergent mechanism underlies the epileptic phenotypes of the major mitochondrial encephalopathies, including Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes (MELAS) (m.3243A>G in MT-TL1), Myoclonic Epilepsy with Ragged-Red Fibers (MERRF) (m.8344A>G in MT-TK), and Leigh syndrome. In these disorders, myoclonic, focal, or generalized seizures reflect cumulative bioenergetic failure and secondary channel dysfunction in cortical and hippocampal circuits [83,84]. A more direct mtDNA–channel link is observed in MT-ATP6 mutations responsible for NARP and maternally inherited Leigh syndrome, which disrupt mitochondrial ATP synthase Complex V or F1Fo-ATP synthase, a complex structurally involved in mPTP formation. This represents a plausible monogenic basis for mPTP dysregulation in epilepsy [65]. Finally, nuclear DNA polymerase gamma catalytic subunit (POLG) variants associated with Alpers-Huttenlocher syndrome extend this paradigm through secondary mtDNA depletion. These variants contribute both to refractory epilepsy and to the well-documented susceptibility to valproate-induced hepatotoxicity [85,86]. Direct human evidence linking common polymorphisms in nuclear-encoded mitochondrial channel genes such as VDAC1/2/3, MCU, MICU1/2, solute carrier family 8 member B1 (gene encoding NCLX) (SLC8B1)/ (SLC8B1/NCLX), and potassium inwardly rectifying channel subfamily J members 8 and 11 (KCNJ8/11) to epilepsy susceptibility remains scarce. Large-scale exome and whole-genome sequencing efforts in epilepsy cohorts have prioritized synaptic ion channel genes such as sodium voltage-gated channel alpha subunit 1 (SCN1A), potassium voltage-gated channel subfamily Q member 2 (KCNQ2), and Gamma-aminobutyric acid type A receptor subunit alpha 1 (GABRA1), whereas mitochondrial channel loci have been comparatively overlooked. Most existing reports consist of small case series or candidate-gene studies without adequate replication. Subtle variation in these genes may still modulate seizure susceptibility, ASM response, or disease progression, although this possibility remains untested. Targeted resequencing and functional validation of mitochondrial channel genes in well-phenotyped epilepsy cohorts therefore represents a logical next step toward integrating mitochondrial bioenergetics into the genetic architecture of epilepsy and refining stratification for personalized therapy.
Accumulating evidence suggests that various ASMs directly or indirectly modulate mitochondrial function and ion channels, thereby enhancing their anticonvulsant and neuroprotective properties through the regulation of energy metabolism, calcium homeostasis, and apoptotic signaling [87]. Several studies have demonstrated that ASMs can interact with mitochondrial transport proteins, modulate membrane potential, and affect antioxidant systems, thereby influencing mechanisms essential for neuronal survival during hyperexcitable states [87,88]. Before examining each drug individually, Fig. 2 provides a schematic reference of the spatial organization of these channels within the outer and inner mitochondrial membranes, depicting how VDAC, MCU, NCLX, mitoKATP, and the mPTP are anatomically and functionally positioned to serve as pharmacological targets during epileptiform activity. Beyond their well-established blockade of voltage-gated sodium channels, phenytoin and carbamazepine indirectly modulate mitochondrial permeability by shifting the balance between pro-apoptotic and anti-apoptotic proteins that regulate VDAC1 function [87]. Modulation of these systems reduces cytochrome c release and attenuates the activation of apoptotic pathways associated with neuronal excitotoxicity induced by prolonged seizures [87,89]. VDAC1 is a critical regulatory node for metabolic exchange between the cytosol and mitochondria, and its pharmacological modulation contributes to stabilizing neuronal bioenergetics during epileptic episodes [74,87]. Ethosuximide, widely used in the treatment of absence epilepsy, has been functionally linked to the attenuation of mitochondrial Ca2+ overload through indirect modulation of the MCU complex, a key mediator of neuronal excitotoxicity [90,91]. Excessive Ca2+ uptake into the mitochondrial matrix via the MCU induces mitochondrial depolarization and excitotoxic neuronal death, whereas its modulation mitigates ROS generation and neuronal hyperexcitability [90,92]. Accordingly, pharmacological modulation of mitochondrial calcium handling represents an important mechanism underlying anticonvulsant efficacy and neuronal metabolic regulation [92,93]. Valproic acid affects multiple mitochondrial functions, including regulation of the cellular redox state, stabilization of mitochondrial calcium homeostasis, and modulation of mPTP opening [87]. Sustained mPTP opening induces dissipation of the mitochondrial membrane potential and release of pro-apoptotic proteins processes that can be modulated by ASMs with antioxidant properties [94,95,96].
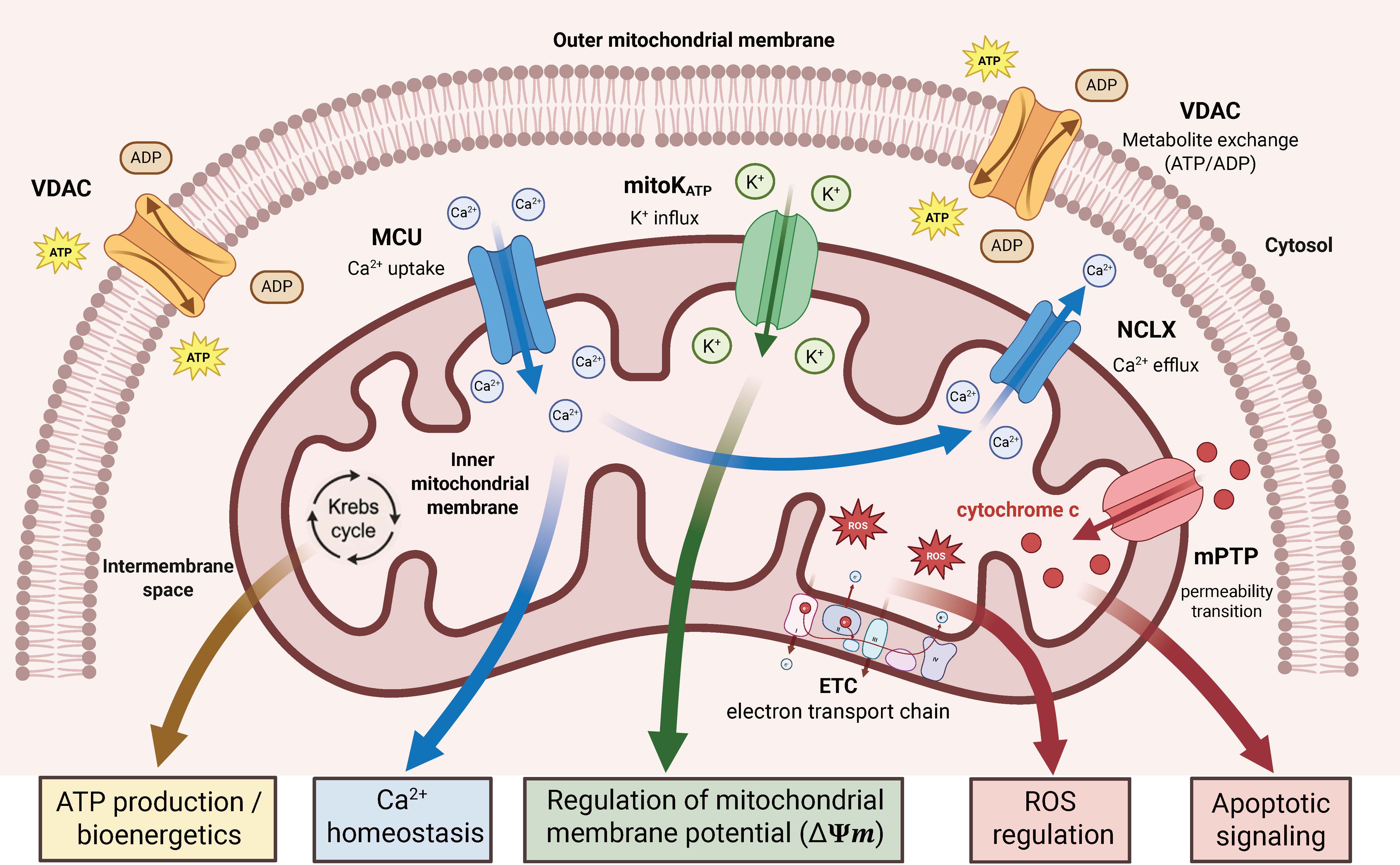 Fig. 2.
Fig. 2.Architecture and functional roles of mitochondrial ion channels in neuronal metabolism. Mitochondrial ion channels regulate the exchange of ions and metabolites across the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), integrating bioenergetics, calcium homeostasis, redox balance, and apoptotic signaling. VDAC, located in the OMM, mediates the exchange of ATP and ADP between the cytosol and intermembrane space, linking cytosolic metabolism to mitochondrial function. Within the IMM, the mitochondrial calcium uniporter (MCU) facilitates Ca2+ uptake into the matrix, stimulating the Krebs cycle and oxidative phosphorylation, while the mitochondrial Na+/Ca2+ exchanger (NCLX) mediates Ca2+ efflux to prevent mitochondrial Ca2+ overload and maintain calcium homeostasis. Mitochondrial ATP-sensitive potassium channels (mitoKATP) regulate K+ influx, contributing to stabilization of mitochondrial membrane potential (ΔΨm). The electron transport chain (ETC) produces reactive oxygen species (ROS), linking oxidative metabolism to redox signaling. Under pathological conditions, opening of the mitochondrial permeability transition pore (mPTP) induces loss of ΔΨm, disruption of oxidative phosphorylation, and release of pro-apoptotic factors such as cytochrome c into the cytosol, promoting apoptotic signaling. Created in BioRender. Pérez-Rubio, R. (2026) https://BioRender.com/w0qfcep.
Furthermore, the antioxidant properties of valproate help reduce ROS generation and preserve mitochondrial structural integrity during seizure-induced oxidative stress [95,97]. Levetiracetam, known for its binding to the synaptic vesicle glycoprotein 2A (SV2A), also modulates intracellular calcium homeostasis by reducing presynaptic Ca2+-dependent neurotransmitter release and thereby enhancing neuronal functional stability [98,99]. This sustained reduction in calcium influx indirectly helps prevent mitochondrial Ca2+ overload and the metabolic dysfunction associated with neuronal hyperexcitability [87]. This mechanism improves the coupling between synaptic activity and mitochondrial metabolism, thereby attenuating chronic neuronal excitability [87,100]. By contrast, activators of mitochondrial ATP-sensitive potassium channels (mitoKATP), such as diazoxide, and potassium channel-opening drugs such as retigabine, induce a mild, protective mitochondrial depolarization that stabilizes membrane potential and enhances cellular respiration [101].
Pharmacological activation of mitoKATP reduces mitochondrial ROS generation and improves neuronal survival following excitotoxic insults or experimentally induced seizures [102,103,104]. In epilepsy models, diazoxide preconditioning has been shown to reduce neuronal cytotoxicity and preserve mitochondrial function through modulation of the inward-rectifier potassium channel (Kir) subunits associated with the mitoKATP channel [103]. These findings indicate that pharmacological targeting of mitochondrial potassium channels represents a complementary neuroprotective mechanism that augments traditional anticonvulsant activity mediated through membrane excitability [75,105]. Current evidence indicates that ASMs modulate not only plasma membrane ion channels but also mitochondrial ion channels, thereby integrating electrical, metabolic, and redox regulation within their anticonvulsant mechanisms [87]. An important translational consideration is whether the concentrations used in the cited preclinical studies match the therapeutic plasma levels reached in clinical practice. For valproate, direct inhibition of mitochondrial cytochrome c oxidase and reductions in oxygen consumption occur in vitro at 0.5–1 mM, which overlaps the upper end of the human therapeutic range (50–100 µg/mL, approximately 0.35–0.70 mM). This supports the translational relevance of its bioenergetic effects [87,106]. However, in vitro studies addressing apoptotic and steatogenic mechanisms in hepatic and neuronal cell lines have often employed 1–10 mM, which clearly exceeds clinical exposure and must be interpreted with caution. Phenytoin and carbamazepine inhibit the mitochondrial respiratory chain at concentrations within or moderately above their therapeutic ranges (40–80 µM for phenytoin; 17–50 µM for carbamazepine), again consistent with clinically achievable levels. Studies on diazoxide and retigabine generally use micromolar concentrations that exceed conventional oral plasma levels, reflecting their use as pharmacological tools to probe mitoKATP and Kv7 channel biology rather than as direct surrogates of clinical pharmacology. Overall, the mitochondrial effects of classical ASMs reported here are largely supported by translationally relevant exposures, particularly for valproate, phenytoin, and carbamazepine. The use of supratherapeutic concentrations in part of the preclinical literature remains a limitation when extrapolating these findings to clinical practice.
Most newer-generation ASMs, including lacosamide, eslicarbazepine acetate, brivaracetam, perampanel, and cenobamate, have not been systematically characterized with respect to direct interactions with mitochondrial ion channels. The available preclinical literature on these agents focuses mainly on their canonical synaptic targets such as voltage-gated sodium channels, SV2A, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. Dedicated electrophysiological or biochemical studies of VDAC, MCU, NCLX, mPTP, or mitoKATP modulation remain scarce. A relevant exception is cannabidiol, whose anticonvulsant activity in Dravet and Lennox-Gastaut syndromes coexists with documented direct modulation of VDAC1 and regulation of intracellular Ca2+ through mitochondrial targeting [107,108]. Indirect mitochondrial effects, such as reduced glutamate-driven Ca2+ overload secondary to AMPA receptor blockade by perampanel or attenuated excitotoxicity through SV2A modulation by brivaracetam, are biologically plausible but remain inferred rather than directly demonstrated. Systematic characterization of mitochondrial channel pharmacology for newer-generation ASMs represents an unmet need and a priority area for future research, particularly because it may identify agents with both seizure-suppressing and bioenergetically protective properties.
The mitochondrial effects of ASMs differ markedly depending on epilepsy type, the organization of the implicated neural networks, and the regional metabolic demands of the brain [19,109,110]. The pathophysiological differences between focal and generalized epilepsies give rise to significant variations in neuronal bioenergetic dynamics, mitochondrial calcium regulation, and susceptibility to oxidative damage, which in turn produce distinct pharmacological responses [34,78,111]. Mitochondrial dysfunction is a pivotal mechanism of progressive neurodegeneration in temporal lobe epilepsy, contributing to the activation of cytochrome c- and caspase-dependent apoptotic pathways in susceptible hippocampal neurons [19,80,112]. Neuropathological and experimental studies indicate that the cornu ammonis area 1 and 3 (CA1 and CA3) hippocampal subfields are particularly susceptible to oxidative stress, energy deficits, and alterations in mitochondrial membrane potential during recurrent seizures [113,114,115]. Ca2+ overload induced by epileptiform activity triggers mPTP opening, leading to dissipation of the mitochondrial membrane potential, release of pro-apoptotic factors, and selective neuronal death [19,23,65].
Pharmacological modulation of VDAC and the mPTP by specific ASMs can improve mitochondrial function, reduce ROS production, and limit the activation of apoptotic pathways associated with progressive epileptogenesis [50,78,116,117]. Such mitochondrial modulation helps maintain neuronal energy homeostasis and mitigate synaptic loss following recurrent seizures in generalized absend seizures [19,34,118]. In primary generalized epilepsies, by contrast, the pathophysiology predominantly involves thalamocortical circuits that generate synchronized rhythmic oscillations dependent on low-voltage-activated (T-type) Ca2+ currents and neuron-glial metabolic coupling [3,109,119,120]. Mitochondria serve as essential dynamic regulators of intracellular Ca2+ in these circuits, buffering calcium transients associated with rhythmic discharges and influencing overall neuronal excitability [12,36,120]. The capacity of mitochondria to take up and release calcium directly influences the stability of thalamocortical rhythms and the suppression of generalized hypersynchronous discharges characteristic of absence seizures and other idiopathic generalized epilepsies [19,109,121]. Mitochondrial calcium regulation shapes ATP production, which is essential for the operation of ion pumps and voltage-gated channels thereby contributing to the maintenance of the excitation-inhibition balance during physiological oscillatory activity [120,122,123]. Disruption of these bioenergetic systems promotes the pathological synchronization of large-scale neural networks and facilitates the bilateral propagation of epileptiform activity [19,78]. These pathophysiological distinctions have significant pharmacological implications, suggesting that the therapeutic efficacy of specific ASMs may partly depend on their capacity to modulate mitochondrial mechanisms relevant to each epileptic phenotype [16,34,109]. ASMs with antioxidant, mitochondrial calcium-stabilizing, or mPTP-modulating properties may offer greater benefit in neurodegenerative focal epilepsies, whereas those that enhance metabolic coupling and Ca2+ regulation may prove more efficacious in oscillatory network-dependent generalized epilepsies [19,78,124]. Collectively, the available evidence indicates that contemporary antiseizure pharmacotherapy should incorporate mitochondrial bioenergetics as a critical consideration in individualized therapeutic strategies for epilepsy [16,19,109]. The pathophysiological and pharmacological contrasts between temporal lobe epilepsy and primary generalized epilepsy discussed throughout this section are summarized and compared in Table 2 (Ref. [3,16,19,36,39,50,57,60,65,78,80,90,101,109,112,113,115,118,119,120,121,122,124]), which highlights how differences in affected neural circuits, mitochondrial channel involvement, regional oxidative vulnerability, and apoptotic consequences translate into distinct therapeutic priorities for mitochondria-targeted antiseizure intervention.
| Feature | Temporal lobe epilepsy | Primary generalized epilepsy | Key references |
|---|---|---|---|
| Predominantly affected neural networks | Hippocampal circuits, particularly CA1 and CA3 subfields | Thalamocortical circuits generating synchronized rhythmic oscillations | [3,109,113,119] |
| Primary mitochondrial mechanism | Mitochondrial outer membrane permeabilization, mPTP opening, and cytochrome c release driving apoptotic neurodegeneration | Impaired mitochondrial Ca2+ buffering of rhythmic calcium transients disrupting thalamocortical oscillatory stability | [19,65,109,121] |
| Most relevant mitochondrial channels | VDAC1, mPTP | MCU, NCLX, mitoKATP | [19,50,60,109,115] |
| Regional oxidative vulnerability | High susceptibility of hippocampal CA1 and CA3 neurons to oxidative stress, energy deficits, and mitochondrial membrane potential alterations | Disruption of neuron–glial metabolic coupling and ATP-dependent ion pump function in thalamocortical networks | [78,113,120,122] |
| Predominant apoptotic consequence | Selective neuronal death via cytochrome c- and caspase-dependent pathways in hippocampal subfields | Pathological synchronization of large-scale networks; neuronal loss less regionally circumscribed | [19,65,80,112] |
| Mitochondrial Ca2+ dysregulation | Sustained Ca2+ overload inducing mPTP opening and mitochondrial depolarization following prolonged seizures | Impaired Ca2+ buffering of physiological transients linked to T-type channel-dependent rhythmic discharges | [36,39,57,121] |
| ASMs with greatest mitochondrial relevance | Phenytoin, carbamazepine (VDAC1 modulation); valproic acid (mPTP/NCLX stabilization) | Ethosuximide (MCU-mediated Ca2+ overload attenuation); levetiracetam (presynaptic Ca2+ reduction); diazoxide (mitoKATP activation) | [19,90,101,109] |
| Bioenergetic failure pattern | Progressive energy deficit secondary to recurrent seizure-induced mitochondrial dysfunction and oxidative damage | Metabolic uncoupling between neuronal activity and mitochondrial ATP production during hypersynchronous discharges | [16,78,109,118] |
| Therapeutic mitochondrial strategy | Antioxidant protection, mPTP inhibition, and VDAC1 modulation to limit apoptotic cascades and preserve neuronal energy homeostasis | Enhancement of mitochondrial Ca2+ regulation and metabolic coupling to stabilize thalamocortical network excitability | [19,109,124] |
Principal mitochondrial mechanisms underlying seizure generation and neuronal injury across two major epileptic phenotypes, highlighting differences in affected neural networks, mitochondrial channel involvement, oxidative vulnerability, apoptotic consequences, and the antiseizure medications most relevant to each context. These distinctions have direct implications for the selection of mitochondria-targeted therapeutic strategies tailored to specific epileptic phenotypes. Abbreviations: ASMs, antiseizure medications; CA1/CA3, cornu ammonis subfields 1 and 3; VDAC1, voltage-dependent anion channel isoform 1.
The anticonvulsant effect emerges from the interplay between traditional neuronal mechanisms and mitochondrial functions that concurrently regulate electrical excitability and brain energy metabolism [8,16,80]. Voltage-gated neuronal ion channels including sodium, potassium, and calcium channels regulate membrane excitability and the generation of hypersynchronous discharges characteristic of epileptic seizures [125,126,127]. Mitochondrial channels, including the MCU and VDAC, simultaneously regulate the energy supply and metabolic homeostasis essential for sustaining high-intensity synaptic activity [53,128]. Mitochondria serve as a critical integrator of anticonvulsant drug efficacy by processing intracellular signals related to calcium dynamics, ATP synthesis, and cellular redox state [129,130]. Several antiseizure medications have been shown to indirectly support mitochondrial function by reducing oxidative stress, preserving the mitochondrial membrane potential, and enhancing oxidative phosphorylation [78,118]. The interplay among Ca2+, ROS, and ATP constitutes a critical bioenergetic axis underlying anticonvulsant efficacy: calcium drives mitochondrial metabolic activation, while the resulting ATP attenuates neuronal hyperexcitability by sustaining ion gradients [81,131]. Excessive calcium influx during epileptic episodes elevates mitochondrial ROS generation, potentially inducing energy failure and promoting seizure spread in the absence of sufficient antioxidant defense [16,19]. Conversely, effective maintenance of redox balance preserves synaptic integrity and modulates energy-dependent neurotransmitter release, thereby contributing to the stabilization of hyperexcitable neural networks [59,132]. The functional integration of calcium signaling, mitochondrial metabolism, and oxidative regulation collectively modulates neuronal excitability, cell survival, and synaptic plasticity processes essential for both short-and long-term anticonvulsant responses [82,133]. The convergence of classical and mitochondrial mechanisms described throughout this section is captured in Table 3 (Ref. [48,71,74,87,89,90,91,92,94,95,98,99,100,101,103,105,128]), which consolidates, for each drug discussed, the relationship between its primary neuronal target, its proposed mitochondrial site of action, and the functional outcome of that interaction in terms of neuronal survival and seizure control.
| Drug | Classical mechanism of action | Mitochondrial target | Proposed mitochondrial mechanism | Functional outcome | Key references |
|---|---|---|---|---|---|
| Phenytoin | Voltage-gated Na+ channel blockade reducing repetitive neuronal firing | VDAC1 | Modulation of Bax/Bcl-2 balance regulating mitochondrial outer membrane permeability | Reduced cytochrome c release and attenuation of apoptotic signaling | [48,74,87] |
| Carbamazepine | Voltage-gated Na+ channel blockade stabilizing neuronal membrane excitability | VDAC1 | Modulation of mitochondrial outer membrane permeability associated with VDAC1 regulation | Stabilization of mitochondrial integrity during excitotoxic stress | [74,87,89] |
| Ethosuximide | Inhibition of T-type Ca2+ channels | MCU | Indirect modulation of mitochondrial Ca2+ uptake limiting mitochondrial Ca2+ overload mitochondrial Ca2+ homeostasis through Ca2+ extrusion | Prevention of mitochondrial depolarization and ROS generation | [90,91,92] |
| Valproic acid | Enhancement of GABAergic transmission and modulation of Na+ and Ca2+ channels | mPTP/NCLX | Antioxidant effects and stabilization of mitochondrial Ca2+ homeostasis preventing mPTP opening | Preservation of mitochondrial membrane potential and reduced oxidative stress | [87,94,95] |
| Levetiracetam | Binding to synaptic vesicle protein SV2A reducing presynaptic Ca2+ influx | Ca2+ handling (indirect) | Reduction of presynaptic Ca2+ influx indirectly preventing mitochondrial Ca2+ overload | Improved coupling between synaptic activity and mitochondrial metabolism | [98,99,100] |
| Diazoxide | Activation of ATP-sensitive potassium channels | mitoKATP | Pharmacological activation of mitochondrial KATP channels producing mild mitochondrial depolarization | Reduction of ROS production and enhancement of neuronal survival | [71,101,103] |
| Retigabine | Activation of Kv7 (KCNQ) voltage-gated potassium channels | mitoKATP | Modulation of mitochondrial potassium flux stabilizing mitochondrial membrane potential | Decreased oxidative stress and improved mitochondrial resilience | [105,128] |
Classical mechanisms of action of selected antiseizure medications alongside their proposed mitochondrial targets, underlying molecular mechanisms, and functional consequences for mitochondrial integrity and neuronal survival. Abbreviations: SV2A, synaptic vesicle glycoprotein 2A; Na+, sodium ion; Ca2+, calcium ion; K+, potassium ion.
The recognition of mitochondrial channels as secondary therapeutic targets has emerged as a new paradigm in the neuropharmacology of epilepsy, as mitochondrial dysfunction directly contributes to neuronal hyperexcitability and susceptibility to seizures [17,45,134]. Experimental data indicate that alterations in mitochondrial calcium homeostasis, ATP synthesis, and mitochondrial membrane potential regulation promote the pathological neuronal synchronization characteristic of seizures [12,77]. Specifically, the mPTP, MCU, and VDAC play critical roles in regulating neuronal survival during epileptiform activity [37,48,58,66]. Compounds specifically targeting mitochondrial function have demonstrated efficacy in reducing seizure severity in animal models by regulating bioenergetics and attenuating neuronal oxidative stress [87,135]. Several established antiseizure medications, including valproate, levetiracetam, and topiramate, achieve their therapeutic effects in part by indirectly modulating mitochondrial metabolism and reducing ROS levels [33]. In addition, mitochondrially targeted antioxidants such as mitoquinone mesylate (MitoQ) and elamipretide (SS-31) have shown neuroprotective properties by preserving respiratory chain integrity, however, it would be interesting to study its effects and limiting seizure-induced apoptotic activation [136,137]. The development of mitochondrially targeted antiseizure agents represents a promising research avenue aimed at selectively restoring neuronal energy balance while preserving physiological neurotransmission [138].
Nevertheless, the same mitochondrial interactions that contribute to ASM efficacy may also explain part of their adverse effect profiles, particularly when therapeutic windows are exceeded or when underlying mitochondrial dysfunction coexists. Valproate exemplifies this dual nature. At therapeutic concentrations it stabilizes NCLX function and limits mPTP opening, yet it also inhibits mitochondrial β-oxidation, sequesters coenzyme A as valproyl-CoA esters, depletes intracellular carnitine, and inhibits α-ketoglutarate dehydrogenase. Together, these actions impair oxidative phosphorylation and underlie its hepatotoxicity, hyperammonemic encephalopathy, and microvesicular steatosis [87,106]. Recessive POLG variants further increase the risk of fatal valproate-induced hepatotoxicity (odds ratio ~23.6) and represent the only ASM–mitochondrial axis with robust human causal evidence, leading to an FDA contraindication in patients with confirmed POLG-related disease [85,86]. Phenytoin and carbamazepine also show dose-dependent inhibition of the mitochondrial respiratory chain in hepatocyte models. Phenytoin behaves as a Complex I inhibitor, whereas carbamazepine acts as a respiratory substrate inhibitor [87,106]. These effects may contribute to chronic phenytoin-associated cerebellar dysfunction and to the rare hepatocellular injury reported with carbamazepine. By contrast, several adverse events of mitoKATP-targeting agents arise mainly from non-mitochondrial targets. Diazoxide-induced hyperglycemia and hypotension, as well as the cutaneous and retinal pigmentation that prompted retigabine’s withdrawal in 2017, illustrate that ASM toxicity cannot be reduced exclusively to mitochondrial mechanisms. Mitochondrial channel modulation therefore acts as a double-edged sword in ASM pharmacology. It is bioenergetically protective within the therapeutic range yet potentially harmful under supratherapeutic exposure, polytherapy, or pre-existing mitochondrial vulnerability. This reinforces the rationale for individualized prescribing and for the development of more selective mitochondrial-targeted strategies.
These approaches specifically target the modulation of mitochondrial Ca2+ handling to prevent calcium overload, which precipitates excitotoxicity and neuronal death during status epilepticus [39,57]. Similarly, pharmacological modulation of the mitochondrial redox state has been proposed as a critical strategy to break the detrimental cycle between oxidative stress and synaptic dysfunction observed in chronic epilepsy [139]. Preservation of the mitochondrial membrane potential has been shown to inhibit cytochrome c release and prevent the initiation of apoptotic cascades associated with progressive neuronal injury [140]. The identification of mitochondrial biomarkers is gaining increasing relevance for understanding the clinical heterogeneity of epilepsy and predicting treatment responses [141]. Alterations in energy metabolites, circulating mitochondrial DNA levels, and oxidative stress markers have been proposed as candidate biomarkers of mitochondrial dysfunction in patients with epilepsy [142,143]. Such biomarkers may allow patient stratification according to metabolic profiles and facilitate the development of personalized therapeutic strategies targeting specific pathophysiological mechanisms [144]. The convergence of mitochondrial pharmacology, metabolic biomarkers, and targeted therapies suggests that the future of antiseizure treatment may center on neuroprotective interventions capable of both controlling seizures and modifying disease progression [20]. Several limitations of the available evidence deserve acknowledgment. Most mechanistic data come from rodent chemoconvulsant models such as kainate, pilocarpine, and pentylenetetrazol, as well as from in vitro hepatic and neuronal cell lines. These approaches probe distinct aspects of mitochondrial physiology but do not fully reproduce the heterogeneity of human chronic epilepsy. Preclinical studies generally use acute drug exposures, whereas clinical ASM use is chronic and often involves polytherapy. In addition, a substantial part of the mitochondrial toxicity data was generated in hepatocyte models rather than in neurons, which complicates extrapolation to brain tissue. Several key findings have been reported by single laboratories without independent replication, and male animals predominate in most preclinical seizure cohorts. Direct human evidence remains largely restricted to case reports and small case series, with POLG-related valproate hepatotoxicity as the only ASM–mitochondrial axis supported by robust human causal data. These limitations do not invalidate the conceptual framework presented here, but they highlight the need for more clinically relevant models, sex-balanced cohorts, and well-phenotyped human studies.
The evidence reviewed suggests that antiseizure medications not only modulate neuronal excitability but also directly regulate mitochondrial function, highlighting mitochondrial ion channels as critical elements of anticonvulsant and neuroprotective mechanisms. The interactions of ASMs with channels such as VDAC, MCU, NCLX, mPTP, and mitoKATP indicate that the regulation of Ca2+ homeostasis, redox balance, and cellular bioenergetics is essential for mitigating neuronal hyperexcitability and preventing damage from recurrent seizures. This integrative perspective helps explain the therapeutic heterogeneity observed across epileptic phenotypes and the clinically documented neuroprotective effects that cannot be attributed solely to synaptic mechanisms. From a forward-looking perspective, positioning the mitochondria as a pharmacological target opens the opportunity to develop therapeutic strategies focused on restoring neuronal metabolic stability, encompassing mitochondrially targeted compounds, selective modulators of mitochondrial calcium handling, and precision antioxidant therapies. Furthermore, the identification of mitochondrial biomarkers may improve patient stratification and accelerate the development of personalized therapies, steering antiseizure pharmacology toward a neuroprotective framework that not only controls seizures but also modifies the course of epilepsy and improves long-term outcomes.
ADP, Adenosine diphosphate; AIF, Apoptosis-inducing factor; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ASMs, Antiseizure Medications; ATP, Adenosine triphosphate; Bak, Bcl-2 homologous antagonist/killer protein; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2 protein; Ca2+, Calcium ion; CBD, Cannabidiol; CoA, Coenzyme A; Complex I, NADH:ubiquinone oxidoreductase (mitochondrial respiratory complex I); CypD, Cyclophilin D; Ca2+-ATPase, Calcium-transporting ATPase; DNA, Deoxyribonucleic acid; ETC, Electron transport chain; F1Fo-ATP synthase, Mitochondrial ATP synthase (Complex V); GABA, Gamma-aminobutyric acid; GABRA1, Gamma-aminobutyric acid type A receptor subunit alpha 1; ILAE, International League Against Epilepsy; IMM, Inner mitochondrial membrane; K+, Potassium ion; KATP, ATP-sensitive potassium channel; KCNJ8/11, Potassium inwardly rectifying channel subfamily J members 8 and 11; KCNQ2, Potassium voltage-gated channel subfamily Q member 2; Kir, Inward-rectifier potassium channel subunit; Kv7 (KCNQ), Voltage-gated potassium channel subfamily Q; MCU, Mitochondrial calcium uniporter; MELAS, Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes; MERRF, Myoclonic Epilepsy with Ragged-Red Fibers; MICU1, Mitochondrial calcium uptake protein 1; MICU2, Mitochondrial calcium uptake protein 2; MILS, Maternally Inherited Leigh Syndrome; MitoQ, Mitoquinone mesylate (mitochondria-targeted antioxidant); mitoKATP, Mitochondrial ATP-sensitive potassium channel; mPTP, Mitochondrial permeability transition pore; MT-ATP6, Mitochondrially encoded ATP synthase membrane subunit 6; mtDNA, Mitochondrial DNA; MT-TK, Mitochondrially encoded tRNA lysine; MT-TL1, Mitochondrially encoded tRNA leucine 1 (UUA/G); Na+, Sodium ion; Na+/K+-ATPase, Sodium-potassium adenosine triphosphatase; NARP, Neuropathy, Ataxia, and Retinitis Pigmentosa; NCLX, Mitochondrial Na+/Ca2+ exchanger; OMM, Outer mitochondrial membrane; POLG, DNA polymerase gamma catalytic subunit; rRNA, Ribosomal ribonucleic acid; ROS, Reactive oxygen species; SCN1A, Sodium voltage-gated channel alpha subunit 1; SLC8B1, Solute carrier family 8 member B1 (gene encoding NCLX); SS-31, Elamipretide (mitochondria-targeted antioxidant peptide); SV2A, Synaptic vesicle glycoprotein 2A; TBI, Traumatic brain injury; tRNA, Transfer ribonucleic acid; T-type Ca2+ channels, Low-voltage-activated calcium channels; VDAC, Voltage-dependent anion channel; VDAC1, Voltage-dependent anion channel isoform 1; VDAC1/2/3, Voltage-dependent anion channel isoforms 1, 2, and 3; ΔΨm, Mitochondrial membrane potential; α-KG, α-ketoglutarate; α-KGDH, α-ketoglutarate dehydrogenase; β-oxidation, Mitochondrial fatty acid β-oxidation.
CR: Conceptualization, Investigation, Original Draft Preparation, Writing & Editing; NS-G: Conceptualization, Investigation, Review & Editing; RP-R: Review, Editing & Figures; JP-V: Writing, Review & Editing, systematic literature search, screening and Data extraction and synthesis; HR-P: Review, Editing & Validation, systematic literature search, screening and Data extraction and synthesis; AL: Writing, Review & Data Curation; MR-O: Conceptualization, Writing, Original Draft Preparation & Validation. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to express our sincere gratitude to the National Institute of Neurology and Neurosurgery for its institutional support. We also deeply appreciate the valuable comments and suggestions from the reviewers, which undoubtedly contributed to significant improvements in our work.
This research received no external funding.
The authors declare no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.