

 , Baocheng Cang 1,†, Yunhu Bai 1, Ding Zhang 1, You Wu 2, Tongwen Sun 3,*
, Baocheng Cang 1,†, Yunhu Bai 1, Ding Zhang 1, You Wu 2, Tongwen Sun 3,* , Xijing Zhang 2,*
, Xijing Zhang 2,*1 Department of Critical Care Medicine, Joint Logistics Force No. 988 Hospital, 450000 Zhengzhou, Henan, China
2 Department of Critical Care Medicine, Xijing Hospital, Air Force Medical University, 710032 Xi’an, Shaanxi, China
3 Department of Critical Care Medicine, The First Affiliated Hospital, Zhengzhou University, 450052 Zhengzhou, Henan, China
†These authors contributed equally.
Abstract
Sepsis-associated encephalopathy (SAE) is a common and severe neurological syndrome induced by sepsis and characterized by brain dysfunction. The incidence of SAE is as high as 76%. After onset, the mortality rate is as high as 60%. Even if patients survive, about 20% have long-term cognitive impairment. At present, the exact pathogenesis of SAE remains unknown. Our results showed that necroptotic protein was obviously elevated in the hippocampus of SAE mice, but the internal mechanism was still unclear. The stimulator of interferon gene (STING) protein has been reported to be a key molecule in the regulation of programmed cell death. However, the critical role of STING in the progression of SAE remains elusive. We explored the mechanism by which STING regulates hippocampal neuronal necroptosis in SAE.
A cannula was embedded into the lateral ventricle of mice, and mixed lineage kinase domain-like protein (MLKL) inhibitor necrosulfonamide (NSA) and STING inhibitor C-178 were injected. The motor ability and memory function of mice after cecal ligation and puncture (CLP) were assessed by the open field test (OFT) and barnes maze test (BMT), respectively. The expression and distribution of S345p-MLKL and S366p-STING proteins in the hippocampal region of SAE mice were analyzed, and the expression levels of S366p-STING/STING, S232p-receptor-interacting protein kinase 3 (RIPK3)/RIPK3, S345p-MLKL/MLKL, and S166p-RIPK1/RIPK1 proteins in the hippocampal tissue of SAE mice were examined. Co-immunoprecipitation (CO-IP) was used to detect the relationship between STING and RIPK3 protein.
The surviving mice after CLP had cognitive dysfunction. The hippocampal tissues of SAE mice showed upregulated expression of S232p-RIPK3/RIPK3, S345p-MLKL/MLKL, and S166p-RIPK1/RIPK1 proteins. Mice in the CLP+NSA group showed better cognitive performance than the mice in the CLP+dimethyl sulfoxide (DMSO) group. The results showed that the expression of S366p-STING protein was elevated in neurons of the hippocampal cornu ammonis 1 (CA1) region in SAE mice, and there was an interaction between STING and RIPK3 proteins. STING inhibitor C-178 inhibited the protein expression level of S232p-RIPK3/RIPK3 and S345p-MLKL/MLKL and alleviated SAE mice with cognitive dysfunction.
STING is implicated in the development of cognitive dysfunction in SAE by regulating necroptosis in hippocampal neurons. Inhibition of the STING pathway reduced necroptosis-related protein expression and partially improved BMT performance. This result provides a new potential therapeutic target for the prevention and treatment of SAE.
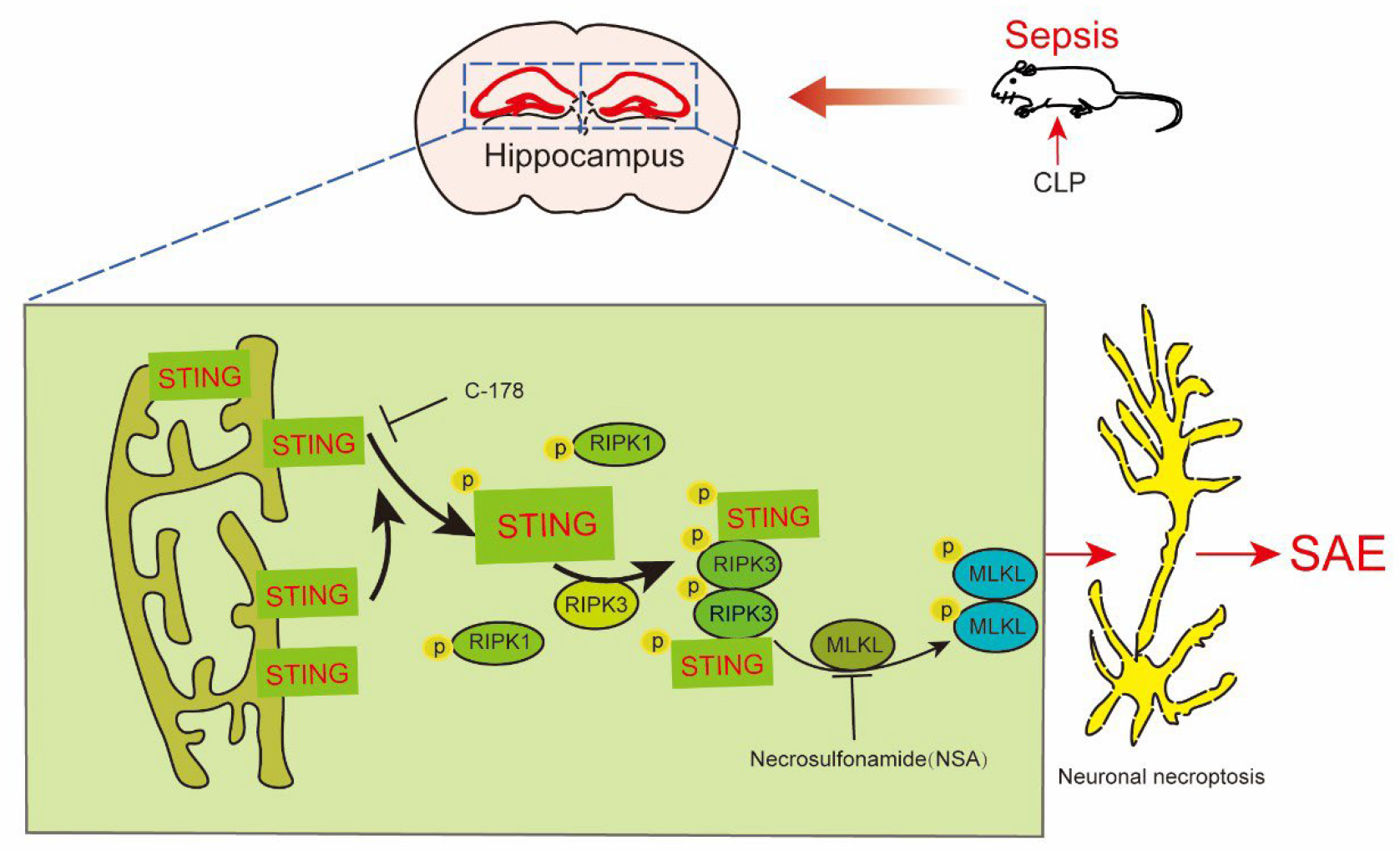
Graphical Abstract
Keywords
- necroptosis
- sepsis-associated encephalopathy
- STING
Sepsis-associated encephalopathy (SAE) is a common and severe neurological syndrome induced by sepsis and characterized by brain dysfunction [1]. SAE has a morbidity rate of about 76% and a mortality rate of up to 60% [2], and even after survival, about 20% of the patients are left with long-term cognitive dysfunction [3]. The pathogenesis of SAE is unclear, and there is a lack of effective prevention and treatment strategies. Neuronal death, particularly necroptosis, has been found to be the core factor leading to brain atrophy, most notably in the hippocampus, in patients with sepsis [4, 5], and is the main cause of cognitive dysfunction in SAE patients [6]. Recent studies have found that necroptosis is closely related to neurodegenerative diseases [7], and the inhibition of the expression of key proteins in necroptosis can effectively improve cognitive dysfunction in neurodegenerative diseases [8, 9, 10]. However, the mechanism of necroptosis in SAE cognitive impairment is currently unclear.
Stimulator of interferon gene (STING) is a key factor in the regulation of programmed cell death [11]. Studies have shown that the STING pathway is significantly initiated in sepsis in peripheral tissues and immune cells of mice, and knockdown of STING can effectively inhibit necroptosis [12, 13]. However, the pathologic mechanism of STING in the development of SAE has not been clarified.
Therefore, the present study found that necroptosis contributes to cognitive dysfunction in SAE mice. Inhibition of STING protein expression reduced the expression of some necroptosis-related proteins and improved cognitive function in SAE mice. Further investigation revealed an interaction between STING and receptor-interacting protein kinase 3 (RIPK3), a key protein in necroptosis. This experiment is expected to provide reliable theoretical support for the treatment of SAE.
Eight-week-old adult male wild-type (WT) C57BL/6J mice were supplied by Viton Lever (Beijing, China) Laboratory Animal Center. All mice were provided with quality inspection certificates.
We provided a single consolidated timeline figure that includes: cecal ligation and puncture (CLP) (time 0), timepoints for hippocampal sampling (6 h, 1–3 days, 7 days, 14 days), the necrosulfonamide (NSA)/C-178 dosing schedule (day of CLP + 3 days), open field test (OFT) timing, and barnes maze test (BMT)/testing days (Supplementary Fig. 1A).
In our experiment, the CLP model was used to induce sepsis. The 2% isoflurane
(20 ml/L, 20240115, RWD Life Science, Shenzhen, Guangdong, China) and 1.5% isoflurane (15 mL/L) were respectively used for anesthesia
induction and maintenance. The abdominal skin of the mice was thoroughly
disinfected before surgery. Along the middle and lower abdomen, a straight line
was drawn for the middle incision (incision of about 1.0 cm), and the skin and the
peritoneum were cut open one by one. A 4-0 surgical suture was used to ligate the midpoint of the cecum,
and a 22-G needle was applied to puncture the cecum, and squeezing the cecum to the
puncture point of the outflow of the bowel content (the size of rice grains).
Layer-by-layer suturing of the skin; disinfection of incisions. We administered
lidocaine (0.1 mL, 0.2%, s.c., D32510281, Jinyao, Xiangyang, Hubei, China) for
pain relief. After completion of the surgical procedure, prewarmed 0.9% normal
saline (30 mL/kg) was administered s.c. to compensate for intraoperative fluid
loss. Postoperatively, mice were placed in a 37 °C incubator until fully
recovered, as indicated by free movement and foraging behavior.
Imipenem/cilastatin (25 mg/kg, s.c., 250620728, Zhuhai Lianbang, Zhuhai,
Guangdong, China) was administered in the perioperative period, as well as at 3
and 6 h postoperatively. Mice in the Sham group did not receive cecal ligation
and puncture, but all other procedures were identical to those in the CLP group.
Exclusion criteria: intraoperative death; incorrect ligation site or failed
puncture; massive intra-abdominal hemorrhage; body weight deviating from the mean
body weight of the same batch by
In our experiment, the OFT was used to detect the recovery of locomotor ability
in mice after surgery (Supplementary Fig. 1B). Mice were placed in a
behavioral chamber for 2 h to habituate. The Anymaze behavioral monitoring system
(60000, Stoelting Company, Wood Dale, IL, USA) was connected to a custom-made
open field test chamber (40
We used the BMT to detect the memory abilities of the mice
(Supplementary Fig. 1C). The adaptation and training phases of the BMT
were conducted 5 days prior to CLP. The a priori primary behavioral
endpoint in the BMT was the latency of mice to first enter the target hole
(escape from the noxious noise) on day 15 after CLP. The BMT was performed and
analyzed by investigators who were blind to the group assignments of the mice.
The BMT steps were as follows. Adaptation stage: The mouse was placed on the maze
platform, and a transparent glass cup was covered. The buzzer (60170, Stoelting Company,
Wood Dale, IL, USA) was turned on to provide a noxious auditory stimulus.
After 1 min, the transparent glass cup was moved to guide the mouse to the target
hole and into the target box, and the noise was terminated. The mouse was kept in
the target box for 1 min and then returned to its cage. Alcohol (75%) was
sprayed on the platform and the target box. Training phase: the duration was four
days. The mouse was positioned on the Barnes maze platform, covered by an opaque
box. The buzzer was turned on to make a noise, and the opaque box was taken away.
The mouse was allowed to freely move on the platform for 2 min. If the mouse
entered the target box, the buzzer was stopped immediately. This behavior was
regarded as a successful escape. Conversely, the mouse was directed to the target
box using a transparent glass cup, and the buzzer was turned off immediately. Day
1, each mouse received 3 training trials; Days 2–4, each mouse received 4
training trials. Training was conducted for a total of 15 trials over 4 days.
Test phase: the mouse was positioned on the Barnes maze platform and covered by
an opaque box. The buzzer was turned on, the opaque box was taken away, and the
mouse explored freely on the platform for 2 min. The latency for the mouse to
enter the target hole for the first time (escape latency) was noted. The time the
mouse stayed in the target quadrant was also noted. Percentage of time spent in
the target quadrant = time spent (target quadrant) / total time
The mixed lineage kinase domain-like protein (MLKL) inhibitor NSA (ab143839, Abcam, Cambridge, UK) and the STING inhibitor C-178 (ab287033, Abcam) were injected into the lateral ventricle via cannula (Supplementary Fig. 1D). The anesthetic for the stereotaxic surgery was 2% pentobarbital sodium (20mg/kg, i.p., 250302, Mindong Lijiesun Pharmaceutical Co., Ningde, Fujian, China). The heads of the mice were sterilized before the operation. Centering on the Bregma point of mice, the trocar position points were: anteroposterior (AP): –0.50 mm; dorsoventral (DV): –1.50 mm; mediolateral (ML): –1.00 mm. Cranial drilling was performed in mice. Trocars were placed into the lateral ventricle, and cerebrospinal fluid was seen to overflow. The trocars were fixed to the cranial bones of the head. The mouse was then placed in an incubator until it was fully awake. CLP was performed one week later. Subsequently, the internal injection cannula, polyethylene (PE) tubing, locking nut, and injector were assembled in advance. The drug was drawn using the syringe pump, and the position of the drug solution was marked on the PE tubing to monitor whether the liquid level decreased during injection. The cannula cap was then removed, the internal injection cannula was slowly inserted into the guide cannula, and secured with the locking nut. The total injection time was 10 min. NSA was injected at a rate of 0.2 µL/min, and C-178 at 0.15 µL/min. After the injection was completed, the cannula was kept in place for approximately 10 min to allow sufficient drug diffusion, followed by slow withdrawal of the internal injection cannula. The cannula cap was reinserted and tightened. Cannula specifications: cannula length (C) 3.5 mm; outer diameter (D) 0.41 mm; gauge inner diameter 1 (G1) 0.5 mm; gauge inner diameter 2 (G2) 0.5 mm. Injector specifications: 0.5–100 µL. During the experiment, 2 mice were excluded due to cannula dislodgement. No mice were excluded because of cannula occlusion or surgical complications. To verify the correct placement of the cannula in the mouse lateral ventricle, needle-track histology was performed post-mortem (Supplementary Fig. 1E).
Euthanasia of experimental mice was carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals issued by the National Institutes of Health (NIH). Mice were euthanized by intraperitoneal injection of an overdose (100 mg/kg) of pentobarbital sodium to achieve rapid and painless death. Complete cardiac and respiratory arrest was confirmed before tissue harvesting to verify the success of euthanasia.
We used WB to test the expression level of specific proteins. First,
hippocampal-protein samples were prepared according to the standard protocol.
Then, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (10%, P0012A, Beyotime, Shanghai, China)
of varying concentrations were selected according to the molecular weight of the
target protein. For electrophoresis, the prepared samples were loaded, and
electrophoresis was performed at 80 V until the 70 kDa band of the protein marker
was visible, after which the voltage was adjusted to 100 V. Transfers were
carried out under constant voltage at 100 V for 2 h. The membrane was blocked
with 5% skimmed milk for 1.5 h, then washed three times with (1
IF was used to detect specific binding between antigen and antibody to detect target proteins in tissue samples. Brain tissue was harvested from CLP and Sham mice on postoperative day 1. Brain slices were blocked with the mixture (donkey serum (5%, ab7475, Abcam) and Triton (0.3%, ab286840, Abcam)) at 37 °C for 1 h. Incubation of primary antibodies: p-STING (1:100, rabbit, AP1369, ABclonal), p-MLKL (1:100, rabbit, ab196436, Abcam), neuronal antibody (1:200, mouse, ab104224, Abcam), ionized calcium-binding adapter molecule 1 (IBA1) antibody (1:200, goat, ab5076, Abcam), overnight at 4 °C.
Secondary antibody buffer containing Alexa Fluor 594 (1:300, S32356, Invitrogen, Carlsbad, CA, USA) or Alexa Fluor 488 (A-11001, Invitrogen, 1:300) was added dropwise. Incubate at 37 °C for 2 h in the dark. Subsequently, 4′,6-diamidino-2-phenylindole (DAPI) solution (1:200, ab104139, Abcam) was added and incubated for 10 min. After thorough washing and coverslipping, the sections were air-dried. Confocal microscopy (FV1200, Olympus, Tokyo, Japan) for photography. We defined the region of interest (ROI) as the region showing co-localization of the target protein and neurons in the hippocampal cornu ammonis 1 (CA1) area. The mean fluorescence intensity in the non-specific staining region adjacent to the ROI was measured as the background value. Corrected fluorescence intensity = mean intensity of ROI - mean intensity of local background. Six mice/group (gp) were used; three sections per mouse and three visual fields per section were analyzed for mean fluorescence intensity. Sections were coded by a third party, and quantification was performed by an investigator who was blind to group allocation.
Statistical analyses were performed using GraphPad Prism 9.0 software (GraphPad
Software Co., San Diego, CA, USA). Experimental data were expressed as
mean
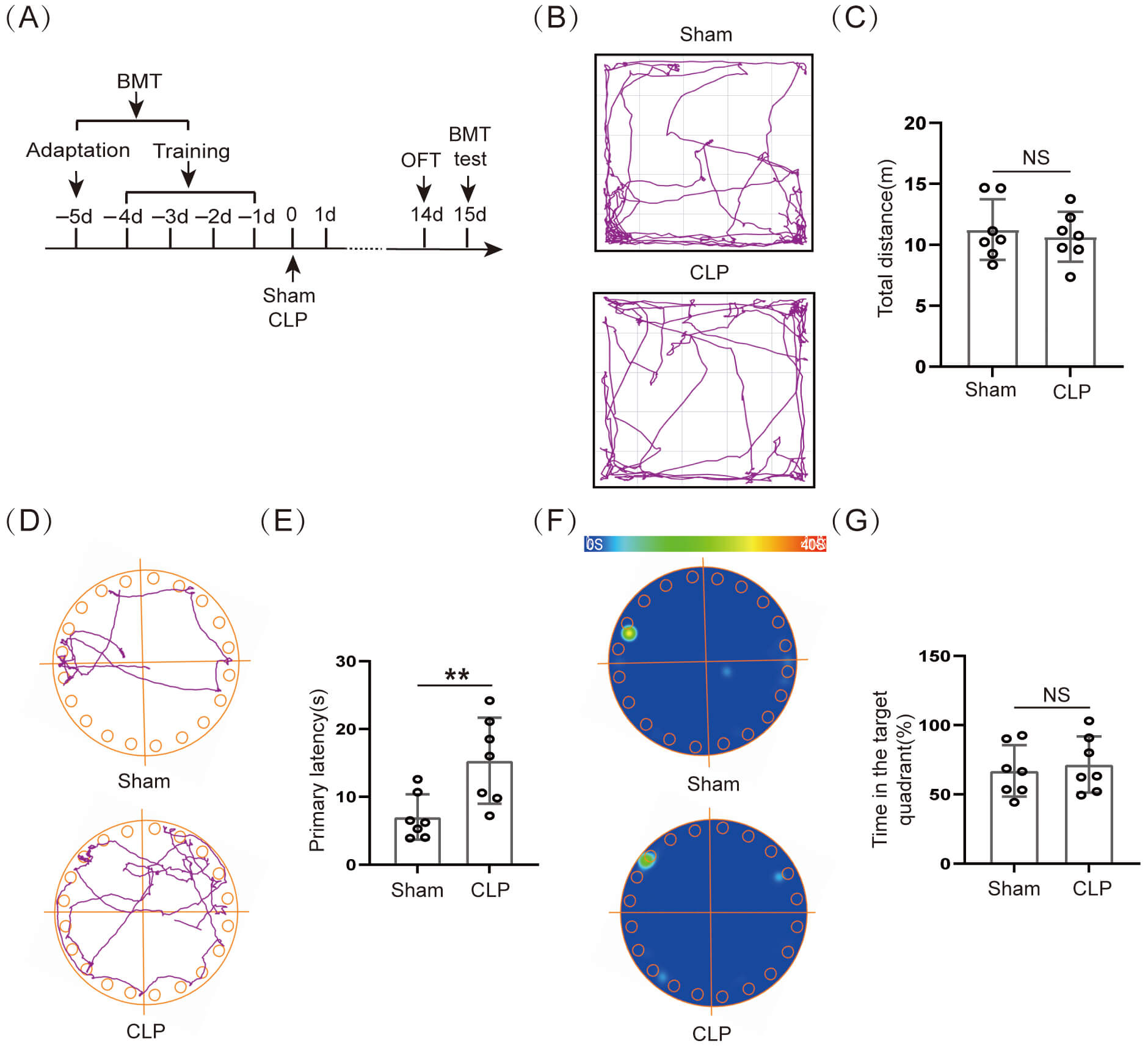
Our experimental results indicated that the 14–day survival rate of CLP mice was 50.0% (7/14) (Supplementary Fig. 1F). The initial weight (%) of the CLP group was obviously lower than that of the Sham group (Supplementary Fig. 1G). The murine sepsis score (MSS) was obviously higher in the CLP group than in the Sham group within 10 days after surgery (Supplementary Fig. 1H). The surviving mice on day 14 after CLP were subjected to behavioral testing (Fig. 1A). First, the OFT data indicated that movement distance did not differ significantly between the two groups (Fig. 1B,C). Our results showed that the voluntary locomotion was not significantly damaged in mice 14 days after CLP, and subsequent behavioral studies at this time could exclude the effect of exercise. The BMT assessed the spatial memory ability of the mice on day 15 after CLP surgery (Fig. 1A). The results suggested that the primary latency time for CLP group mice escape the noxious stimulus was obviously longer than for Sham group mice (Fig. 1D,E), and there was no obviously significant difference in the time that the CLP group mice stayed at the target quadrant (Fig. 1F,G). This suggested that long-term spatial memory function was significantly impaired in mice after CLP.
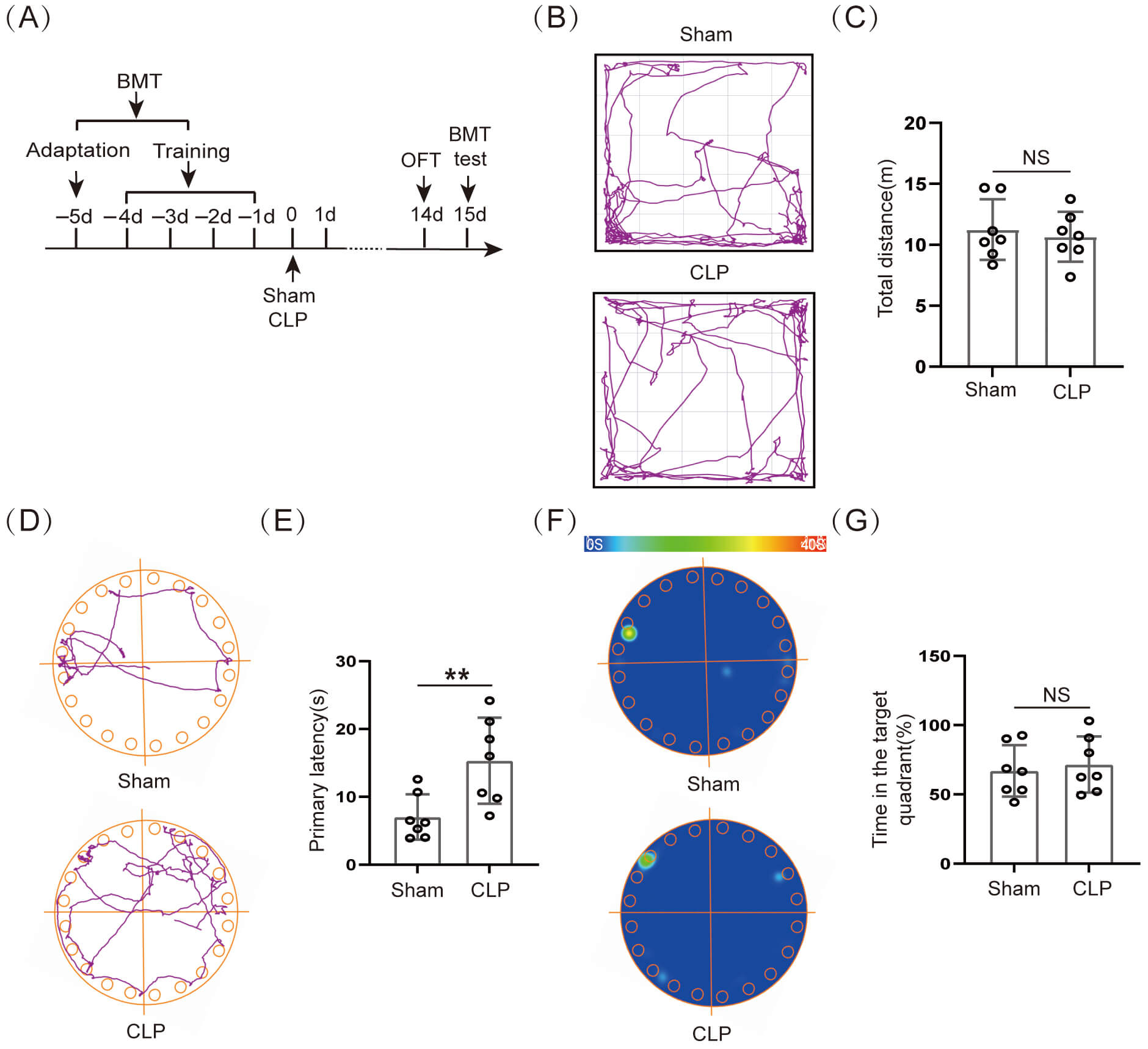 Fig. 1.
Fig. 1.
Cognitive dysfunction occurred in septic mice. (A)
Schematic timeline of BMT and OFT. (B) Trajectory diagram of OFT. (C) Statistical
results of total distance (n = 7, NS: not significant). (D) Trajectory
diagram of BMT and (E) Statistical results of primary latency (n = 7, **p
A study has reported that neuronal necroptosis was closely related to neurodegenerative diseases [7]. If so, then how does necroptosis change in hippocampal neurons of septic mice? The WB was performed to ascertain the expression level of necroptotic proteins in hippocampal tissues of septic mice at 6 h, 1 day, 2 days, 3 days, 7 days, and 14 days. Our data showed that the expression of S166p-RIPK1/RIPK1, S232p-RIPK3/RIPK3, and S345p-MLKL/MLKL proteins in the hippocampal tissues of septic mice was significantly elevated at 1, 2, and 3 days (Fig. 2A–D; The original Western blot images can be found in the Supplementary Materials-Original WB). These results demonstrated that necroptosis occurred in the hippocampal tissues of septic mice. The S345p-MLKL protein was immunofluorescently stained with neurons (NeuN). The IF intensity of S345p-MLKL in the hippocampal CA1 region co-localized with mouse hippocampal neurons on day 1 after CLP surgery was significantly different (Fig. 2E,F). In addition, our IF results demonstrated significant neuronal loss in the hippocampal CA1 region of mice on the third day after CLP surgery (Supplementary Fig. 1I). This suggested that the necroptosis protein S345p-MLKL was expressed in neurons in the hippocampal CA1 region of SAE mice.
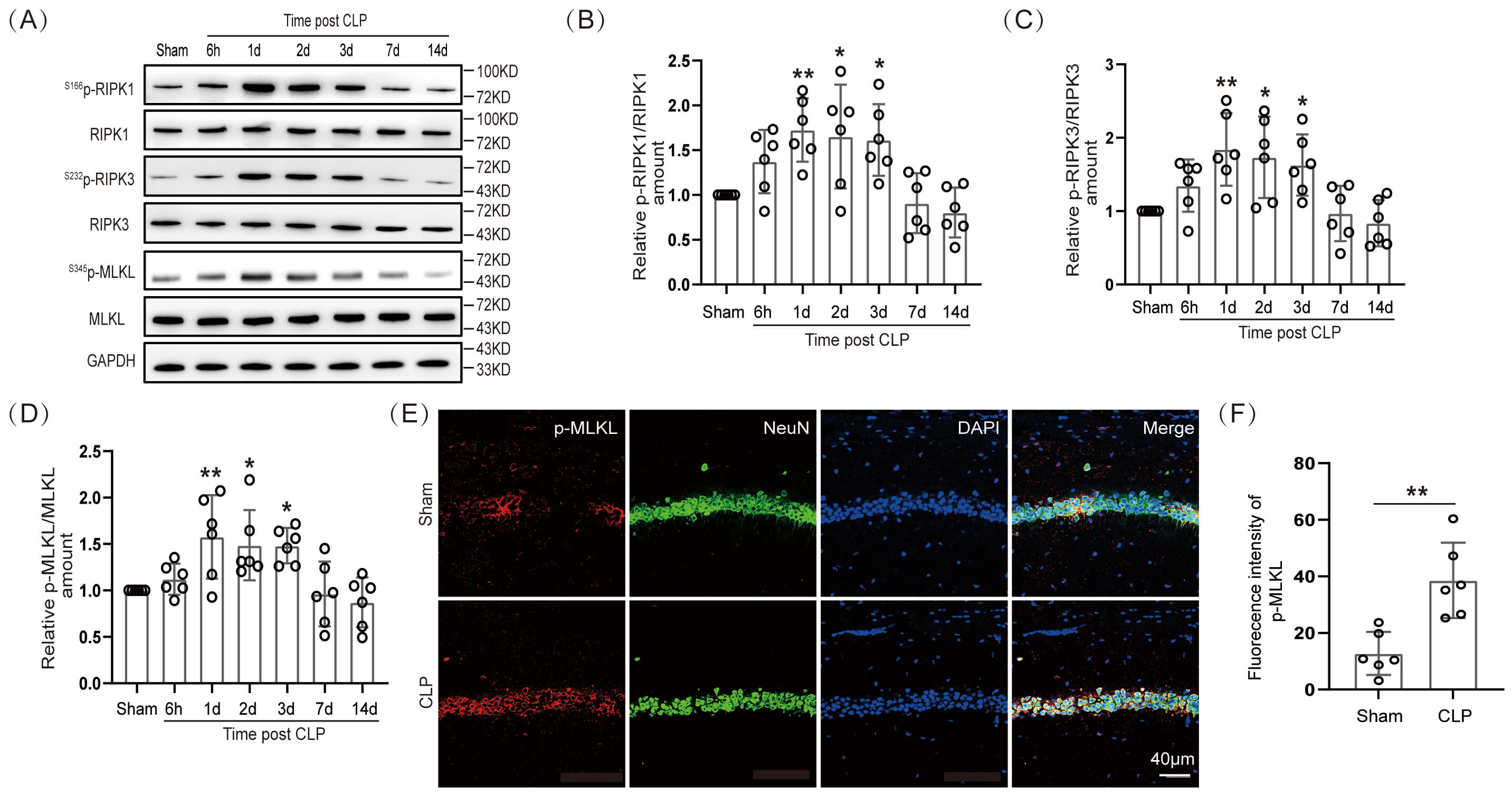 Fig. 2.
Fig. 2.
Necroptosis arose in hippocampal neurons of SAE mice.
(A) WB and (B–D) statistical results of necroptotic proteins
(n = 6, *p
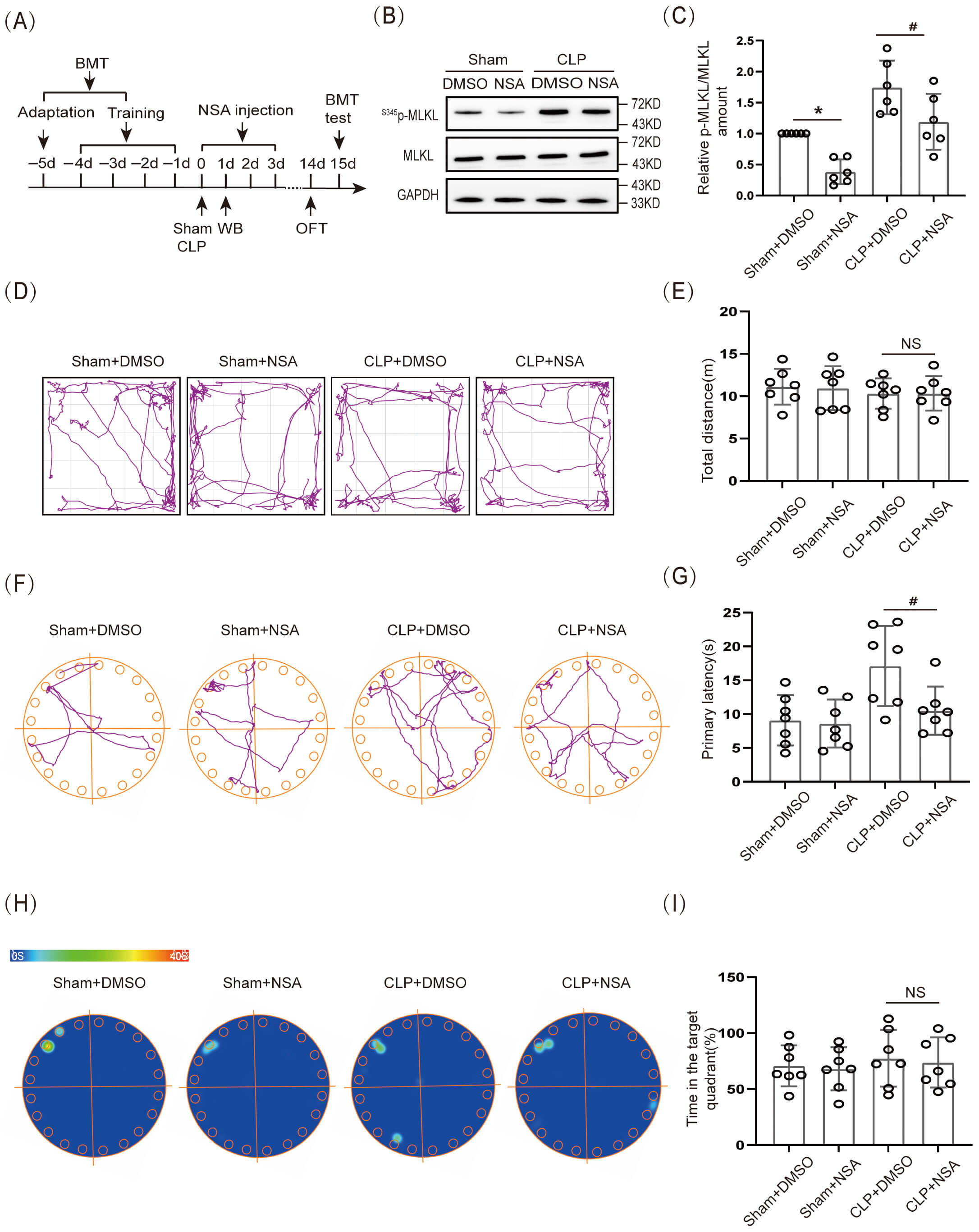
The effect of necroptosis on cognitive dysfunction in SAE mice was further verified by inhibiting S345p-MLKL protein expression. In this experiment, a cannula was embedded in the lateral ventricle of 8-wk-old WT mice. MLKL inhibitor NSA (2 µL; 1 mg/mL) was injected through the cannula on the day of CLP, and the drug was administered for 3 consecutive days (Fig. 3A). First, WB was used to verify the effectiveness of the NSA. The results showed that S345p-MLKL/MLKL protein expression in the hippocampal tissue of the Sham+NSA group was obviously lower than that in the Sham+ dimethyl sulfoxide (DMSO) group, and S345p-MLKL/MLKL protein expression in the hippocampal tissue of the CLP+NSA group was obviously lower than that of the CLP+DMSO group (Fig. 3B,C; The original Western blot images can be found in the Supplementary Materials-Original WB). Further, the OFT results demonstrated that movement distance did not differ significantly among all groups (Fig. 3D,E). The BMT results demonstrated that the CLP+NSA group had an obviously shorter primary latency time to find the target hole (escape) than did the CLP+DMSO group (Fig. 3F,G), whereas the difference in the time spent in the target quadrant was not significant (Fig. 3H,I).
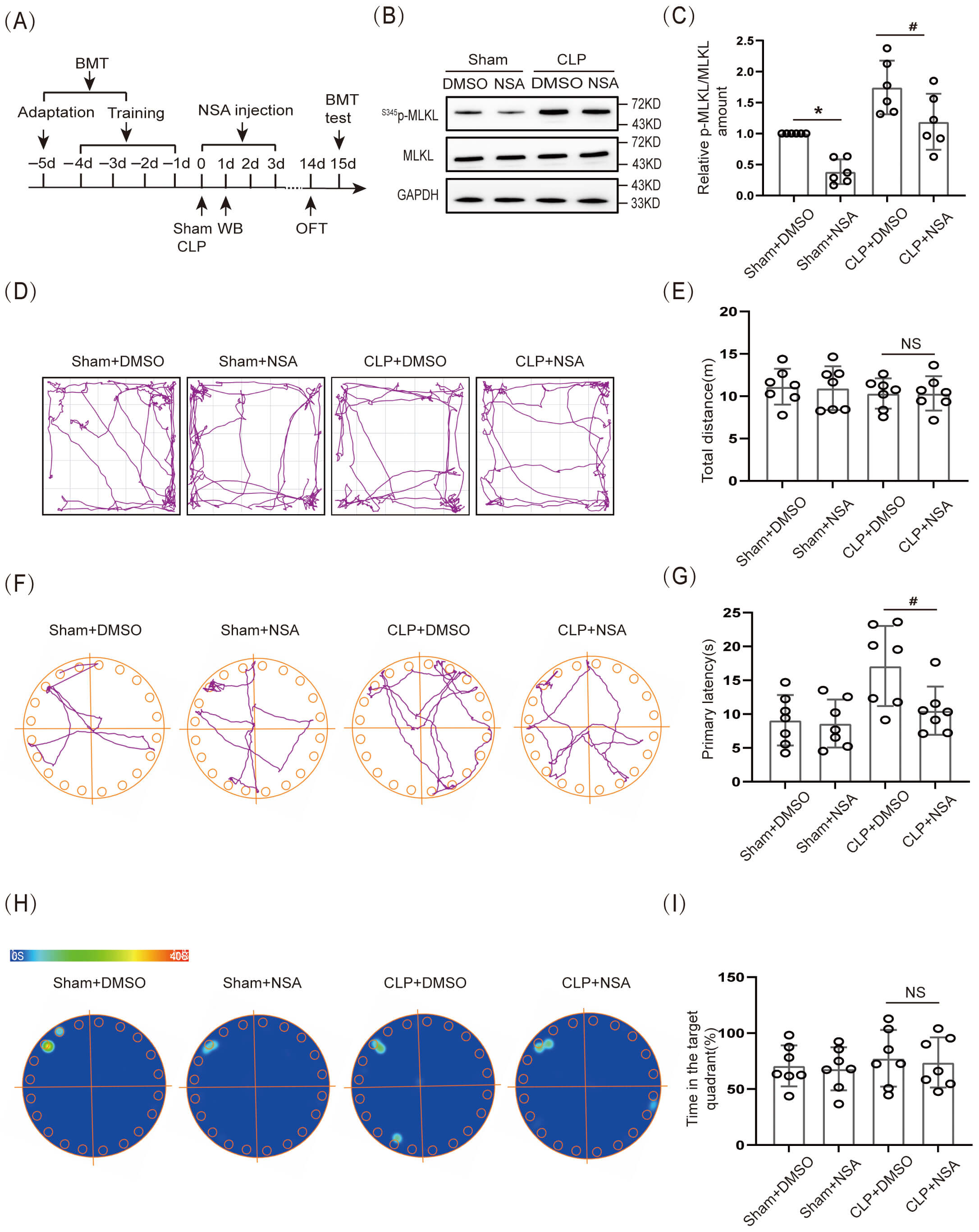 Fig. 3.
Fig. 3.
Necroptosis caused cognitive dysfunction in SAE mice.
(A) Schematic timeline of BMT, NSA injection, and OFT. (B) WB and (C) Statistical
results of S345p-MLKL/MLKL (n = 6, *p
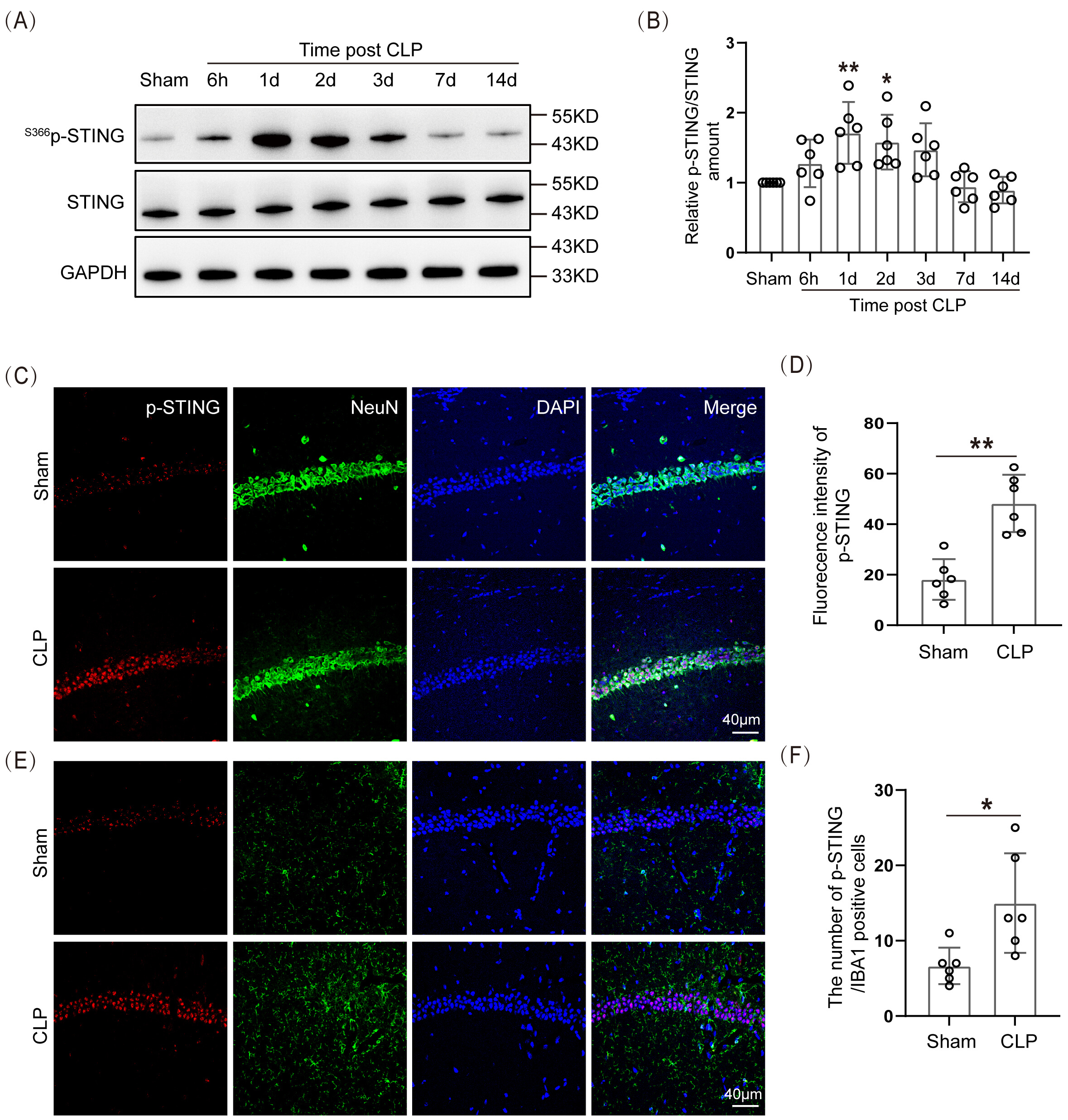
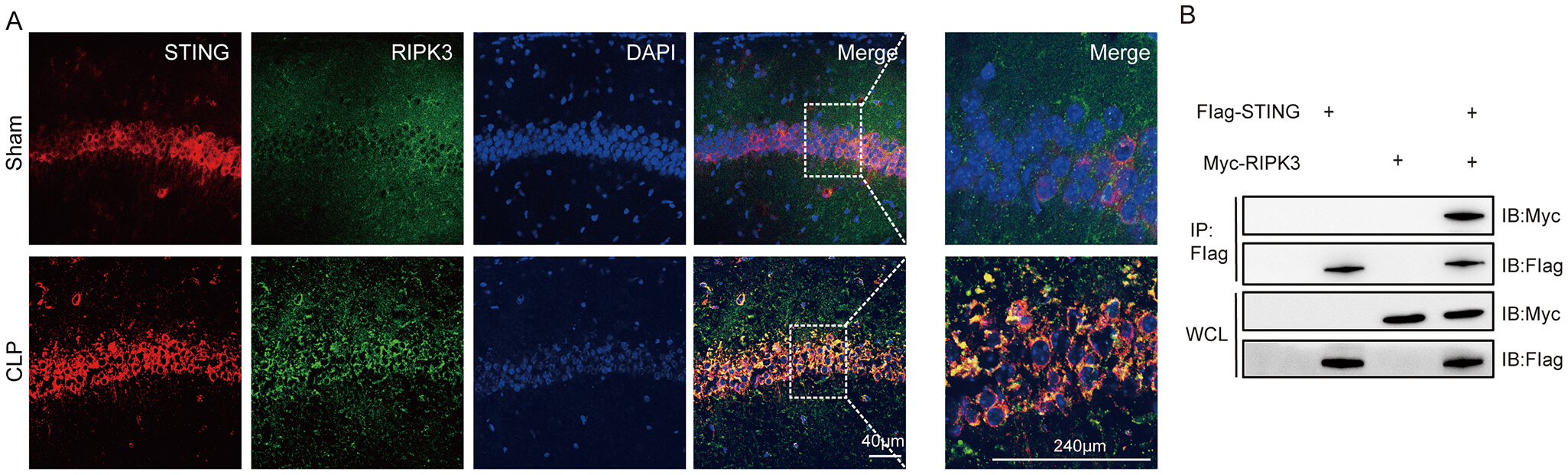
The data indicated that the expression of S366p-STING/STING proteins in the hippocampal tissue of the SAE mice was significantly higher 1 day and 2 days after modeling than in the Sham group (Fig. 4A,B; The original Western blot images can be found in the Supplementary Materials-Original WB). The IF staining of S366p-STING protein with neurons and microglia was performed. The data demonstrated that S366p-STING in the hippocampal CA1 region was mainly co-localized with hippocampal neurons in CLP mice, and there was a statistically significant difference in their IF intensities (Fig. 4C,D). There was a small amount of co-localization with microglia in the hippocampal CA1 region (Fig. 4E,F). This suggested that p-STING had elevated expression in neurons of the hippocampal CA1 region in SAE mice.
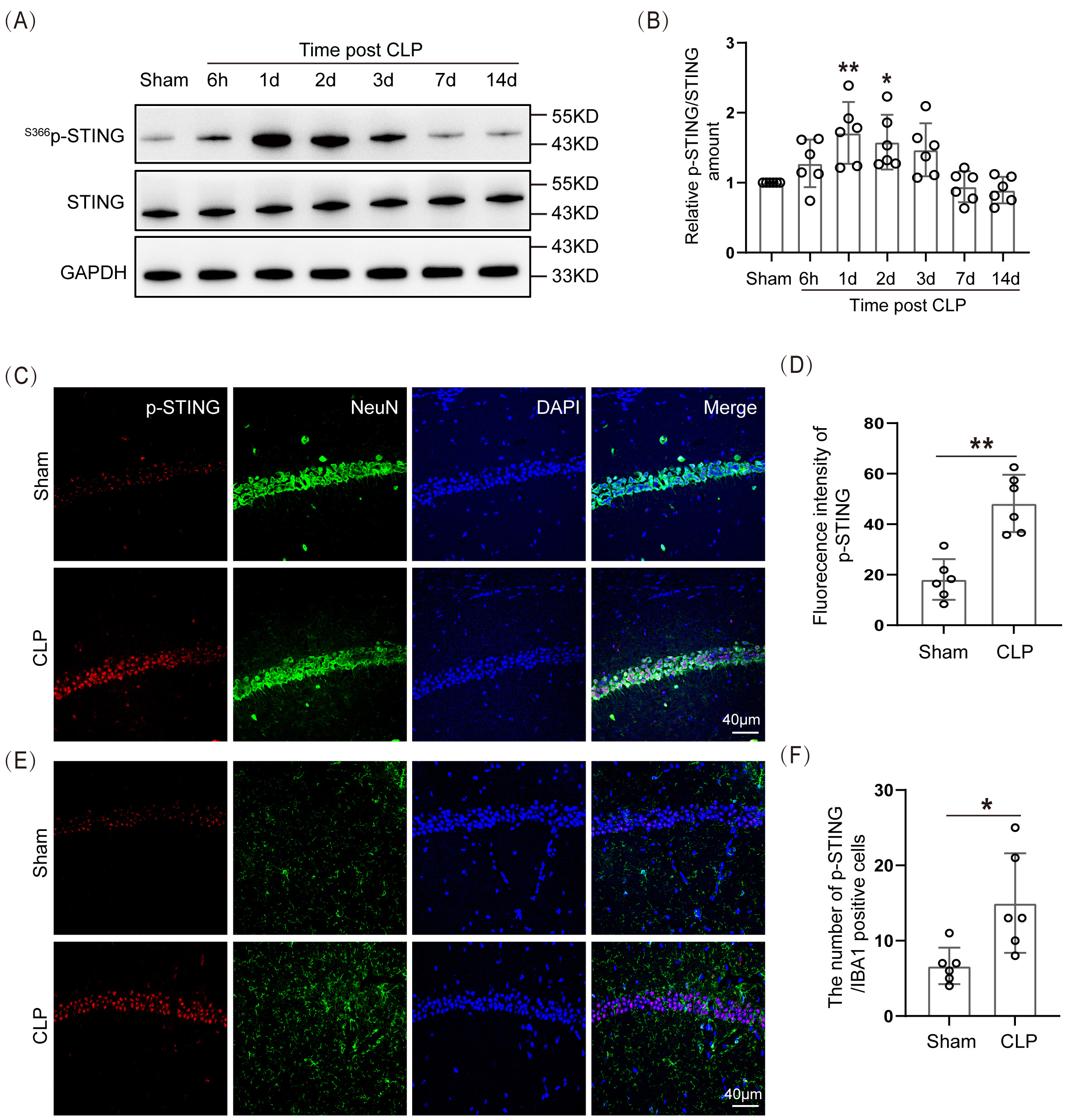 Fig. 4.
Fig. 4.
Spatiotemporal expression of STING protein increased in the
hippocampus of SAE mice. (A) WB and (B) Statistical results of
S366p-STING/STING protein expression levels (n = 6, *p
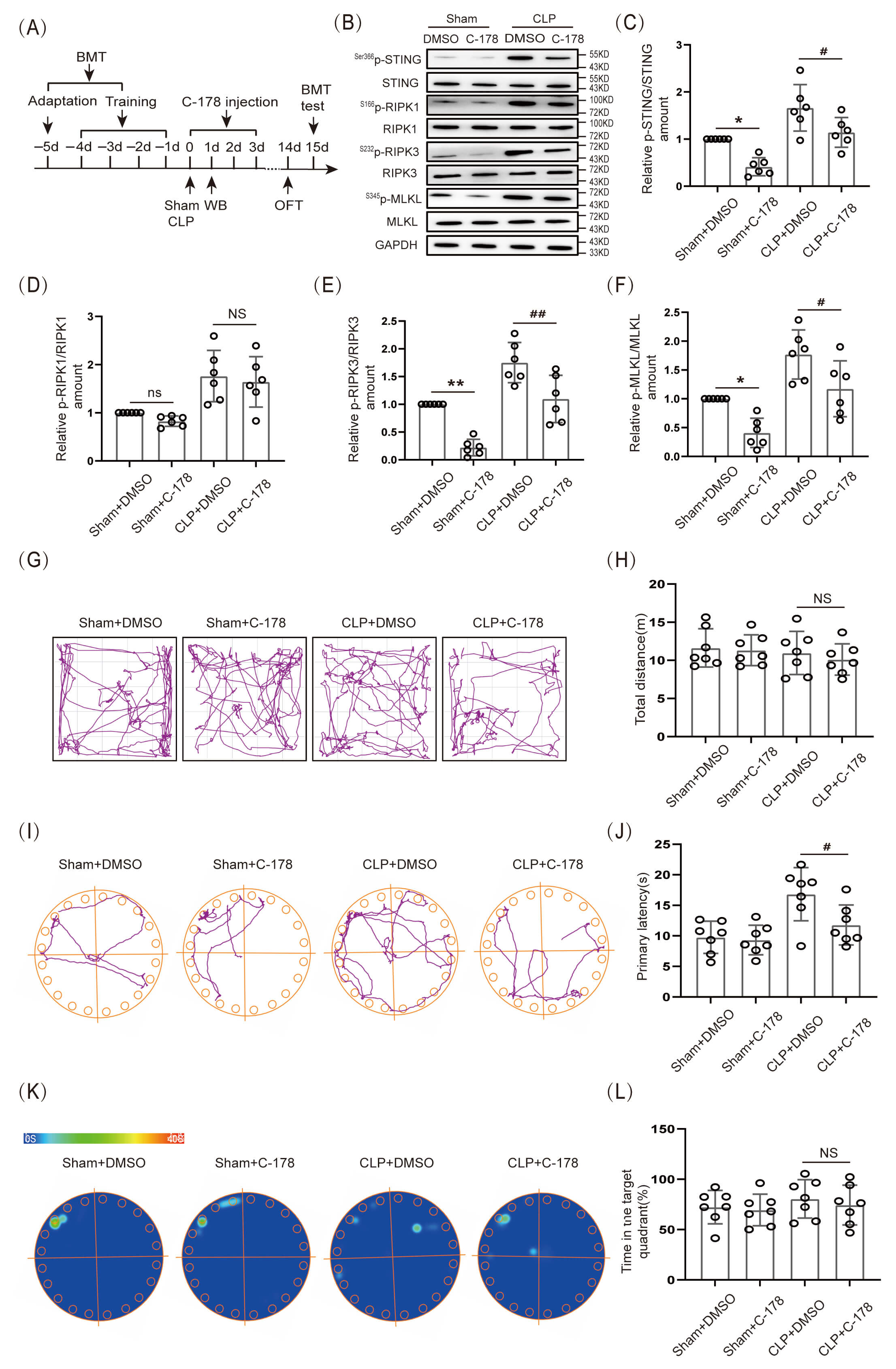
In the present experiment, 1.5 µL of STING inhibitor C-178 (1 mg/mL) was injected on the day of CLP, and the drug was administered for 3 consecutive days (Fig. 5A). The data indicated that the expression level of S366p-STING/STING, S232p-RIPK3/RIPK3 and S345p-MLKL/MLKL protein in the hippocampal tissue of CLP+C-178 group mice was obviously lower than that in the CLP+DMSO group (Fig. 5B,C,E,F; The original Western blot images can be found in the Supplementary Materials-Original WB), however, there was no significant decrease in S166p-RIPK1/RIPK1 protein expression (Fig. 5B,D). The results indicated that inhibition of STING down-regulated the level of S232p-RIPK3/RIPK3 and S345p-MLKL/MLKL proteins, however, it could not down-regulate the expression of S166p-RIPK1/RIPK1 protein. In addition, OFT data indicated that there was no significant difference in the locomotor ability of mice among the groups (Fig. 5G,H). BMT results demonstrated that the primary latency time to find the target hole (escape) was obviously shorter in the CLP+C-178 group mice than in mice of the CLP+DMSO group (Fig. 5I,J), but the difference in the time spent in the target quadrant was not statistically significant (Fig. 5K,L). Those results indicated that inhibition of STING partially improved BMT performance in SAE mice.
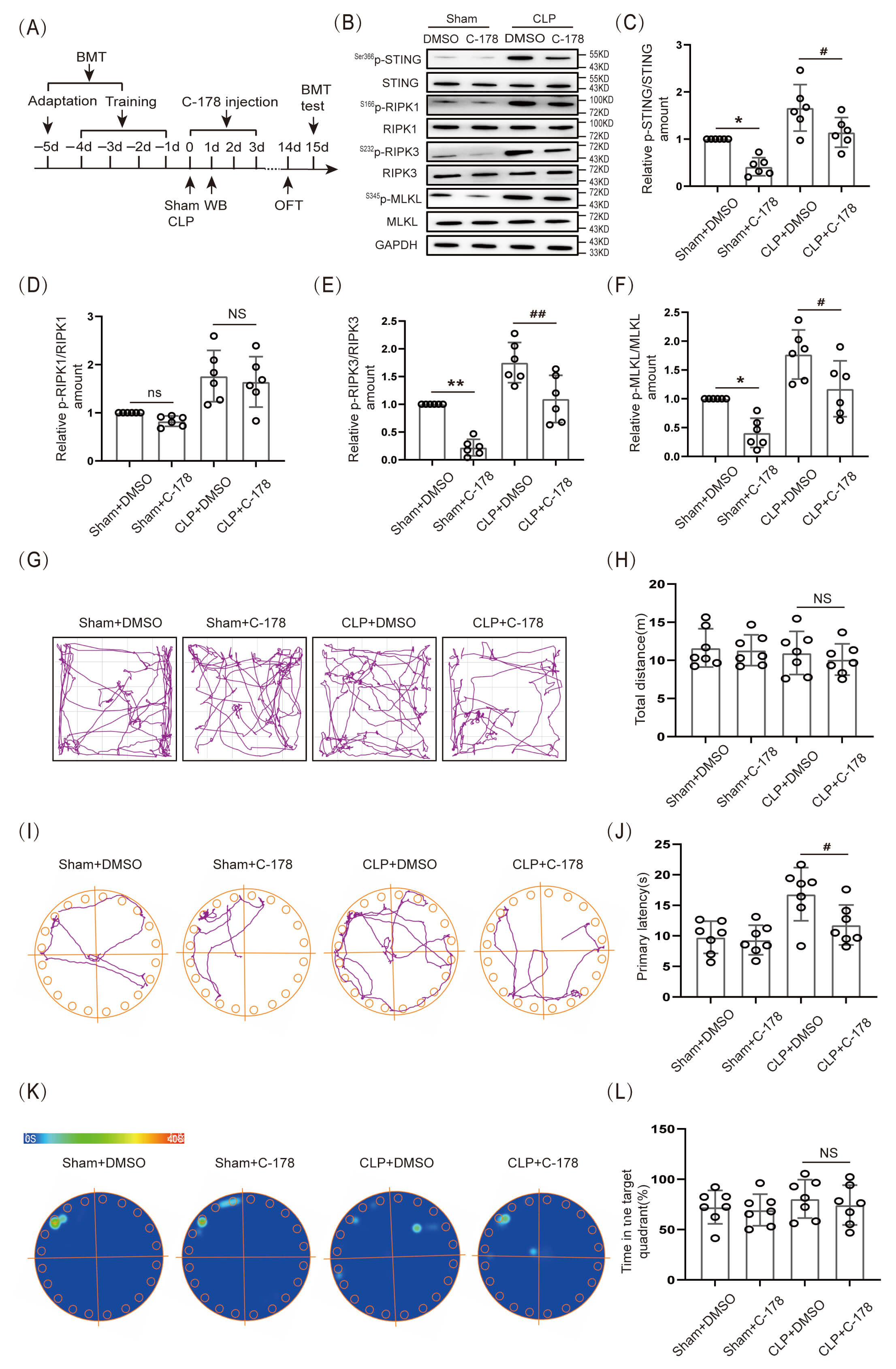 Fig. 5.
Fig. 5.
Inhibition of STING reduced the expression of associated
necroptosis proteins and improved cognitive function in SAE mice. (A) Schematic
timeline of BMT, C-178 injection, and OFT. (B) WB. (C–F) Statistical results of
S166p-RIPK1/RIPK1, S232p-RIPK3/RIPK3 and S345p-MLKL/ MLKL protein
expression levels after C-178 inhibitor (n = 6, *p
In this experiment, immunofluorescence and Co-immunoprecipitation (Co-IP) tests were used to detect the interaction between STING and RIPK3 protein. The results of IF showed that STING and RIPK3 proteins co-localized abundantly in neuronal cells in the hippocampal CA region of SAE mice (Fig. 6A). Co-IP results showed that STING interacted with RIPK3 protein (Fig. 6B; The original Western blot images can be found in the Supplementary Materials-Original WB) in neurons.
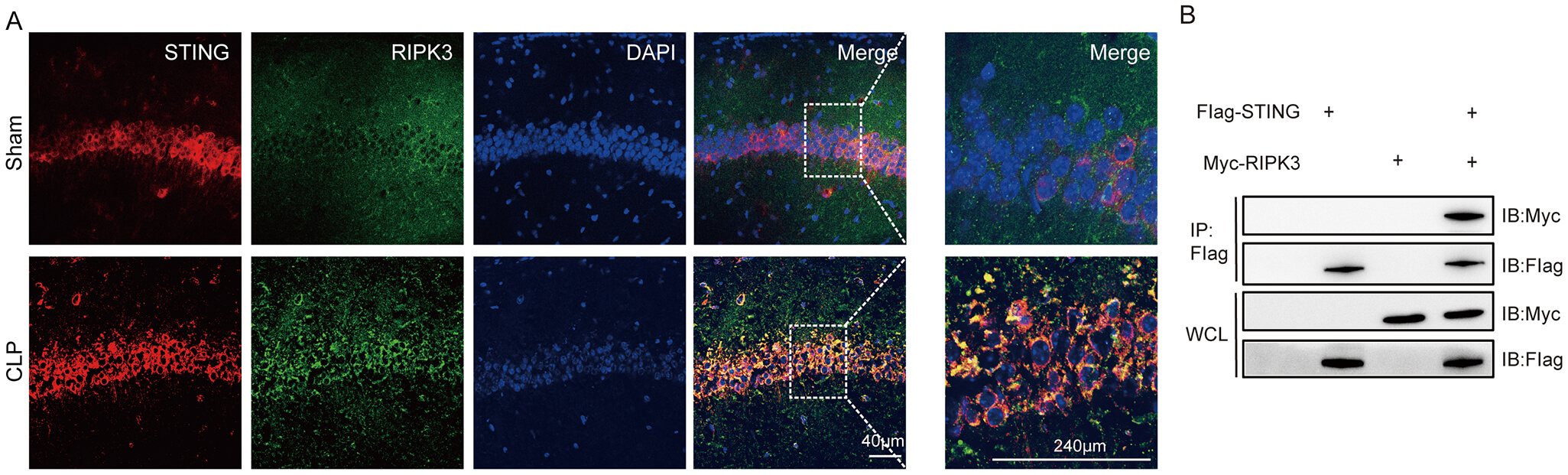 Fig. 6.
Fig. 6.
STING interacted with the RIPK3 protein. (A) IF of STING co-localized with RIPK3 (scale bar = 40 µm, 240 µm). (B) Co-IP of STING and RIPK3 proteins in lysates of hippocampal tissues. Co-IP, Co-immunoprecipitation; WCL, whole cell lysates; IB, immunoblotting; IP, immunoprecipitation.
In this study, we first experimentally verified that SAE mice developed cognitive dysfunction. According to the literature [14], the BMT behavioral paradigm can be used to reflect cognitive impairment. Our results showed that the latency to find the target hole (to escape a noxious stimulus) was significantly longer in septic mice. In previous studies [15], the latency and time to explore new objects in sepsis mice were obviously longer than in the Sham group mice, indicating that sepsis induced memory impairment. Therefore, the results of our experiments suggested that cognitive dysfunction occurs in SAE mice.
If so, what caused cognitive dysfunction to occur in SAE mice? We found that imaging and histological examination results demonstrated that the hippocampal volume and cell number were evidently reduced in the SAE group [16, 17]. This suggested that hippocampal neuronal death may be a potential mechanism leading to the occurrence of cognitive dysfunction in SAE. A study has reported that neurons of the hippocampal CA1 region play an important role in learning and memory [18, 19]. The reason for this is that the hippocampal CA1 region is rich in pyramidal neurons and pyramidal axial-dendritic synapses, and has high blood flow and energy requirements [20, 21]. Therefore, during sepsis, neurons in the hippocampal CA1 region are prone to die, causing cognitive dysfunction.
Programmed cell death is a genetically controlled mode of autonomous and orderly cell death that comes in various forms, such as pyroptosis, necroptosis, apoptosis, and ferroptosis [22]. Previous findings suggested that programmed cell death plays an important role in central nervous system (CNS) disorders, but the mechanism is not clear. Recent studies have shown that neuronal necroptosis is strongly related to neurodegenerative diseases [7], and inhibition of necroptosis can improve cognitive dysfunction in neurodegenerative diseases [8, 9, 10]. However, the mechanism of necroptosis in cognitive impairment in SAE is currently unclear.
Our results indicated that necroptotic proteins S166p-RIPK1/RIPK1, S232p-RIPK3/RIPK3, and execution protein S345p-MLKL/MLKL were significantly elevated in the hippocampus of SAE mice. In addition, IF results suggested that the level of S345p-MLKL was obviously elevated in neurons in the hippocampal CA1 region. These results suggested that sepsis induced necroptotic signaling in hippocampal neurons, and phosphorylation of RIPK1 and RIPK3, which led to phosphorylation of MLKL. Phosphorylated p-MLKL binds to cell membranes to form “holes” that lead to cytoplasmic leakage, cell swelling, and even cell death. Dead cells release inflammatory factors, which further exaggerate necroptosis and form a vicious circle [23, 24, 25]. Our experimental results indicated that hippocampal neurons in SAE mice incurred necroptosis. So, would inhibition of necroptosis attenuate the occurrence of cognitive dysfunction in SAE mice? We were surprised to find that the latency to escape was significantly shorter in MLKL-inhibited SAE mice. This suggested that necroptosis mainly contributed to cognitive dysfunction in SAE mice. However, the mechanism was still unclear.
STING proteins are also known as TMEM173, MITA, MPYS, NET23, ERIS, etc. [26].
STING was first reported in 2008 by Ishikawa and Barber [27]. It
consists of 379 amino acids and is anchored to the endoplasmic reticulum membrane
by the N-terminus. The C-terminus includes the C-terminal tail and ligand-binding
domains [27, 28, 29]. Ligand binding to STING leads to its dimerization and
phosphorylation. Then, S366p-STING may regulate programmed cell death and
inflammatory responses via the TANK-binding kinase 1
(TBK1)/interferon regulatory factor 3 (IRF3)/nuclear factor-kappa B
(NF-
However, some limitations existed in our study. First, the specific molecular mechanism of STING interaction with RIPK3 needs to be further elucidated. Second, further validation of the safety and efficacy of STING inhibitors in patients with clinical SAE is needed.
STING participates in the development of cognitive dysfunction in SAE by regulating necroptosis in hippocampal neurons. The STING inhibitor C-178 showed the potential to partially improve Barnes maze performance in SAE and suggested new avenues for clinical treatment.
Data and materials relevant to support the current study can be obtained from our corresponding authors.
XFG, BCC and YHB initiated the research and wrote the manuscript. DZ analyzed the data and performed the experiments. YW, TWS and XJZ participated in the study design, interpreted the experimental results, and revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was approved by the committee on ethics of medicine of the 988th hospital of joint logistics support forces of PLA with approval number (No. 988YY20240023LLSP). Our animal experiments adhered to the protocols outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Not applicable.
This research was funded by joint construction project of medical science and technology tackling plan of Henan province, grant number LHGJ20240976. This research was funded by general program of natural science foundation of Henan province, grant number 252300421400.
The authors declare no conflicts of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN50074.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.