


 , Anna Futyma 1,2, Olga Kokorniak 1,2, Aleksandra Szczech 1,2, Bartosz Słowikowski 3, Joanna Poszwa 1, Oliwia Szymanowicz 1,4, Ulyana Goutor 1, Paweł P. Jagodziński 3, Wojciech Kozubski 5, Jolanta Dorszewska 1,*
, Anna Futyma 1,2, Olga Kokorniak 1,2, Aleksandra Szczech 1,2, Bartosz Słowikowski 3, Joanna Poszwa 1, Oliwia Szymanowicz 1,4, Ulyana Goutor 1, Paweł P. Jagodziński 3, Wojciech Kozubski 5, Jolanta Dorszewska 1,*
1 Laboratory of Neurobiology, Department of Neurology, Poznan University of Medical Sciences, 60-355 Poznan, Poland
2 Student Scientific Society, Poznan University of Medical Sciences, 60-355 Poznan, Poland
3 Department of Biochemistry and Molecular Biology, Poznan University of Medical Sciences, 60-355 Poznan, Poland
4 Doctoral School, Poznan University of Medical Sciences, 60-355 Poznan, Poland
5 Department of Neurology, Poznan University of Medical Sciences, 60-355 Poznan, Poland
Abstract
Parkinson’s disease (PD) is the second most common degenerative disease of the central nervous system, characterized by both motor and non-motor disorders. Non-motor disorders include dementia, depression, and anxiety. The prevalence of dementia increases with disease progression, affecting 24–31% of PD patients and up to 75% after 10 years. Furthermore, approximately 50% of PD patients suffer from depression, while anxiety affects around 31% of this population. Molecular factors, including genetic factors, are currently implicated in the manifestation of neuropsychiatric disorders. It is believed that dementia and affective disorders in PD may result from both disturbed homeostasis of proteins such as α-synuclein, amyloid beta, and phosphorylated tau, as well as increased inflammatory processes and impaired neurotransmission. Chronic microglial activation, elevated levels of proinflammatory cytokines, and oxidative stress contribute to the severity of neuroinflammation, which in turn contributes to neuronal dysfunction and synaptic abnormalities. While the effect of pathological proteins in PD primarily involves impairment of dopaminergic neurotransmission, disturbances in the serotonergic, noradrenergic, cholinergic, and glutamatergic systems are also observed. Furthermore, genetic risk factors have been identified in PD patients, including monogenic causes, which are rare in unselected populations. Identifying causative molecular targets in PD is essential for developing therapies for this neurodegenerative disease. In this review, we present current views on the involvement of molecular factors in the development of psychiatric disorders in patients with PD, suggesting that they may have diagnostic and predictive value in the future.
Keywords
- molecular factors
- dementia
- depression
- anxiety
- Parkinson’s disease
Parkinson’s disease (PD) is a complex, progressive neurodegenerative disorder primarily characterized by resting tremor, bradykinesia, rigidity, postural instability [1], and numerous additional motor symptoms (Table 1, Ref. [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17]). However, beyond these most recognizable motor symptoms, PD, especially in later stages, is frequently associated with a broad spectrum of neuropsychiatric problems, such as depression, anxiety, cognitive impairment, psychosis, mood swings, and apathy [1, 18]. These non-motor symptoms (NMS) significantly affect patients’ quality of life and may even precede motor manifestations (Table 1). Notably, in some studies, participants reported that non-motor fluctuations caused even greater disability [19]. It may be suggested that neuropsychiatric complications are associated with disease development rather than being mere consequences of disability [20]. Understanding the molecular mechanisms underlying neuropsychiatric symptoms in PD remains an important challenge. Emerging evidence suggests that these manifestations are connected to widespread neurodegenerative changes beyond the dopaminergic system, involving alterations in multiple neurotransmitter systems, neuroinflammatory processes, genetic predispositions, and dysregulated cellular signaling pathways [21, 22]. The interaction among these molecular mechanisms may contribute to the heterogeneity of neuropsychiatric phenotypes observed among PD patients.
| Motor symptoms | Non-motor symptoms |
| ● Bradykinesia/akinesia | ● Psychiatric symptoms (depression, anxiety, apathy, cognitive slowing, cognitive deficits, dementia, psychosis, hallucinations) |
| ● Muscular rigidity | |
| ● 4–6 Hz resting tremor | |
| ● Postural instability | ● Sleep disorders |
| ● Postural deformities (e.g., camptocormia, antecollis, scoliosis) | ● Sensory symptoms and pain (tingling, burning sensations, neuralgic pain) |
| ● Gait disturbances | ● Olfactory dysfunction |
| ● Loss of spontaneous movements (e.g., decreased gesturing, blinking, hypomimia) | ● Autonomic dysfunction (excessive sweating, orthostatic hypotension, sexual dysfunction, constipation, urinary incontinence, esophageal dysmotility) |
| ● Impaired fine motor skills (e.g., micrographia) | |
| ● Oral motor impairments (disturbed speech, swallowing and saliva control) | ● Executive dysfunction, deficits in speech, visuospatial abilities, and memory |
| ● Dystonia |
One of the primary factors associated with neuropsychiatric symptoms in PD is
the dysfunction of multiple neurotransmitter systems. It is commonly known that
dopaminergic neurodegeneration in the substantia nigra is central to PD
pathophysiology [2, 22]. In addition, alterations in cholinergic, serotonergic,
noradrenergic,
| Neurotransmitter system | Dysregulation in Parkinson’s disease | Mechanism |
| Dopamine | ↓ downregulated | Degeneration of substantia nigra neurons |
| Acetylcholine | ↓ downregulated | Degeneration of basal forebrain cholinergic neurons |
| Serotonin | ↓ downregulated | Loss of SERT binding in the dorsal raphe nuclei |
| Noradrenaline | ↓ downregulated | Degeneration in locus coeruleus neurons |
| GABA | ↑ upregulated | Increased inhibitory output from globus pallidus internus/striatum |
| Glutamate | ↑ upregulated | Hyperactivity in the subthalamic nucleus and cortex |
GABA,
Neuroinflammation has also been recognized as an important factor contributing to neuropsychiatric disturbances in PD. Chronic activation of microglia, increased levels of pro-inflammatory cytokines, and oxidative stress influence neuronal dysfunction and synaptic abnormalities, thereby exacerbating both motor symptoms and NMS [25, 26]. Elevated inflammatory markers in the cerebrospinal fluid and postmortem brain tissue of PD patients have been correlated with severity of depression and cognitive decline, emphasizing the role of immune dysregulation in the disease’s neuropsychiatric manifestations [21, 27, 28].
Furthermore, genetic predisposition plays a role in the onset of
neuropsychiatric symptoms in PD. Nucleotide variants present in genes such as the
Given the multifactorial and complex nature of neuropsychiatric disorders in PD, a comprehensive understanding of their molecular basis is essential for developing targeted therapeutic strategies. Current pharmacological treatments, including selective serotonin reuptake inhibitors (SSRIs), dopamine agonists, and acetylcholinesterase inhibitors, offer symptomatic relief but often fail to address the pathophysiological causes [34]. Therefore, novel interventions aimed at modulating neuroinflammation, restoring neurotransmitter balance, and preserving mitochondrial function hold promise for improving neuropsychiatric outcomes in PD.
This review provides an in-depth analysis of the molecular mechanisms underlying neuropsychiatric symptoms in PD, highlighting recent advances in our understanding of neurotransmitter dysregulation, neuroinflammation, genetic susceptibility, and cellular dysfunction. By elucidating these complex interactions, this review may help pave the way for innovative therapeutic approaches that can improve the quality of life for individuals with PD.
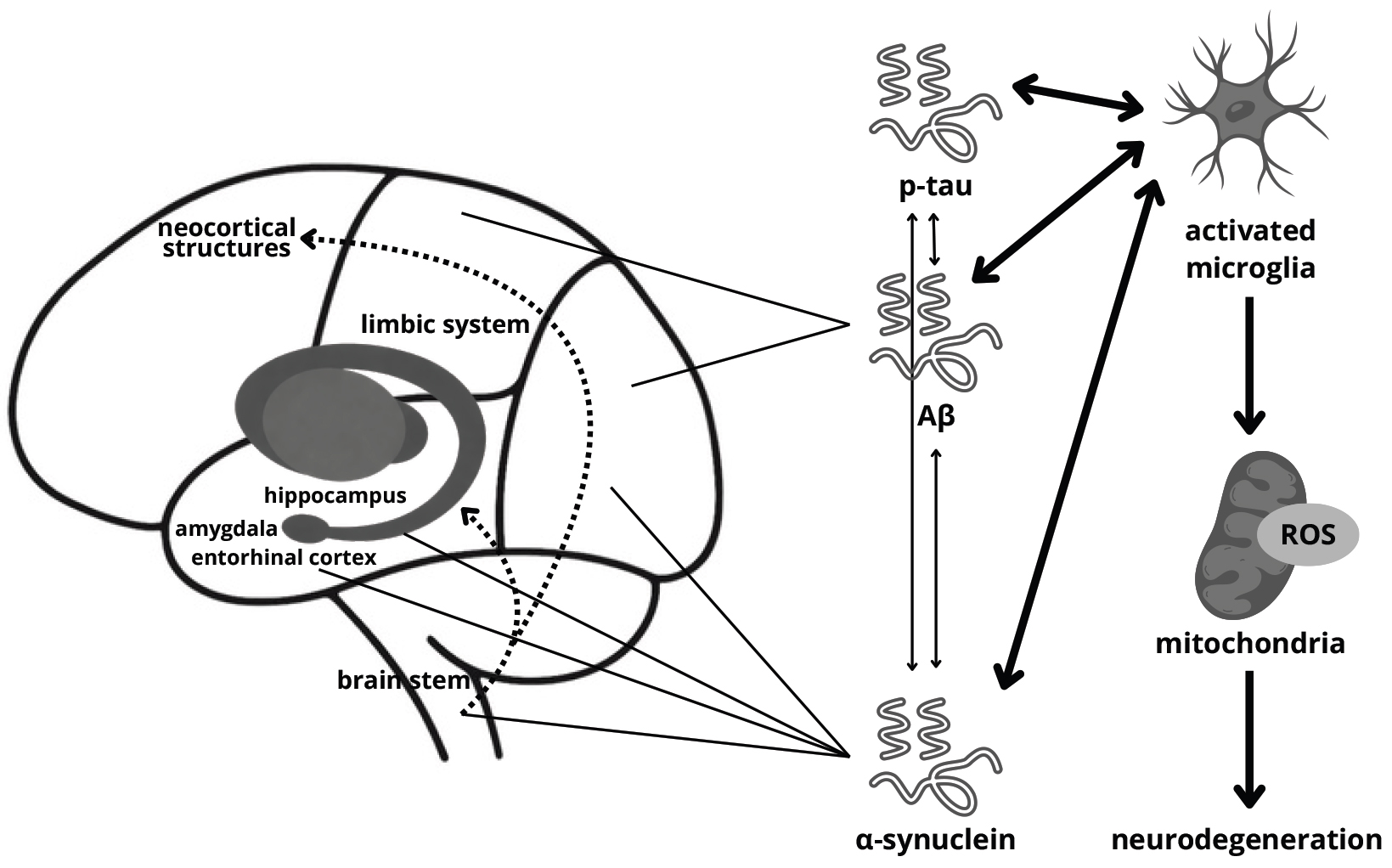
Parkinson’s disease dementia (PDD) may develop via various pathophysiological pathways. Proposed mechanisms include protein misfolding, synaptic dysfunction and loss, neurotransmitter activity, microglial and astroglial alterations, adenosine receptor activation, cerebral network disruption, neuroinflammation, aberrant mitochondrial function, and retrograde signaling (Fig. 1) [35, 36].
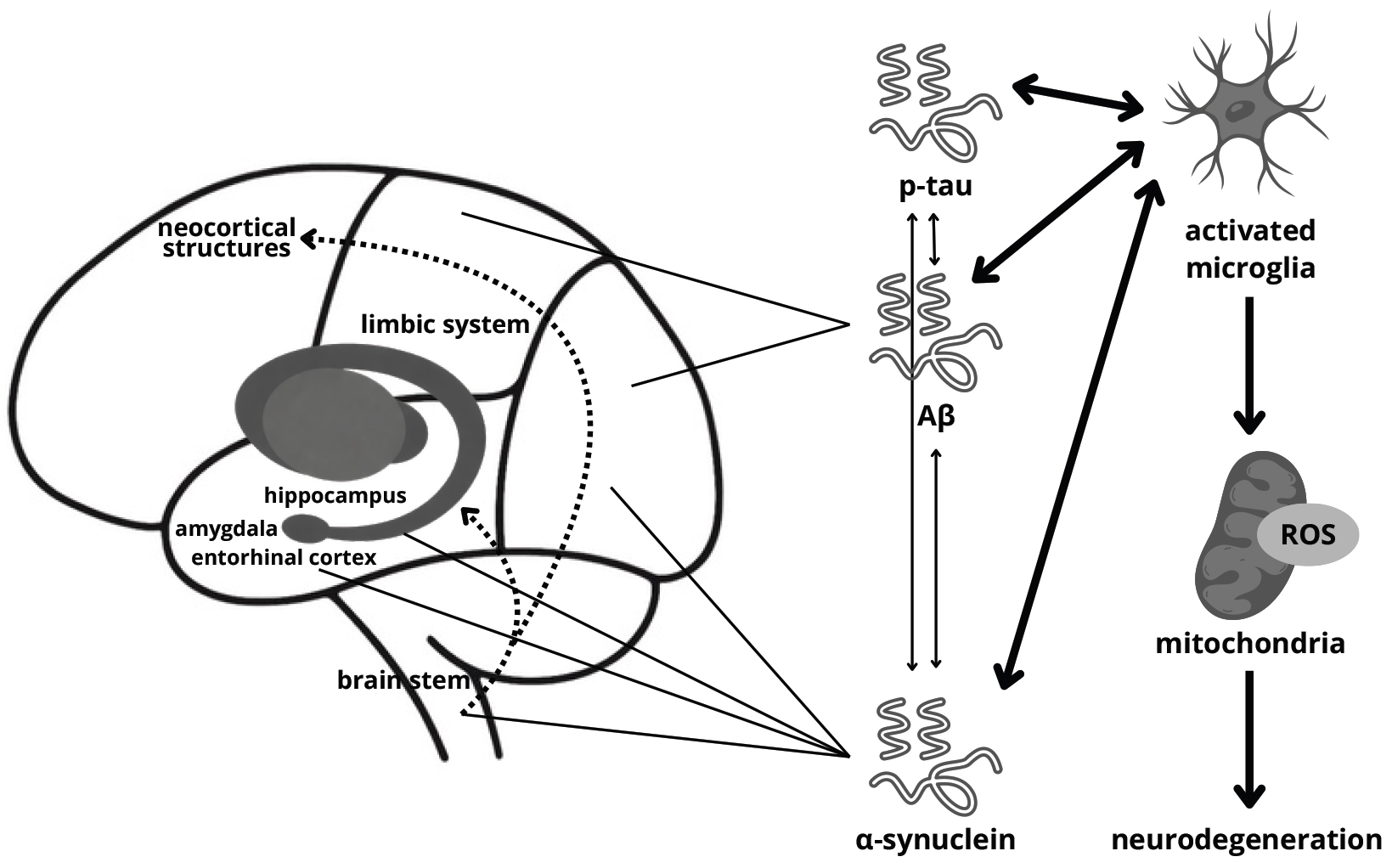 Fig. 1.
Fig. 1.
Probable mechanism of dementia development in
Parkinson’s disease. ROS, reactive oxygen species; p-tau, phosphorylated tau;
A
The major pathological protein of PD is
Interestingly, studies show that many patients affected by PDD present
Alzheimer’s disease (AD) pathology—accumulation of amyloid beta (A
Although pathological proteins are implicated in PDD pathogenesis, growing
evidence suggests that additional factors are involved in the development of
cognitive decline in PD [60]. A recent study shows that the PDD pathomechanism
may include oxidative stress, Toll-like Receptor (TLR) 4/9 activation, impaired
DNA binding, and cytosolic DNA sensing. Additionally, aberrant regulation of
interferon (IFN) signaling triggers damage to mitochondrial DNA (mtDNA), which
spreads in an infectious-like pattern involving extracellular vesicles and
Ribosomal Protein S3 (rpS3). Evidence indicates that interferon-beta
(IFN-
Cognition in PD may also be influenced by neurotransmitter functioning [73, 74]. In vivo observations indicate that PDD patients exhibit cholinergic and dopaminergic deficit profiles in the brain [75, 76]. Similarly, postmortem examinations show decreased choline acetyltransferase (ChAT) activity in PDD [77, 78]. Moreover, there is a significant elevation in muscarinic acetylcholine receptors in specific brain regions in PDD [79]. Evidence indicates that serotonin and noradrenaline pathways may also be disturbed, contributing to alterations in neural networks (Table 2) [74, 80, 81, 82].
Aberrant functioning of synapses and impaired synaptic plasticity are other factors implicated in PDD etiology [83, 84]. Synaptic plasticity contributes to neuronal connections, altering the structure of dendritic spines and synapses, axonal modification, and synaptogenesis [85]. Neurotrophins are important molecular mediators of synaptic plasticity and include multiple molecules: immunological factors and biogenic amines described above, as well as growth factors [83, 86]. Deregulation of growth factors: epidermal growth factor (EGF), nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and glial-cell-derived neurotrophic factor (GDNF) seems to be a part of PDD pathology related to synaptic dysfunction [83, 87, 88, 89, 90]. Evidence suggests that synaptic markers such as presynaptic vesicle protein Ras-related protein (Rab3A) and synaptosomal-associated protein, 25 kDa (SNAP25), neurogranin, zinc transporter 3 (ZnT3), postsynaptic density protein 95 (PSD95), synaptopodin, and Dynamin-1 participate in vesicle recycling, vesicle docking, postsynaptic signaling, synaptic release and regulation of divalent zinc cation (Zn2+), regulation of postsynaptic neurotransmitter receptors, cellular scaffolding, neurotransmitter reuptake, or receptor internalization, and play a role in PDD pathogenesis [91, 92, 93, 94].
Similarly to PDD, the pathogenesis of affective disorders in PD remains poorly
understood [95, 96, 97]. Emerging evidence indicates that common pathological
mechanisms, including
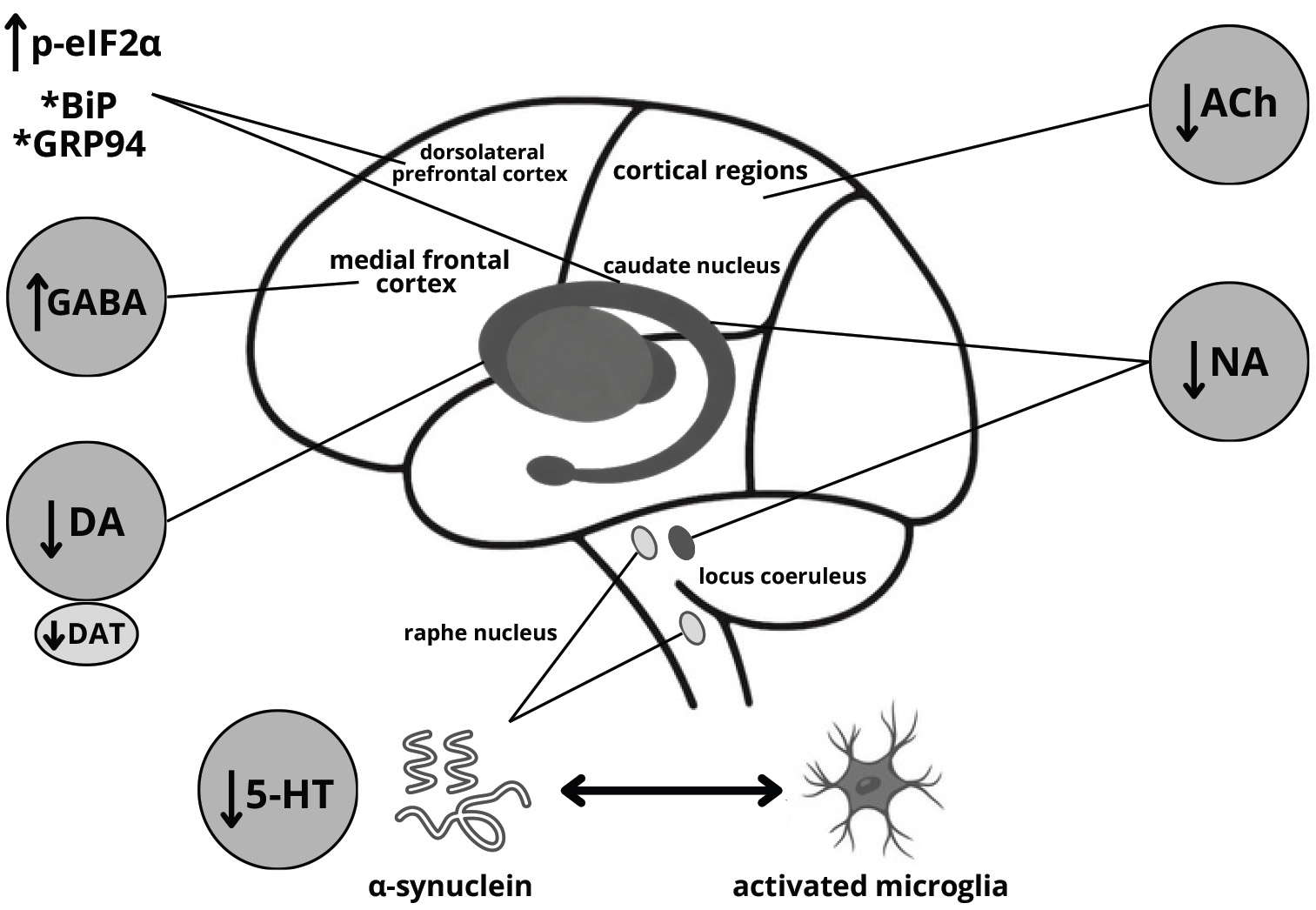 Fig. 2.
Fig. 2.
Probable mechanism of depression development in
Parkinson’s disease. *levels altered in a region-dependent manner. DA, dopamine;
DAT, dopamine transporter; Ach, acetylcholine; NA, noradrenaline; 5-HT,
serotonin; p-eIF2
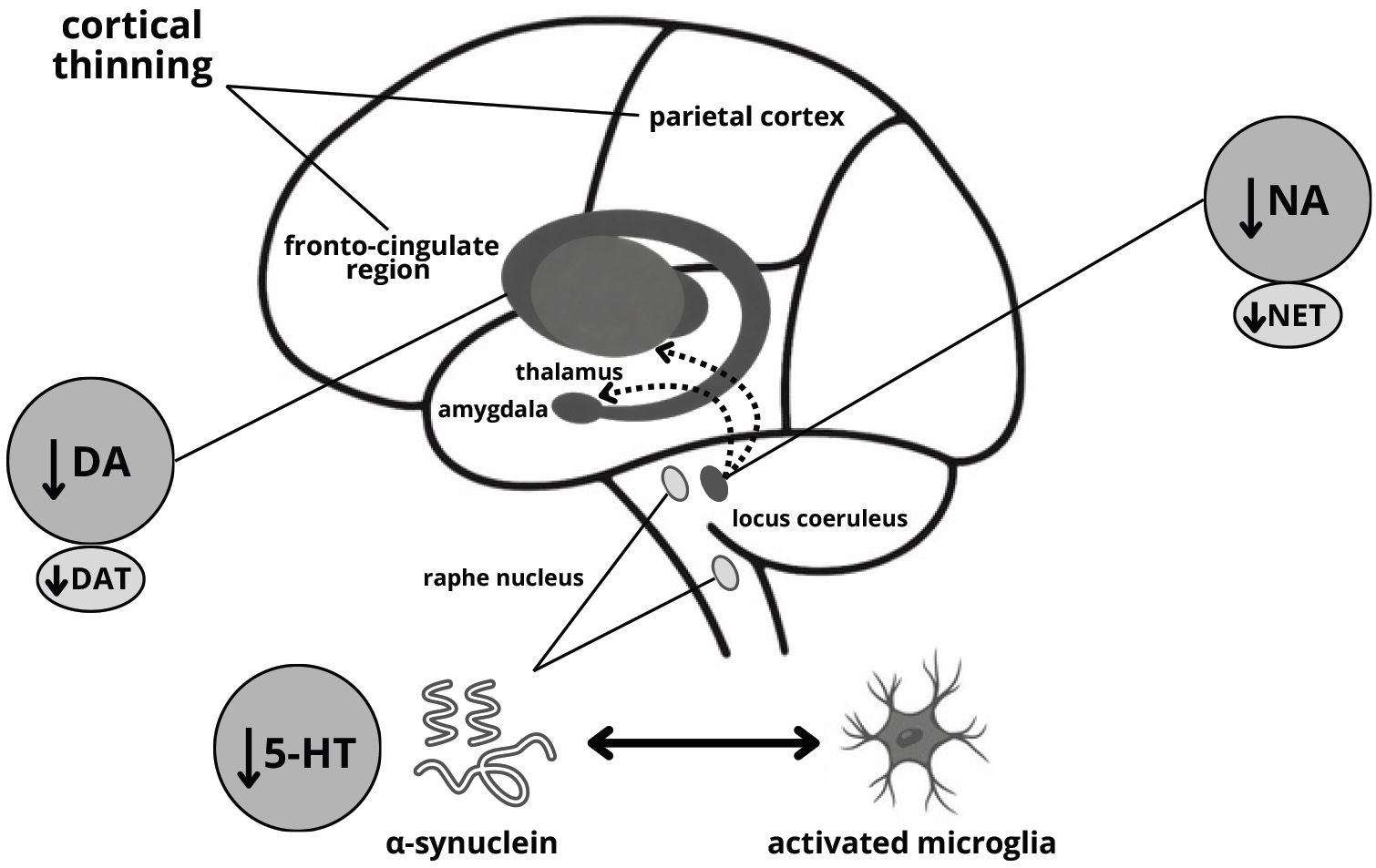
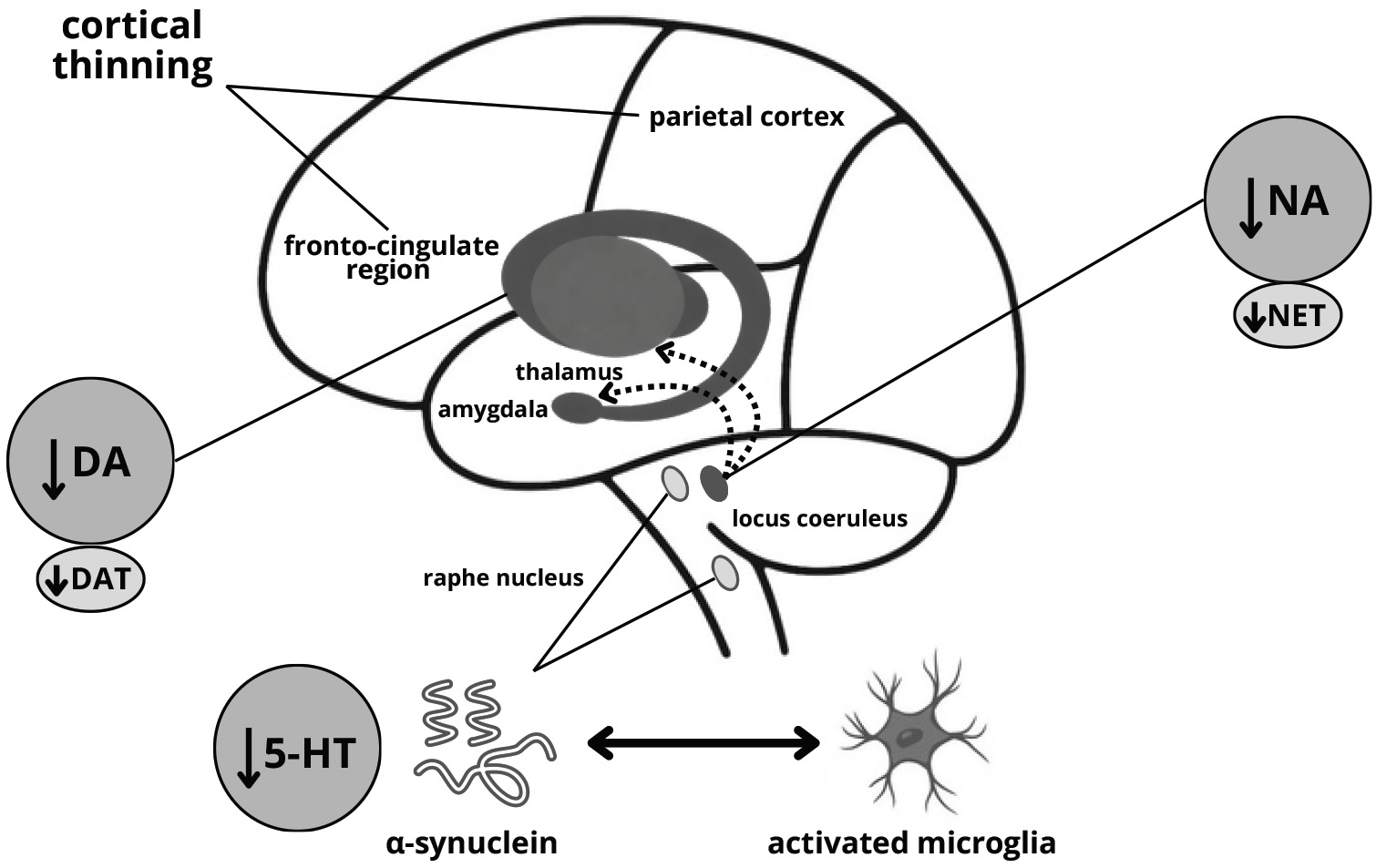 Fig. 3.
Fig. 3.
Probable mechanism of anxiety development in Parkinson’s disease. NET, norepinephrine (noradrenaline) transporter.
2.2.1.1 Protein Pathology
In addition to its well-established role in motor dysfunction,
2.2.1.2 Neuroinflammation
Emerging evidence suggests that neuroinflammation contributes to the development
of depressive symptoms in PD and is particularly associated with altered function
and connectivity of the posterior cingulate cortex and insula, as well as the
amygdala, hippocampus, precuneus, and frontal cortex [103, 104]. Recent research
shows that PD depression is associated with neuroglial activation-induced
dopaminergic neuron apoptosis marked by increased levels of soluble triggering
receptor expressed on myeloid cells 2 (sTREM2) and chitinase-3-like protein 1
(YKL-40) in the cerebrospinal fluid [105]. While these findings suggest potential
diagnostic and prognostic value, it is important to note that analytical factors
such as assay platform, pre-analytical handling of CSF, and inter-laboratory
standardization were not fully assessed, and further validation is needed to
confirm clinical utility. Moreover, therapeutic approaches in PD models indicate
that reducing ionized calcium-binding adapter molecule 1 (IBA-1) and GFAP
positive cells, as well as phospho-nuclear factor kappa-light-chain-enhancer of
activated B cells (NF-
2.2.1.3 Neurotransmitter Activity, Synaptic Function
Alterations in dopaminergic, serotonergic, cholinergic, and noradrenergic systems are important parts of PD depression etiology. Reports show that PD depression is linked with a specific loss of noradrenaline and dopamine innervation in the limbic system [114]. Moreover, rat models show that presynaptic dopamine receptors participate in PD depression regulation [115]. Dopamine transporter density and availability in PD depression differ in various brain regions and the results remain inconclusive; however, a decreasing tendency is predominant [116, 117, 118, 119, 120, 121]. Concomitantly, Maillet et al. [122] showed that serotonergic disruption plays a prominent role in de novo PD depression. Moreover, reports indicate that serotonergic dysfunction occurs early in PD development; however, its correlation with NMS is not confirmed [123, 124]. Indeed, reports regarding the activity of serotonin receptors are inconclusive; some suggest standard activity in early PD without depression, whereas others dysregulated [125, 126]. Politis et al. [127] conclude that higher serotonin transporter binding in raphe and limbic structures correlates with depression in PD; however, serotonin receptor density differs between specific brain regions, as another study shows [128]. This complex interplay of serotonergic and dopaminergic systems is further influenced by cholinergic and noradrenergic systems [113, 120]. Degeneration of the cholinergic system is associated with PD pathology [129, 130]. Evidence shows that cholinergic cortical denervation is also linked with PD depression and tends to be more evident in this disorder [131]. When it comes to the noradrenergic pathway in PD depression, studies show decreased noradrenaline levels, neuronal loss in locus coeruleus, which is a primary source of this neurotransmitter, and loss of noradrenaline innervation in the limbic system [98, 114, 120, 132]. Interestingly, there is also GABAergic dysfunction observed in PD depression, predominantly affecting the medial frontal cortex via elevated GABA+ levels [133]. Furthermore, similarly to PDD, neurotrophic alterations are implicated in the pathophysiology of depression in PD, leading to impaired synaptic plasticity. Emerging evidence demonstrates that BDNF levels decrease in PD depression patients [134, 135, 136]. Moreover, a therapeutic activation of the BDNF-involved pathway in a PD depression model alleviated depressive symptoms [137]. However, a meta-analysis shows that PD patients have reduced BDNF levels irrespective of depression status [138], highlighting the need for further research to determine whether BDNF alterations are specifically linked to depressive symptoms or reflect general neurodegenerative processes in PD.
2.2.2.1 Protein Pathology
Accumulation of pathological proteins, particularly
2.2.2.2 Neuroinflammation
Similar inflammatory mediators are implicated in both anxiety and depression;
however, in anxiety they tend to promote hyperreactivity of fear-related neural
circuits—particularly those involving the locus coeruleus and amygdala—rather
than contributing primarily to low mood [140, 141]. In animal models,
interventions that reduce microglial activation—such as botulinum neurotoxin A
or baicalein, which also reduces
2.2.2.3 Neurotransmitter Activity, Synaptic Function
Anxiety in PD involves dysfunctions of serotonergic, dopaminergic, and noradrenergic neurotransmission and its neurobiological basis appears to be closely related to fear and stress circuitry. Retrospective studies link anxiety with disease progression and withdrawal of dopaminergic medication, while postmortem analyses show degeneration of the amygdala, a key structure for fear responses that receives mesolimbic dopaminergic input. However, dopaminergic medication does not consistently alleviate anxiety as it does motor symptoms, and anxiety can precede motor deficits by decades, appearing even in medication-naïve patients at diagnosis. These observations suggest that anxiety in PD arises from early alterations in brainstem structures involved in stress responses, including the serotonergic raphe nuclei and noradrenergic locus coeruleus, as well as from alterations in large-scale network connectivity that may involve cognitive circuits before clinical symptom onset. Genetic factors, family history of anxiety, and sex-specific influences further modulate susceptibility. Collectively, this evidence indicates that anxiety in PD reflects complex interactions between multiple neurotransmitter systems and network-level synaptic dysfunction, rather than a single pathway [143]. Neurochemical imaging studies provide further evidence for specific circuit-level alterations. PET using [11C]RTI-32, a marker of dopamine and noradrenaline transporter binding, revealed that anxiety severity in PD patients inversely correlates with transporter availability in the amygdala, locus coeruleus, and thalamus. These findings suggest that loss of dopaminergic and noradrenergic innervation in limbic and brainstem structures contributes directly to anxiety symptoms. Together with serotonergic deficits in the raphe nuclei, these alterations highlight a network-level dysfunction across multiple neurotransmitter systems, including dopamine, noradrenaline, and serotonin, which underlie the synaptic and circuit disruptions responsible for heightened anxiety in PD [100, 140, 144].
Symptoms such as depression, anxiety, apathy, psychosis, and cognitive impairment can manifest at any stage of PD and often contribute to greater disability than motor dysfunction [145, 146]. While these symptoms arise from complex interactions between neurodegeneration, neurotransmitter imbalances, and environmental factors [147], there is growing evidence that genetic predisposition plays a critical role in their development. Numerous genetic variations influence susceptibility to neuropsychiatric manifestations in PD, either through direct effects on brain function or by modifying the underlying disease pathology [148, 149]. It is important to note that rare, highly penetrant mutations, such as those in SNCA and PRKN, are typically associated with familial or early-onset forms of PD and thus explain neuropsychiatric manifestations only in a small subset of patients. In contrast, sporadic PD, which represents the majority of cases, is influenced by a polygenic risk profile and common genetic variants that modestly affect susceptibility to neuropsychiatric symptoms.
The SNCA gene, which encodes
Another gene involved in the pathogenesis of PD is the PRKN gene. The PRKN gene, encoding the Parkin protein, is one of the key genes associated with the autosomal recessive form of early-onset PD (ARJP). Mutations in this gene, including deletions, duplications, and point mutations, lead to dysfunction of the ubiquitin-proteasome system, which results in the accumulation of damaged proteins and intensification of neurodegenerative processes [157]. In addition to motor symptoms, patients with PRKN mutations can exhibit neuropsychiatric changes, including depression, apathy, and various degrees of cognitive impairment [158]. One of the mechanisms underlying these symptoms is the role of Parkin in mitochondrial regulation and protection of neurons against oxidative stress [159]. Oxidative stress can lead to damage to brain structures responsible for the control of emotions and executive functions [160].
Beyond individual gene mutations, genome-wide association studies (GWAS) have identified several loci associated with psychiatric symptoms in PD. Many of these genetic risk factors overlap with those implicated in major depressive disorder, schizophrenia, and bipolar disorder, supporting the idea that neuropsychiatric manifestations in PD can share common biological mechanisms with primary psychiatric disorders [161]. For example, polymorphisms in genes regulating serotonin receptors, such as 5-hydroxytryptamine receptor 2A (HTR2A), have been linked to increased risk of depression and psychosis in PD [162]. The HTR2A gene encodes the serotonin 2A receptor (5-HT2A), which plays a key role in the regulation of serotonergic neurotransmission and is an important factor modulating cognitive functions, mood, and perception [163]. A study has shown that polymorphisms in HTR2A may influence the susceptibility to the development of neuropsychiatric disorders in the course of PD [164]. One of the best-studied variants is the rs6311 (–1438G/A) polymorphism in the promoter region of the gene, which may affect the expression level of the 5-HT2A receptor. Carriers of the A allele of this polymorphism are more likely to experience visual hallucinations and other psychotic symptoms in PD, suggesting that increased 5-HT2A receptor expression may lead to hypersensitivity to serotonergic signals and impaired perceptual processing [165, 166].
Another gene of interest is the BDNF gene, which encodes brain-derived neurotrophic factor, a key regulator of neuronal survival, synaptic plasticity, and emotional stability [167]. Variants in the BDNF gene have been associated with depression in PD [134]. Since BDNF plays a critical role in maintaining dopaminergic and serotonergic neurotransmission, genetic alterations affecting its expression or function may lead to increased susceptibility to mood disorders and cognitive dysfunction [168, 169]. One of the most important polymorphisms in the BDNF gene is rs6265 (Val66Met), in which valine (Val) is replaced by methionine (Met) at position 66 of the protein. This change affects BDNF secretion and its transport in the neuron, leading to reduced neurotrophic activity [170]. The Val66Met polymorphism has been associated with a greater risk of depression and severe cognitive deficits in patients with PD [171]. Research indicates that carriers of the Met allele have a greater tendency to develop mood and anxiety disorders, as well as a worse adaptive capacity of the brain in response to neurodegenerative processes [172].
Apart from the described genes, there are many others, such as leucine-rich repeat kinase 2 (LRRK2), vacuolar protein sorting 35 (VPS35), ATPase cation transporting 13A2 (ATP13A2), microtubule-associated protein tau (MAPT), and tyrosine hydroxylase (TH), which also play an important role in the pathogenesis of PD, influencing both neurodegenerative processes and the development of neuropsychiatric symptoms. In sporadic PD, these effects are typically mediated by common variants and polygenic risk factors, rather than rare, high-penetrance mutations like those in SNCA and PRKN genes associated with familial PD. Understanding the genetic basis of neuropsychiatric disorders in PD has significant implications for personalized medicine. Identifying genetic risk factors may allow for early detection of individuals at higher risk of developing psychiatric symptoms.
A comprehensive understanding of the molecular mechanisms underlying dementia and affective disorders in Parkinson’s disease is essential for improving early diagnostic strategies and developing targeted therapeutic interventions.
Alterations in glucose metabolism, which can be assessed using
18F-fluorodeoxyglucose PET (FDG-PET), are indicative of cognitive decline
and are widely used to detect functional brain changes in AD [173]. In PD,
FDG-PET similarly reveals frontal hypometabolism even in cognitively normal
patients, suggesting early functional alterations preceding overt cognitive
impairment. In one study, 3 of 29 cognitively normal PD patients and 3 of 17 PD
patients with mild cognitive impairment (MCI), who exhibited hypometabolism in
the parietal and occipital lobe areas, developed dementia within 3 years of the
study. Thus, changes in brain metabolism may be a potential predictor of the
onset of dementia in PD patients [174]. Magnetic resonance imaging (MRI) studies
indicate structural differences in the brains of patients with PDD and Lewy body
dementia (DLB). PDD is characterized by bilateral atrophy of frontal areas, while
in DLB, the atrophy involves parietal and occipital areas. In addition, resting
state functional MRI (rs-fMRI) indicates in PDD an impaired synchronization of
neuronal activity in the frontal lobes, while in DLB it shows impaired neuronal
connectivity in the posterior parts of the brain. These findings point to
different mechanisms underlying PDD and DLB and may help differentiate between
these types of dementia [175]. Other rs-fMRI studies have shown that 22 of 32
patients with PDD have reduced functional connectivity of the mediodorsal
thalamus with the posterior cingulate cortex (PCC) compared to cognitively
unimpaired PD subjects. This is particularly relevant to cognitive impairment, as
the PCC plays a key role in attention, memory and information processing [176].
Inflammatory processes are also involved in the pathogenesis of PDD. They are
associated with the activation of microglia and astrocytes, leading to the
release of proinflammatory cytokines and chemokines. One of the factors that
stimulates their expression is glia maturation factor (GMF). Studies indicate
there is increased GMF expression in the substantia nigra as well as increased
activation of microglia and astrocytes in PDD patients (p
Current therapeutic approaches in PDD primarily focus on symptomatic relief
through cholinesterase inhibitors, N-methyl-D-aspartate (NMDA) receptor
antagonists, and emerging combination therapies that target multiple pathways
involved in cognitive decline. It has been described that olfactory dysfunction,
in the form of hyposmia, can increase the risk of dementia in PD patients [178].
The Donepezil Application for Severe Hyposmic Parkinson’s Disease (DASH-PD) study
was conducted to investigate the potential of cholinesterase inhibitors
(donepezil) to prevent the onset of dementia in PD patients at increased risk of
dementia. 4-year donepezil therapy did not affect the risk of developing dementia
in PD patients, but had some beneficial effects on general cognitive function
compared to placebo [179]. However, another study described the effect of
combined therapy with donepezil and Di-Huang-Yi-Zhi (DHYZ), an herbal formula
with a potential protective effect against A
The detection of depression and anxiety in PD is particularly challenging due to
symptom overlap between affective features and motor or disease-related
manifestations, including psychomotor slowing, sleep disturbances, fatigue, and
cognitive impairment. Clinical diagnosis is primarily based on structured
interviews and validated rating scales, such as the Hamilton Depression Rating
Scale (HAMD), the Beck Depression Inventory (BDI) or HAMA-14 [186]. However, no
specific biomarker has yet been established for routine clinical use. Emerging
evidence indicates that specific inflammatory markers may contribute to the
pathophysiology of depression and anxiety in PD. Notably, plasma IL-17A has shown
a association with both PD depression in females (rho = 0.075, p
Treatment of depression and anxiety in PD currently relies on a combination of pharmacological and non-pharmacological approaches. First-line treatment typically involves SSRIs, including citalopram, sertraline, paroxetine, and fluoxetine, which are widely used because of their favorable safety profile and good tolerability, although evidence from randomized controlled trials (RCTs) shows limited rates of full remission and occasional exacerbation of motor symptoms. On the other hand, serotonin–noradrenaline reuptake inhibitors (SNRIs), such as venlafaxine and duloxetine, which increase both synaptic 5-HT and noradrenaline, have demonstrated efficacy in reducing mood-related symptoms without worsening motor function. Furthermore, tricyclic antidepressants (TCAs), including desipramine and nortriptyline, may offer stronger antidepressant effects than SSRIs in PD, though their use requires caution due to cardiovascular side effects. In treatment-resistant depression (TRD), monoamine oxidase inhibitors (MAOIs) are used due to their effect not only on 5-HT, noradrenaline, but also dopamine. Selegiline and safinamide can provide both motor and antidepressant benefits, with selective and reversible MAO inhibition offering additional therapeutic potential [191].
Non-pharmacological treatments play a crucial role in managing depression and anxiety in PD, complementing pharmacotherapy and addressing symptoms that may not fully respond to medication. Cognitive-behavioral therapy (CBT) has shown efficacy in reducing affective symptoms in patients with PD, demonstrating sustained improvements in HAMD and HAMA scores following structured CBT interventions [192]. Despite these benefits, access to therapy can be limited by patient mobility and availability of trained clinicians, highlighting the need for tailored, delivery-adapted programs in this population. Interestingly, remote or CBT has also demonstrated positive effects, offering a promising solution to overcome these barriers [193]. Despite CBT, other non-invasive brain stimulation (NIBS) techniques, such as repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS), have emerged as promising non-pharmacological interventions for cognitive decline and affective symptoms in PD, by modulating cortical excitability, network connectivity, and neuroplasticity. In case of TRD, electroconvulsive therapy (ECT) remains the most effective option, promoting neuroplastic changes via increased BDNF expression, particularly in the hippocampus. Evidence from systematic reviews and meta-analyses shows that ECT improves depressive and motor symptoms without major cognitive worsening, although transient delirium and autonomic complications may occur. Together, NIBS and ECT represent complementary approaches for managing depression in PD, though further research is needed to optimize safety and long-term efficacy [194]. Furthermore, recent studies on fast-acting antidepressants, such as ketamine, suggest that their efficacy may be primarily mediated through modulation of mitochondrial metabolism rather than classical post-synaptic N-methyl-D-aspartate receptor (NMDAr) antagonism [195]. This mechanism could be relevant not only for neuronal mitochondria but also for astrocytic mitochondria, where NMDAr on the inner mitochondrial membrane may play a role [196]. Ketamine is proposed to exert a “preconditioning” effect that enhances mitochondrial function, potentially influencing processes involved in depression, dementia onset, and broader motor and non-motor features of PD [197]. Further research is needed to clarify these mechanisms. Additionally, ketamine has been shown to increase local melatonin production [198], which may contribute to autonomic regulation deficits observed in PD, likely linked to impaired vagal nerve activity [197]. Vagal nerve-mediated anti-inflammatory effects appear to depend on the ability to upregulate melatonin locally, as supported by both clinical [199] and preclinical [200] studies. These findings suggest that gut-mediated melatonergic pathways may be closely involved in PD pathophysiology and its autonomic dysregulation.
Overall, while combined treatment with antidepressants and non-pharmacological approaches continues to form the cornerstone of management, the current therapeutic landscape does not yet fully reflect the complex molecular interplay among proteinopathy, neuroinflammation, and synaptic dysfunction in PD-related depression and anxiety. Future advances in biomarker-guided diagnostics and mechanism-based therapies may enable more personalized and disease-modifying treatment strategies.
In recent years, cerebral organoids have become a valuable platform for modeling PD pathomechanisms (Table 3, Ref. [45, 47, 48, 49, 55, 61, 64, 70, 78, 87, 89, 101, 102, 103, 104, 105, 106, 108, 109, 111, 112, 113, 114, 115, 119, 121, 122, 123, 124, 125, 126, 127, 128, 131, 132, 133, 134, 138, 147, 149, 150, 152, 162, 166, 167, 176, 180, 181, 182, 183, 184, 185, 186, 187, 188, 189, 190, 191, 192, 201]). These three-dimensional models recapitulate human brain architecture and cell interactions, bridging in vitro and in vivo studies [202, 203, 204, 205, 206, 207, 208, 209, 210, 211, 212, 213, 214], and allow investigation of molecular mechanisms and testing of novel treatments [206, 207, 215, 216, 217]. While organoids do not directly replicate dementia or affective disorders, they enable the study of markers associated with cognitive decline, depression, and anxiety in PD. Complete cerebral organoids, as well as region-specific models (e.g., midbrain, striatum), can be generated from patient-derived induced pluripotent stem cells (iPSCs), carrying disease-relevant mutations [115, 207, 218, 219, 220, 221].
| Symptom | Biomarker | In vivo studies | Human studies | In vitro studies/organoid studies |
| Dementia | SNCA/ |
in murine models, A53T BAC-SNCA transgenic mice and |
in PDD, |
cerebral and midbrain organoids carrying SNCA A53T mutation or triplication effectively model |
| PRKN | - | exons 4–6 deletion was associated with dementia [152] | midbrain organoids with PRKN mutation modeling astrocytic dysfunction [191] | |
| Immunologic abnormalities | IFN- |
in PDD, T cell infiltration in the amygdala and substantia nigra and increased microglial activation—negatively correlated with Mini-Mental State Examination scores—indicate region-specific immune involvement [45, 55] | stem cell-derived human midbrain organoids offering a platform to study T cell interactions with midbrain neuronal tissue [192] | |
| GFAP | - | an elevated number of GFAP reactive astrocytes was detected in PDD patients in areas of their brains affected by neurodegeneration [64] | - | |
| PIAS2 | elevated expression of PIAS2 deteriorated mitochondrial function and caused cognitive impairment in mice [70] | - | - | |
| cholinergic disturbances | - | in PDD, increased muscarinic acetylcholine receptor density in the occipital lobes and decreased choline acetyltransferase activity are associated with cognitive impairment [78, 176] | - | |
| BDNF | - | BDNF Val66Met variant was analyzed regarding cognitive impairment in PD; Val/Val alleles were linked to poorer delayed recall of information [87] | - | |
| EGF | - | low EGF levels were prognostic of cognitive decline [89] | - | |
| Affective disorders | BDNF | lower BDNF levels in PD patients with depression [131, 132, 133, 162] | activation of the BDNF/TrkB/CREB pathway attenuates depressive-like behaviors in mice, whereas homozygous Val66Met variant carriers show increased anxiety-like behaviors resistant to antidepressants [134, 167] | - |
| BDNF valine-to-methionine substitution at codon 66 (Val66Met) genetic variant was associated with a greater risk of depression, anxiety and severe cognitive deficits [166] | ||||
| dopaminergic disturbances | DA depletion and pre-synaptic D4 receptors play a role in the onset and regulation of PD-related depression in rat models [102] | alterations in dopaminergic and noradrenergic innervation in PD are associated with depression and anxiety, with studies showing region-specific changes in DAT and [11C]RTI-32 binding, including lower DAT availability in limbic and striatal regions correlating with higher affective symptom scores [101, 103, 104, 105, 106, 108] | human midbrain organoids modeling dopaminergic neurons provide a platform to study DA dysfunction, including PINK1 and LRRK2 mutation–induced disruptions in the tyrosine hydroxylase–dopamine pathway leading to neuronal death [180, 181, 182, 183] | |
| serotonergic disturbances | - | serotonergic system alterations in PD are linked to depression and anxiety, with PET studies showing both correlations and region-specific changes in SERT availability, while non-depressed patients show no significant differences compared to controls [109, 111, 112, 113, 114, 115] | - | |
| cholinergic disturbances | - | depressive symptoms were correlated with cortical cholinergic denervation [119] | - | |
| GABA disturbances | - | elevated GABA+ levels in the medial frontal cortex of PD patients with depression and anxiety [121] | - | |
| cortisol | - | elevated cortisol levels in PD patients are associated with both depression and anxiety, and correlate with depression severity following levodopa administration [138, 201] | - | |
| immunologic abnormalities | modulation of microglial and astrocytic activation (IBA-1, GFAP) and pro-inflammatory cytokines (IL‑1 |
higher baseline CSF sTREM2 and YKL-40 levels predict faster progression of depression [122] | ||
| TNF‑ |
- |
SNCA,
Mutations in SNCA, including triplication [209, 210, 211, 212, 213, 214] and A53T variant [203, 206], can be studied in organoid models, as can PRKN variants linked to dementia, illustrating astrocytic dysfunction [213]. Organoids also allow exploration of immune system involvement [214, 222], metabolic disturbances [221, 223], and PD-associated mitochondrial and lysosomal dysfunction due to mutations in PINK1, GBA, Parkinson disease protein 7 (DJ1), and DnaJ homolog subfamily C member 6 (DNAJC6) [224, 225, 226]. Assembloid models further facilitate investigation of synaptic dysfunction and neurotransmitter imbalances, including serotonin and dopaminergic disturbances relevant to depression and anxiety in PD [202, 203, 205, 227]. Transplantation studies of iPSC-derived midbrain organoids into PD rodent models showed increased dopaminergic neurons and functional improvements, highlighting therapeutic potential [228, 229].
Ultimately, the employment of organoids in PD studies holds promise and hopefully will accelerate advancements in our understanding of neuropsychiatric manifestations in PD. They account for a versatile platform for studying molecular intricacies and lay the ground for the development of novel avenues in treatment that would enable personalized therapeutic modalities.
Understanding the molecular basis of neuropsychiatric disorders in PD patients, including changes in multiple neurotransmitter systems, neuroinflammatory processes, genetic predispositions, and dysregulated cell signaling pathways, can facilitate accurate diagnosis of PD and enable early pharmacological intervention. Moreover, it should be remembered that the treatment of PD patients should be aimed not only at reducing motor symptoms but also at maintaining good mental performance, including cognitive performance as well as depression and anxiety. Impairment of these functions can affect both the deterioration of the patient’s daily functioning and their social or professional life. Due to the high rate of neuropsychiatric disorders in the course of PD and the increasingly longer life of PD patients, the development of sensitive and specific neuropsychological tools for early diagnosis and effective treatment is becoming a significant clinical challenge.
Writing and literature collection of the manuscript — WO, AF, OK, AS; Writing of the manuscript and design, and draw the figures — BS, OS, JP; Editing and language correction — BS, UG; Conception or design of the work, final manuscript review — PPJ, WK, JD, and UG. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We are grateful for the support of Poznan University of Medical Sciences.
This research received no external funding.
The authors declare no conflicts of interest. Jolanta Dorszewska is serving as one of the Guest Editors and Editorial Board members of this journal. We declare that Jolanta Dorszewska had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Bettina Platt.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.