

 , Pengwei Zhuang 1,2,3,4,*
, Pengwei Zhuang 1,2,3,4,*
1 State Key Laboratory of Chinese Medicine Modernization, Tianjin University of Traditional Chinese Medicine, 301617 Tianjin, China
2 Haihe Laboratory of Modern Chinese Medicine, Tianjin University of Traditional Chinese Medicine, 301617 Tianjin, China
3 First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, 300193 Tianjin, China
4 National Clinical Research Center for Chinese Medicine Acupuncture and Moxibustion, 300193 Tianjin, China
Abstract
Diabetic cognitive impairment (DCI) affects approximately 25%–35% of patients with diabetes and is characterized by progressive cognitive decline. Dysfunction of mitochondria—the energy factories within neurons—is considered a potential pathogenic factor of DCI, involving processes such as oxidative stress, calcium overload, autophagic dysfunction, and genetic mutations, ultimately disrupting normal neuronal function. Maintaining mitochondrial quality and function is critical for neuronal health. Recent studies have shown that there are multiple ways in which cells can communicate signals, such as extracellular vesicles (EVs), tunneling nanotubes and gap junctions, which can repair and replace damaged mitochondria within receptor cells. Notably, EV-mediated mitochondrial transplantation has demonstrated significant potential by transferring healthy mitochondria to impaired neurons and restoring energy metabolism and antioxidant defences, thereby offering novel therapeutic strategies for intervening in DCI progression with valuable clinical translation potential. This review systematically elucidates multimodal signalling strategies targeting mitochondrial homeostasis, with a focused analysis on the role of EV-mediated mitochondrial transplantation in restoring neuronal energy balance, providing a theoretical foundation for the development of innovative DCI interventions.
Graphical Abstract
Keywords
- diabetes mellitus
- type 2
- cognitive dysfunction
- neurons
- mitochondria
- cell communication
Diabetes is a chronic metabolic disorder characterized by persistent hyperglycaemia and insulin resistance (IR). With changing dietary patterns and the advent of an aging society, the prevalence of diabetes among adults has been increasing annually and is projected to reach 642 million by 2040 [1]. Accumulating evidence indicates that type 2 diabetes mellitus (T2DM) is not only a global metabolic epidemic but also a significant modifiable risk factor for both Alzheimer’s disease (AD) and vascular dementia (VaD) [2, 3]. Clinically, this risk manifests as a significantly higher incidence of cognitive impairment in diabetic patients. Such impairment, induced or exacerbated by diabetes, is collectively termed diabetic cognitive impairment (DCI) and has now become a major central nervous system (CNS) complication of diabetes [4, 5, 6]. It is estimated that DCI affects 25%–35% of patients with diabetes and diabetic patients have a 1.5- to 2.5-fold increased risk of cognitive decline compared with individuals without diabetes [7, 8]. Unfortunately, the pathological and molecular mechanisms underlying DCI remain elusive, and effective clinical treatments are currently lacking.
The pathological mechanisms through which diabetes leads to cognitive decline
are complex, with chronic hyperglycemia-induced indirect damage to the central
vascular system constituting a critical hub [9]. Specifically, chronic
hyperglycemia promotes the accumulation of advanced glycation end products
(AGEs), exacerbates oxidative stress, and triggers systemic low-grade
inflammation, thereby impairing cerebrovascular endothelial function,
accelerating cerebral small vessel disease, and disrupting blood-brain barrier
(BBB) integrity [10]. These changes lead to pathological consequences such as
cerebral hypoperfusion, microinfarcts, and white matter hyperintensities, which
represent the core pathological basis of VaD. Simultaneously, this compromised
vascular environment also provides the “soil” for the development of AD
pathology. Furthermore, the study has found that diabetic brain tissues exhibit
accumulations of pathological proteins similar to those in AD, such as
amyloid-beta (A
Mitochondria are the energy factories within cells, and their proper function is
crucial for cell survival and metabolism. Although the brain accounts for only
2% of total body weight, it consumes 20% of the body’s energy [13]. Neurons,
the primary functional units of the CNS, have continuous and high-energy demands.
However, their capacity to generate adenosine triphosphate (ATP) via cytoplasmic
glycolysis is limited, making them highly dependent on the mitochondrial ATP
supply and extremely sensitive to mitochondrial dysfunction. Recent research
published in Science indicates that mitochondrial dysfunction is a central
mechanism in type 2 diabetes. Defects in mitochondrial quality control, such as
mitochondrial DNA (mtDNA) depletion, impaired mitochondrial dynamics, and
abnormal autophagy, trigger electron transport chain dysfunction [14]. This
subsequently activates the integrated stress response, leading to metabolic
dysfunction [14]. Neuronal mitochondrial damage can cause abnormal central energy
metabolism and disrupted cellular signalling, thereby contributing to various
neurodegenerative diseases [15]. In DCI, disturbances in glucose and lipid
metabolism disrupt mitochondrial functional integrity, including induction of the
mitochondrial permeability transition pore (mPTP) opening (resulting in
uncontrolled substance exchange) [16], loss of mitochondrial membrane potential
(
Under physiological conditions, neuronal mitochondria convert intracellular nutrients into cellular energy and perform neuroprotective functions through intercellular transfer. However, in a glucose and lipid metabolism-disordered environment, the self-repair capacity of neuronal mitochondria is limited. Severe mitochondrial damage further amplifies defects in the classic mitochondrial quality control system, thereby mobilizing more active mitochondrial transfer. This process supplements exogenous healthy mitochondria while clearing endogenous damaged mitochondria, ultimately promoting organismal recovery [20]. Consequently, both central and peripheral mitochondria are recruited to aid neurons. Numerous studies report that mitochondria transfer primarily between cells by tunneling nanotubes (TNTs) [21], extracellular vesicles (EVs) [22], gap junctions [23], and free mitochondria [24, 25], as well as direct extrusion and internalization. Additionally, mitochondria can be transferred between organs via EVs to counter intense energetic stress [26]. Multimodal signalling can restore neuronal mitochondrial function by regulating mitochondrial transfer. However, under metabolic disorder mitochondria in astrocytes—the largest glial cells proximal to central neurons—undergo functional collapse and lose their neuroprotective role [27]. Moreover, dysfunction in peripheral IR target organs prevents them from supporting central neurons to reverse cognitive impairment. Therefore, exogenous mitochondrial intervention may be effective for restoring neuronal mitochondrial function, increasing mitochondrial quantity, and maintaining systemic mitochondrial quality control.
EV-mediated mitochondrial transplantation has recently attracted widespread attention as an emerging strategy. A hypothesis has emerged on the basis of intercellular and interorgan mitochondrial transfer, that proposes that mitochondrial implantation can ameliorate energy deficiency in target cells. Intriguingly, a novel therapeutic paradigm—mitotherapy—may overcome major bottlenecks in traditional treatments for mitochondria-related diseases [28]. Extensive basic and preclinical studies have reported that in vitro mitochondrial implantation effectively restores biological functions in target cells [29, 30]. EVs can adapt to rapidly changing environments, increase transfer efficiency, reduce allogeneic immunogenicity, and cause fewer complications. Thus, EV-wrapped mitochondrial transplantation is proposed to be a promising tool for treating neurodegenerative diseases.
Neuronal mitochondria are the energy factories within cells that generate ATP
through oxidative phosphorylation and participate in various biological
functions, such as intracellular calcium signalling, autophagy, and
neurotransmitter release. The high energy demands of biological processes and
limited glycolytic capacity render neurons critically dependent on mitochondria
[31]. As shown in Fig. 1, mitochondrial dysfunction is an important initiating
factor in the pathological cascade of DCI. Once mitochondria are damaged, it
triggers a series of cellular dysfunctions including oxidative stress, calcium
dyshomeostasis, impaired autophagy, and decreased genetic stability. The
persistence of these abnormalities collectively undermines the survival basis and
normal function of neurons, ultimately leading to progressive decline in
cognitive ability. Basic research has indicated that mitochondrial dysfunction
readily affects central neurons in mice with diabetes [32, 33]. Concurrently,
diabetes is often accompanied by hyperglycaemia, oxidative stress, and energy
disruption—all of which exacerbate neuronal mitochondrial damage and death,
subsequently leading to cognitive dysfunction [34, 35]. Clinical study also
report that hyperglycaemia and hyperinsulinaemia can accelerate the formation of
neuritic plaques and cognitive impairment [36]. Furthermore, mitochondrial
dysfunction is a significant factor in oxidative stress [32], calcium
dyshomeostasis [37], autophagy impairment [28], gene mutations [38], IR [39],
cerebrovascular alterations [40], Tau hyperphosphorylation [41], and A
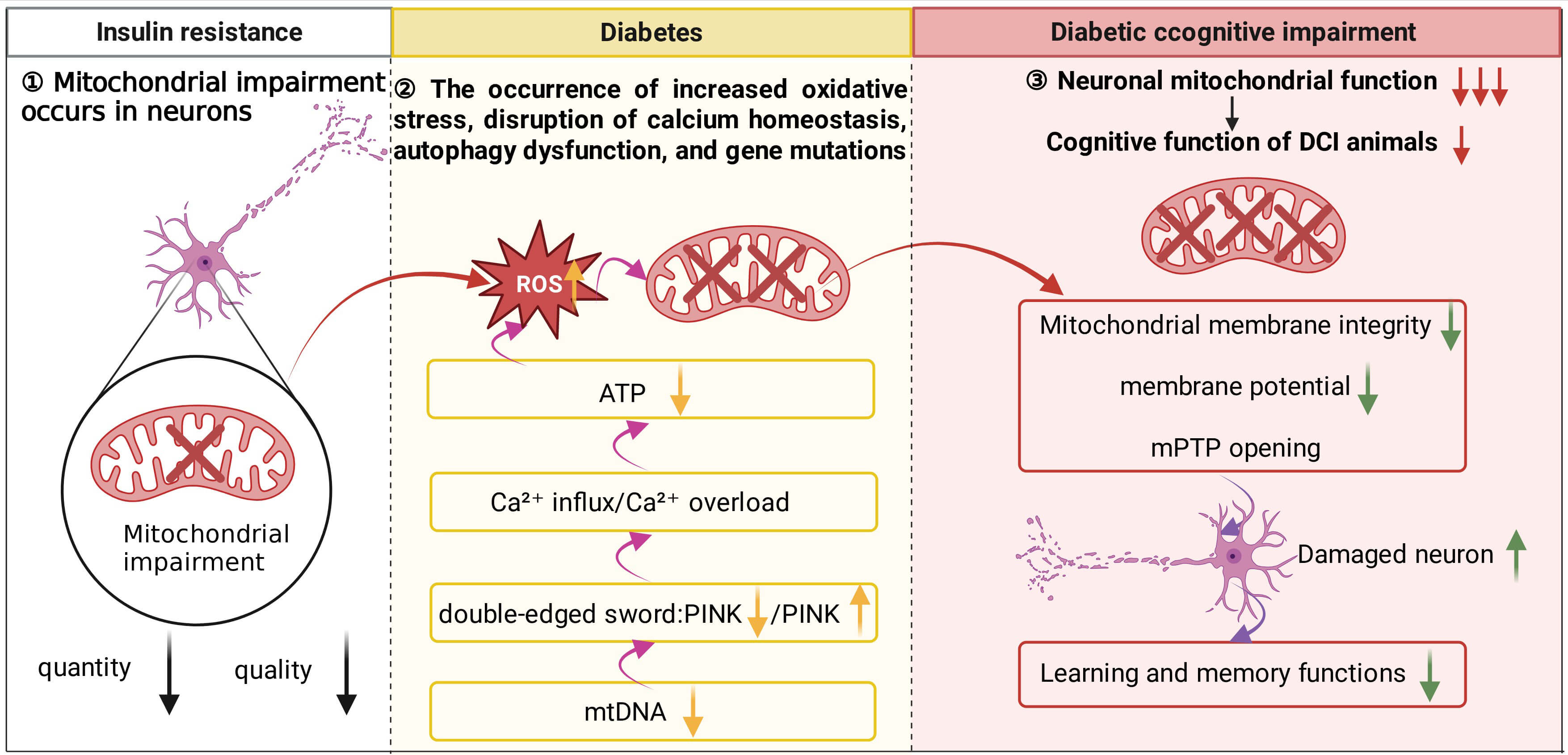
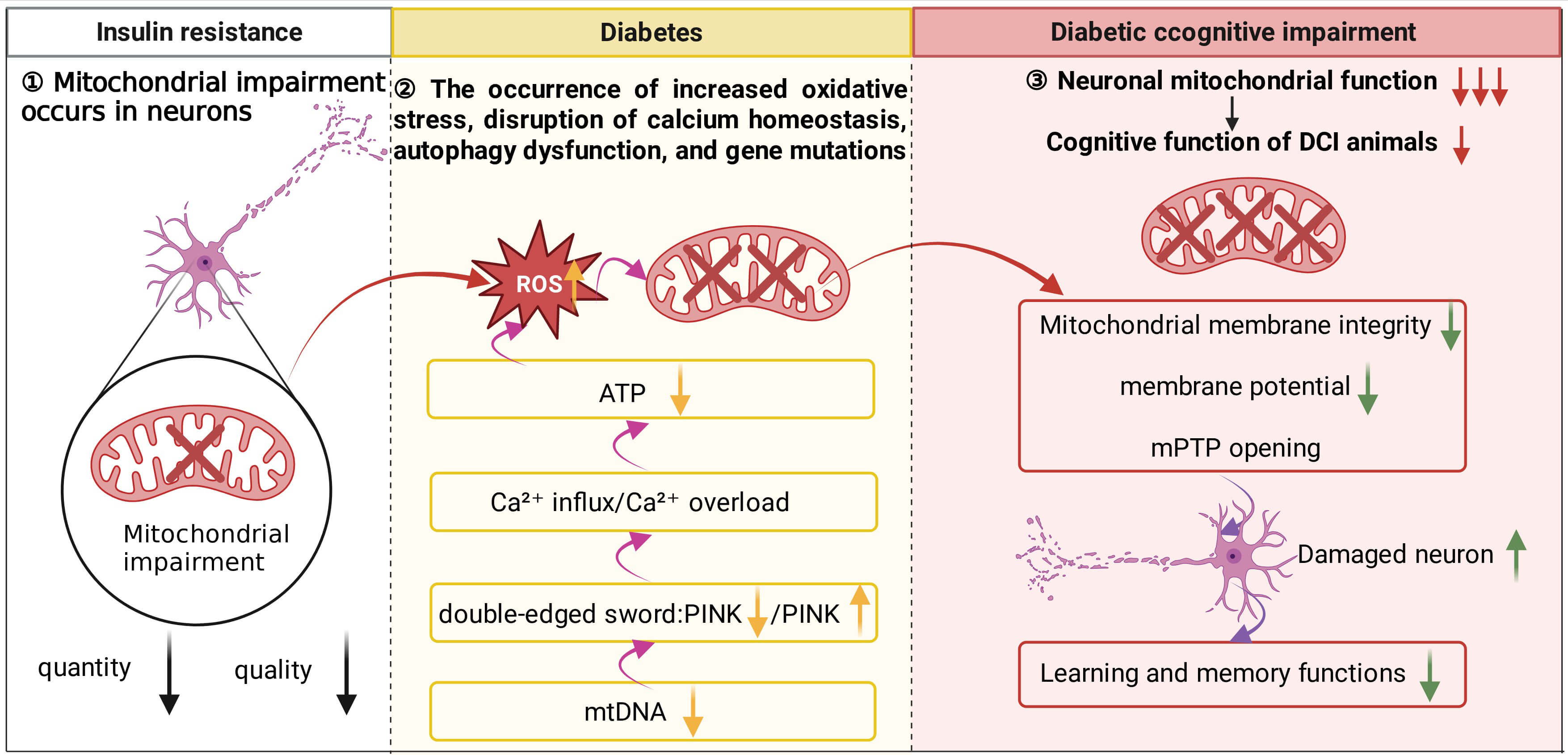 Fig. 1.
Fig. 1.
Relationships between neuronal mitochondrial damage, diabetes,
and cognitive impairment. During the initial stage of IR, mitochondrial damage
is already present in neurons. As the condition progresses to the type 2 diabetes
stage, impairment and loss of mtDNA further disrupt mitochondrial biogenesis and
autophagy, leading to a marked decline in overall mitochondrial quality and
oxidative phosphorylation capacity. At this point, excessively activated
mitophagy exacerbates neuronal injury. Specifically, loss of PINK1 function
disrupts mitochondrial Ca2+ homeostasis, resulting in calcium overload,
which in turn reduces ATP production and promotes substantial ROS generation.
Excessive ROS not only compromises mitochondrial membrane integrity and reduces
Clinical study has indicated that mitochondrial damage occurs in the body before
IR progresses to type 2 diabetes [39]. Notably, upon progression to type 2
diabetes, mitochondrial oxidative phosphorylation contributes to the exacerbation
of IR [46]. Concurrently, hyperglycaemia and IR trigger oxidative stress by
disrupting the balance between reactive oxygen species (ROS) production and the
antioxidant defence system, leading to the accumulation of destructive ROS that
damage lipids, proteins, and DNA and ultimately impair cellular structure and
function [47, 48]. Oxidative stress impairs mitochondrial membrane integrity and
fluidity, leading to reduced
Under physiological conditions, mitochondria serve as the primary
energy-producing organelles within cells, generating ATP through oxidative
phosphorylation [53], with calcium ions acting as key regulatory factors [54].
However, diabetes disrupts intracellular calcium homeostasis, leading to
increased mitochondrial calcium influx and the accumulation of mitochondrial ROS
[44]. This subsequently affects cytoskeletal remodelling, cell adhesion, and the
cell cycle. Conversely, calcium overload (excessive Ca2+ uptake by
mitochondria) inhibits oxidative phosphorylation, impairs ATP production,
generates excessive ROS, and triggers the opening of the mPTP [16]. The mPTP is a
nonselective channel whose aberrant opening causes excessive release of nearly
one hundred substances, including Ca2+, ROS, nicotinamide adenine
dinucleotide (NAD+), and cytochrome C. The overload of both ROS and
cytochrome C can induce apoptosis. Glucose-stimulated insulin secretion (GSIS) by
pancreatic
Mitophagy is a specialized autophagic pathway dedicated to clearing damaged or superfluous mitochondria, thereby preventing oxidative stress and apoptotic signalling and supporting the maintenance of neuronal health [62]. Its mechanism may involve neuron-specific PTEN induced putative kinase 1 (PINK1) mRNA cotransport with mitochondria, facilitating PINK1 translation in dendrites and axons to enable the removal of damaged organelles [63]. Autophagy acts as a “double-edged sword”. PINK1 deficiency induces mitochondrial Ca2+ overload, increases ROS levels, and promotes mPTP opening, ultimately leading to neuronal apoptosis [64]. Furthermore, diabetes disrupts neuronal mitophagy activity, slowing mitochondrial clearance and consequently exacerbating mitochondrial dysfunction and cellular damage. Park et al. [28] reported that compared with diabetic wild-type (WT) mice, diabetic mice with neuron-specific Drp1 knockout exhibited increased engulfment of enlarged mitochondria by large autophagosomes, accompanied by aggravated synaptic and cognitive impairment. In summary, mitophagy represents a critical cellular biological process in neurons, and its dysregulation is closely associated with the onset and progression of cognitive dysfunction.
mtDNA is a key determinant of cellular energy metabolism and homeostasis. Its damage and depletion lead to mitochondrial dysfunction, affecting critical processes including energy metabolism, oxidative stress, apoptosis, and autophagy [65]. Diabetes induces mtDNA damage and depletion, compromising mitochondrial genetic stability and gene expression [66]. Such damage, referred to as gene mutations—including point mutations and deletions—disrupts mitochondrial morphology and quantity, leading to swelling, fragmentation, and outer membrane rupture, ultimately causing damage to cellular membranes and organelles. Additionally, mtDNA defects impair mitochondrial biogenesis and autophagy, significantly reducing mitochondrial mass and oxidative phosphorylation capacity and ultimately diminishing mitochondrial activity. Collectively, these alterations disrupt neuronal energy metabolism, oxidative stress responses, calcium signalling, autophagy, and apoptotic pathways, resulting in neuronal dysfunction and death that culminates in cognitive impairment.
In summary, diabetes impairs neuronal mitochondrial structure and function through multiple pathways, driving the neuronal damage and death that underlies cognitive dysfunction. Consequently, protecting and restoring neuronal mitochondrial function represents a crucial therapeutic strategy for treating diabetes-associated cognitive impairment.
EVs are small, lipid bilayer-enclosed vesicles (30–1000 nm) secreted by living
cells [67]. On the basis of their size and biogenesis pathways, they can be
further classified into the following subtypes: Exosomes [68, 69, 70], microvesicles
[71], and mitovesicles [72, 73, 74, 75]. EVs regulate signalling in target cells and
facilitate intercellular communication [76] by transferring diverse cargoes such
as mitochondria [77, 78], nucleic acids [79, 80, 81, 82, 83, 84, 85], proteins [81, 86, 87, 88, 89, 90], and lipids
[75, 91]. As illustrated in Fig. 2a, EVs serve as a primary pathway for
intercellular mitochondrial transfer, with the precise sorting of mitochondrial
components relying on two distinct mitochondrial-derived vesicle (MDV) pathways.
The interactions between mitochondria and EVs can be divided into two categories.
One is the transfer of substances from mitochondria to EVs, mitophagy, a
selective autophagy process, maintains mitochondrial quality through the
encapsulation of damaged or superfluous mitochondria into autophagosomes for
lysosomal degradation. Cells selectively target damaged mitochondrial components
for lysosomal degradation to prevent the release of inflammatory contents into
EVs. This relies on mitochondrial-derived vesicles (MDVs), small vesicles that
transport mitochondrial proteins to other organelles. Precise sorting requires
two distinct MDV pathways: sorting nexin 9 (Snx9)-dependent MDVs (a subset that
regulates mitochondrial antigen presentation) deliver mitochondrial proteins to
EVs [92]. MDVs carrying damaged components are targeted for lysosomal degradation
via a Parkin-dependent process. Cells selectively regulate mitochondrial protein
packaging into EVs to prevent release of damaged components [77]. However, not
all engulfed mitochondria are degraded; some autophagosomes evade the lysosomal
pathway and transform into EVs containing intact or fragmented mtDNA/proteins.
These EVs harbouring neuronal mitochondrial components can be taken up by
adjacent (astrocytes, microglia, and endothelial cells) or distant cells,
influencing recipient cell mitochondrial function and gene expression, or
triggering inflammatory/immune responses. The other direction involves cargo
transfer from EVs to mitochondria: EVs that transfer cargo to mitochondria can
benefit recipient cells by restoring/enhancing mitochondrial function. For
example, astrocyte-derived exosomes (AS-Exos) protect against traumatic brain
injury (TBI)-induced oxidative stress and neuronal apoptosis by activating
nuclear factor erythroid 2-related factor 2 (Nrf2) signalling and alleviating
neurobehavioural deficits, cognitive impairment, and brain oedema in rats [93].
Astrocytes release functional intact mitochondria, via a calcium-dependent
mechanism involving CD38/cyclic ADP-ribose signalling to neighbouring neurons
[94]. Nigrostriatal astrocyte-derived EVs rescue neuronal mitochondrial complex I
function impaired by the neurotoxin 1-methyl-4-phenylpyridinium (MPP+) [95].
Conversely, this transfer can be detrimental. For instance, A-SMase in the
microglia of adult 5xFAD mice controls the A
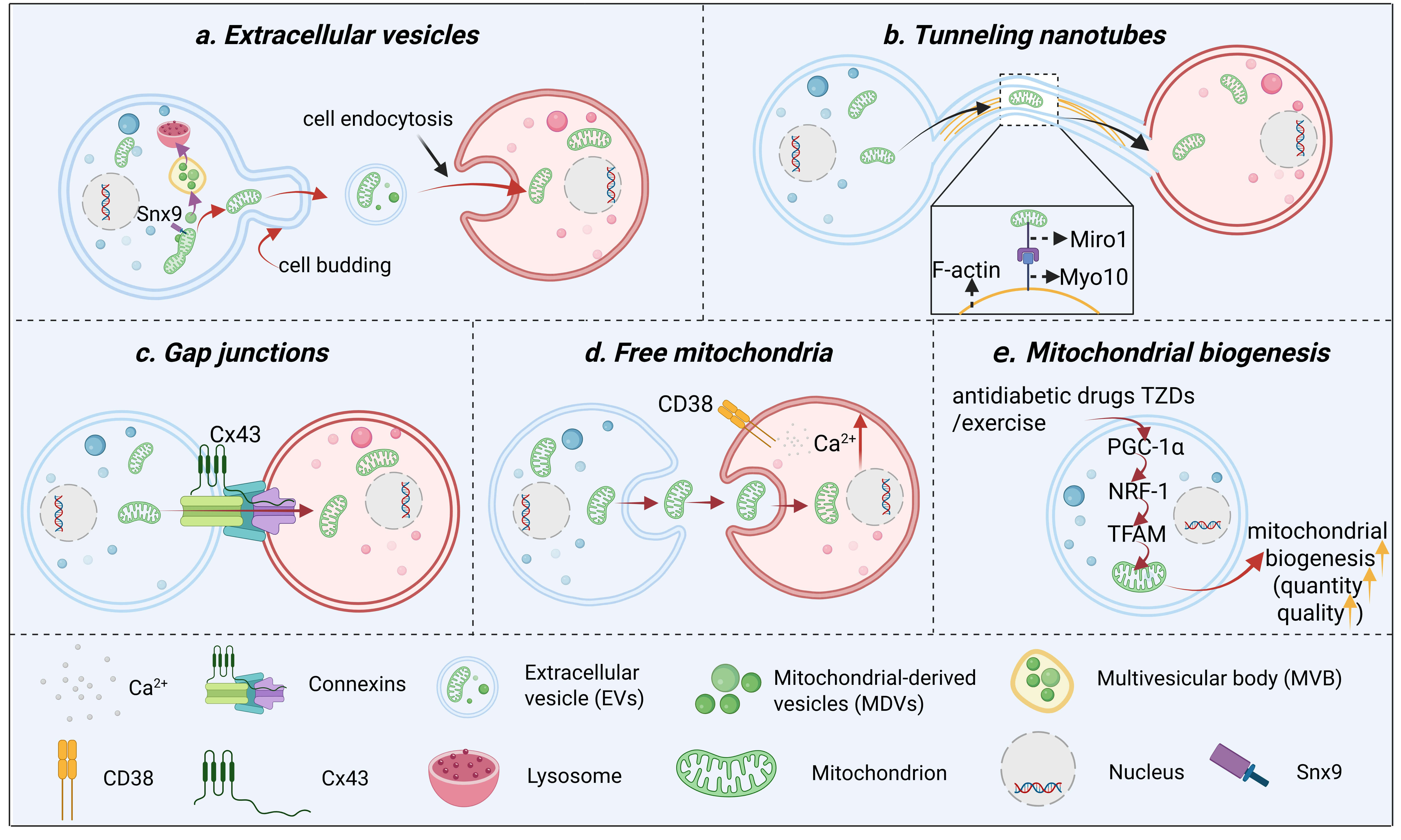
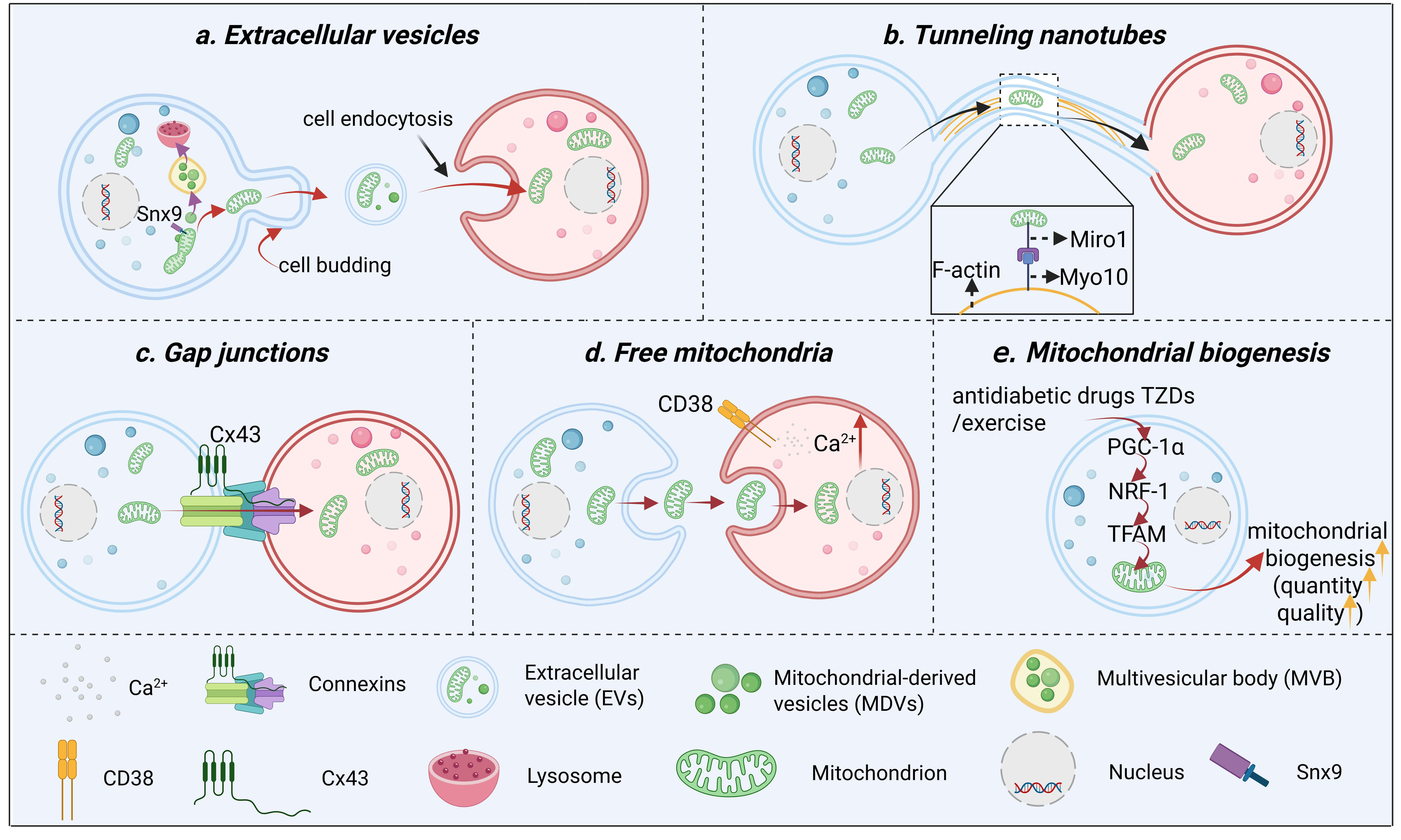 Fig. 2.
Fig. 2.
The main mode of mitochondrial transfer between cells. (a) EVs
serve as the primary pathway for intercellular mitochondrial transfer, with the
precise sorting of mitochondrial components relying on two distinct
mitochondrial-derived vesicle pathways: The Snx9-dependent pathway delivers
mitochondrial proteins to EVs, which are subsequently internalized into recipient
cells via endocytosis to achieve functional transfer; conversely, multivesicular
bodies (MVBs) carrying damaged mitochondrial components are targeted to lysosomes
for degradation, thereby eliminating abnormal constituents. (b) Through TNTs,
mitochondria are transferred from donor cells to recipient cells along actin
filaments within these membrane-bound channels, a process that requires the
presence of Miro1 and Myo10. (c) Via gap junctions mediated by connexins such as
Cx43, mitochondria, along with small molecules and ions, can be directly
transferred between adjacent cells through membrane contact sites. (d) In free
mitochondrial transfer, donor cells release isolated mitochondria via exocytosis,
which are subsequently internalized by recipient cells through endocytosis—a
process dependent on the CD38-mediated increase in the intracellular Ca2+
concentration. (e) mitochondrial biogenesis, antidiabetic drugs TZDs and exercise
promote mitochondrial biogenesis via the PGC-1
TNTs, first reported in 2004, are ultrafine, F-actin-rich cytoplasmic bridges
between cells [101]. Fig. 2b illustrates how they mediate the transfer of
mitochondria between cells. Specifically, mitochondria travel along the actin
filaments within these channels in a process that is dependent on the proteins
mitochondrial Rho GTPase 1 (Miro1) and Myosin X (Myo10) [20]. TNTs facilitate the
transfer of various organelles and molecules, including mitochondria [102, 103, 104, 105],
between adjacent cells and participate in multiple biological functions such as
oxidative phosphorylation, calcium signalling, autophagy, and gene expression.
Interactions between mitochondria and TNTs can influence mitochondrial function
and state, thereby modulating neuronal responses and adaptation. These
interactions may also impact the onset and progression of diabetes and its
complications. TNTs may serve as a pathway for mitochondrial dissemination. Zheng
et al. [106] demonstrated that TNTs can transfer healthy mitochondria
from astrocytes to neurons, mitigating neuronal damage. Concurrently, Chakraborty
et al. [21] proposed that
Gap junctions are direct intercellular channels composed of membrane proteins (connexins). As depicted in Fig. 2c, via gap junctions mediated by connexins such as Connexin 43 (Cx43), mitochondria, along with small molecules and ions, can be directly transferred between adjacent cells through membrane contact sites. These channels allow the passage of small molecules and ions between adjacent cells, enabling cellular synchronization and coordination. The interaction between gap junctions and mitochondria can modulate mitochondrial function and state, thereby regulating neuronal energy metabolism, calcium balance, the antioxidant stress response, and neuroprotection. This crosstalk has also been implicated in diabetes. Gap junctions may act as regulators of mitochondrial ATP in diabetic conditions; under high-glucose conditions, mitochondrial structural proteins are damaged—reducing ATP production—while decreased connexin expression leads to increased ATP leakage, collectively accelerating the onset and progression of diabetes and its complications [108].
Cells can release certain types of free mitochondria, similar to the EV-mediated release of mitochondria through exocytosis [109]. As illustrated in Fig. 2d, in free mitochondrial transfer, donor cells release isolated mitochondria via exocytosis, which are subsequently internalized by recipient cells through endocytosis—a process dependent on the CD38-mediated increase in intracellular Ca2+ concentration [110]. However, these released free mitochondria are not cleared by the original cells; instead, they are predominantly recognized and phagocytosed by neighbouring cells via phagocytes, which differs from EV-mediated mitochondrial transfer [109]. In obesity, mitochondrial transfer from adipocytes to macrophages is impaired. Rather, adipocytes release mitochondria into the bloodstream to induce a protective antioxidant response in the heart [111]. Therefore, the release of free mitochondria has unique pathophysiological significance and potential organ-protective functions in metabolic diseases, thereby offering novel perspectives and therapeutic strategies for intervention.
In the context of diabetes, targeting upstream signalling pathways to activate
mitochondrial biogenesis represents a viable therapeutic strategy. Furthermore,
enhancing endogenous mitochondrial biogenesis has emerged as a crucial direction
for treating diabetic cognitive impairment, with studies demonstrating
significant potential for restoring mitochondrial renewal capacity through such
interventions. Fujisawa et al. [112] demonstrated in vitro that
antidiabetic drugs thiazolidinediones (TZDs) such as pioglitazone, activate the
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
(PGC-1
Under physiological conditions, intercellular mitochondrial transfer contributes
to biological development, energy coordination, and the clearance of harmful
cellular components. However, in a disordered glucose and lipid metabolism
environment, the clearance of damaged neuronal mitochondria is slowed.
Concurrently, mitochondrial function decreases in central astrocytes, leading to
a loss of their neuroprotective role [27]. In this context, pharmacological
strategies aimed at increasing autophagosome-mitochondria colocalization and
promoting autophagic flux (e.g., by enhancing autophagosome-lysosome fusion) are
proposed to facilitate the clearance of damaged mitochondria in target cells,
thereby achieving cytoprotection [114]. Recently, a novel therapeutic
paradigm—mitotherapy—has emerged to address the major limitations of
traditional treatments for mitochondrial-related diseases [28]. Notably,
mitotherapy has been applied in clinical settings for patients with multisystem
involvement (including neurological disorders) due to mitochondrial damage and
myocardial ischaemia [30, 115, 116]. Patients not only exhibited improved energy
metabolism but also confirmed the safety of the therapy [117]. In vivo
and in vitro studies by Yang et al. [118] suggested that the
molecular mechanism underlying mitotherapy may involve the activation of neuronal
autophagy through the NAD+/silent information regulator (SIRT) signalling
pathway. This activation upregulates autophagy-related proteins (such as FOXO3,
BNIP3, and LC3), promoting the clearance of A
| Mitochondrial source | Target cells/organs | Administration method | Mitochondrial concentration/range | Therapeutic outcomes |
| Healthy mitochondria isolated from mouse brain tissue | A |
Co-incubation | 1.5 × 106/mL | Reversed oxidative stress and promoted autophagy [118] |
| Healthy mitochondria isolated from mouse brain tissue | Brain of alzheimer’s disease (AD) model mice induced by A |
Tail vein injection | 3 × 106/0.2 mL per mouse | Improved cognitive ability and enhanced autophagy [118] |
| Mitochondria from allogeneic liver or autologous muscle | Cortical neurons/astrocytes/microglia | Co-incubation | 7 × 106/mL | Rescued neuronal apoptosis and upregulated astrocytic brain-derived neurotrophic factor (BDNF) [119] |
| Mitochondria from allogeneic liver or autologous muscle | Injured cortex of traumatic brain injury mice | Cortical injection | 1.2–1.4 × 106/mL | Restored mitochondrial function, promoted neuronal survival, improved spatial memory and cognitive function in mice [119] |
| Platelet-derived mitochondria | Brain of db/db mice | Intracerebroventr-icular injection | 1 × 105 particles | Increased mitochondrial quantity, restored mitochondrial function, reduced oxidative stress and neuronal apoptosis, decreased A |
| Mitochondria from human hepatocellular carcinoma cells (HepG2 cells) | SH-SY5Y cells induced by neurotoxin MPP+ | Coincubation | 1.56–50 µg/mL | Increased cell viability and ATP content, elevated glutathione (GSH) levels and mitochondrial complex I activity, exhibited anti-apoptotic and anti-necrotic effects [120] |
| Mitochondria from human hepatocellular carcinoma cells (HepG2 cells) | Brain of parkinson’s disease (PD) mice induced by neurotoxin MPP+ | Tail vein injection | 0.5 mg/kg | Improved behavioural deficits, enhanced electron transport chain activity, reduced ROS levels, prevented apoptosis and necrosis [120] |
| Rat pheochromocytoma cells (allogeneic) and human osteosarcoma cybrids (xenogeneic source) | PC12 cells induced by the neurotoxin 6-hydroxydopamine (6-OHDA) | Cell-penetrating peptide 1 (Pep-1)-mediated coincubation | 105 µg/0.2 mL | Antioxidant stress and anti-apoptotic effects [121] |
| Rat pheochromocytoma cells (allogeneic) and human osteosarcoma cybrids (xenogeneic source) | Medial forebrain bundle (MFB) of PD rats induced by neurotoxin (6-OHDA) | Medial forebrain bundle injection (Pep-1-mediated) | 1.05 µg per mouse | Reduced dopaminergic neuron loss and improved oxidative DNA damage [121] |
| Rat brain synaptosomes | Human neuroblastoma cells (the human neuroblastoma cell line LAN5) | Coincubation (synaptic delivery system) | 2.5 × 107–10.2 × 107 particles/100 µL | Replaced or supplemented damaged mitochondria [122] |
| Bone marrow mesenchymal stem cells (BM-MSCs) | Streptozotocin (STZ)-induced renal proximal tubular epithelial cells (PTECs) | Coincubation | 3.5 × 103 cells/cm2 | Inhibited ROS production and suppressed apoptosis [123] |
| Bone marrow mesenchymal stem cells (BM-MSCs) | Renal tubular epithelium in diabetic nephropathy (DN) | Tail vein injection | Mitochondria isolated from 1 × 10⁶ BM-MSCs | Suppressed apoptosis by inhibiting ROS production [123] |
First, EVs can adapt to dynamically changing environments. Given the diverse
cell types and constant communication within the CNS, EVs may reach locations
inaccessible to cells and can deliver functional molecules or therapeutics to
target cells or participate in recipient cell signalling pathways [124, 125].
Second, EVs ensure efficient mitochondrial transfer with low alloreactivity.
Owing to their small size and solubility, secreted EV cargo can cross the BBB. As
illustrated in Fig. 3, exogenous EVs enter the CNS via the bloodstream and
deliver functional mitochondria to neurons. In this process, the soluble
N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex
mediates the fusion of EVs with the neuronal membrane, enabling efficient
transfer [126]. This mechanism not only significantly increases intracellular ATP
levels but also promotes mitochondrial biogenesis (via PGC-1
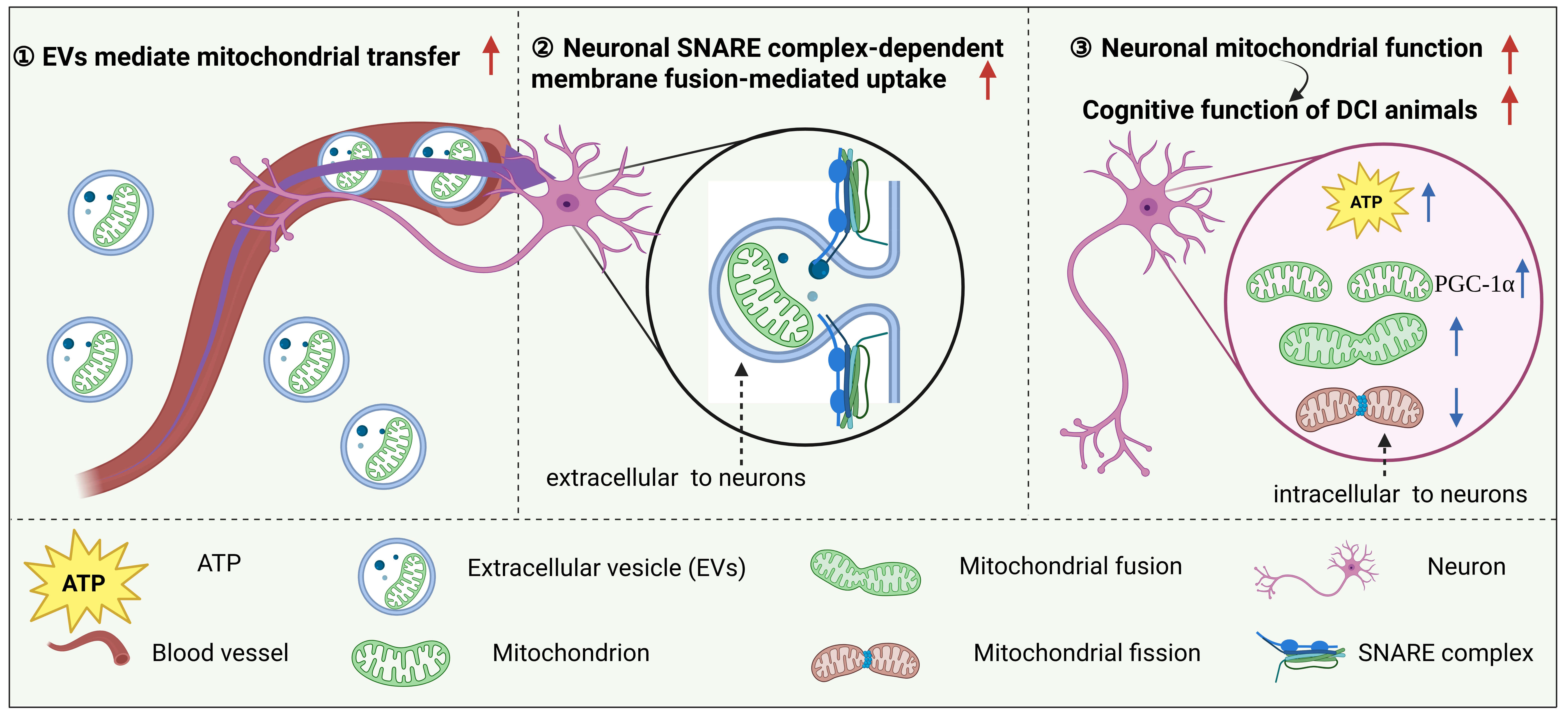
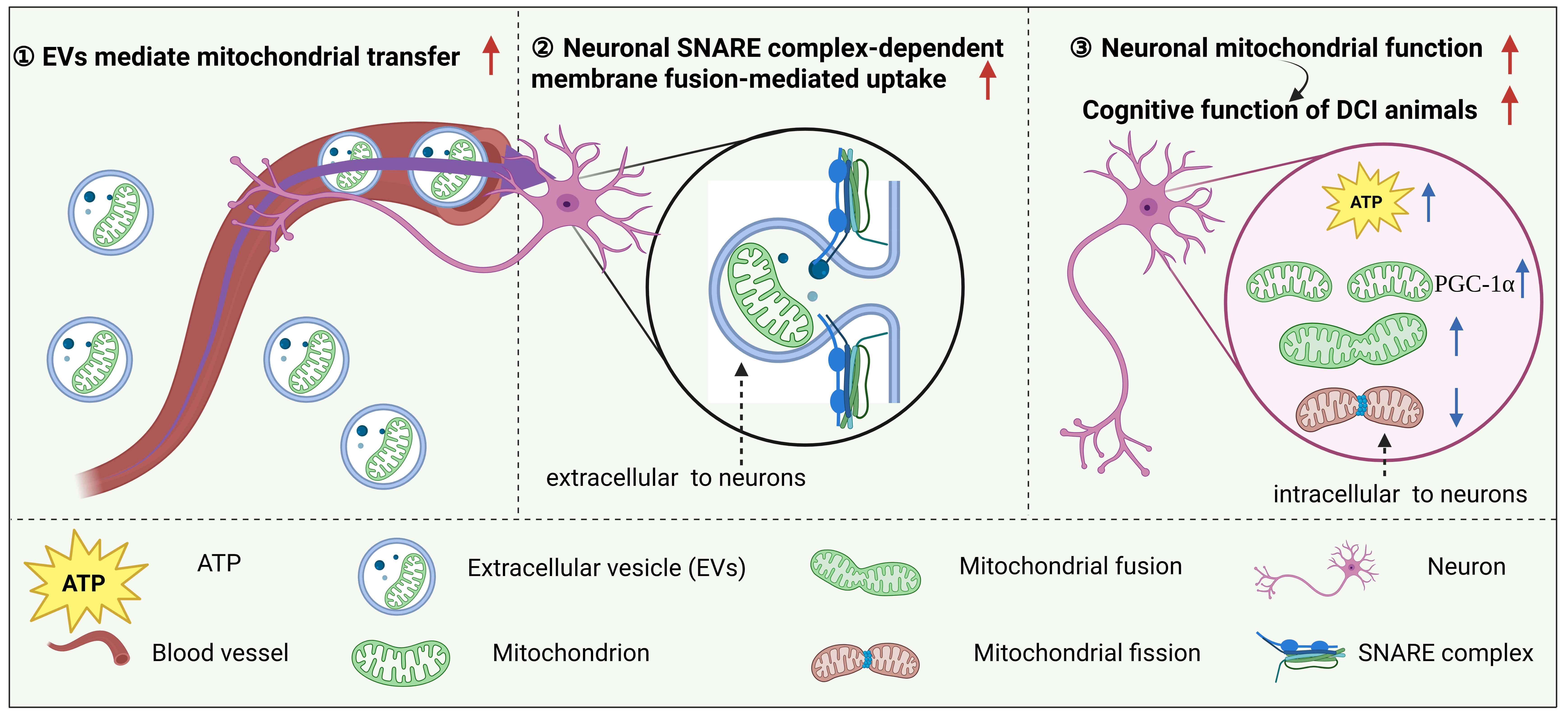 Fig. 3.
Fig. 3.
EV-mediated mitochondrial transplantation may serve as a medium
to ameliorate the progression of DCI. Exogenous EVs enter the CNS via the
bloodstream and deliver functional mitochondria to neurons. In this process, the
SNARE complex mediates the fusion of EVs with the neuronal membrane, enabling
efficient transfer of mitochondria into recipient neurons. This mechanism not
only significantly increases intracellular ATP levels but also promotes
mitochondrial biogenesis (via PGC-1
| Mitochondrial source | Target cells/organs | Administration method | Mitochondrial concentration/range | Therapeutic outcomes |
| Human cerebral microvascular endothelial cell line D3 medium/large EV (hCMEC/D3 m/lEV) | Oxygen-glucose deprived hCMEC | Coincubation | 10, 25, 50 µg/well incubated for 24 h, 48 h, 72 h | Increased ATP levels and mitochondrial function [78] |
| hCMEC/D3 m/lEV | Ischaemic stroke mice | Intravenous injection | 200 µL m/lEV | Reduced cerebral infarct volume in mice [78] |
| Palmitate-treated adipocyte small EV (sEV) | Cardiac ischaemia/reperfusion injury mice | Retro-orbital injection | 1 × 109 sEV | Induced cardiac ROS burst and protected heart [26] |
| Bone marrow-derived mesenchymal stem cell (BMSC) MVs | Acute lung injury (ALI) lungs | Intranasal instillation | 2 × 105/40 µL phosphate-buffered saline (PBS) | Upregulated alveolar ATP [129] |
Under diabetic conditions, mitochondrial regulation demonstrates remarkable heterogeneity across different brain regions (e.g., hippocampus vs. cortex) and cell types (e.g., neurons vs. glial cells), establishing a multi-level regulatory network that directly influences regional vulnerability in DCI. This sophisticated regulatory system operates from the organ level down to specific brain subregions and individual cell types, collectively determining disease progression patterns.
Current evidence confirms significant mitochondrial characteristic differences across tissues at multiple levels: at the organ level, analysis of mitochondrial protein fractional synthesis rates in skeletal muscle, brain, and liver demonstrates that aerobic capacity and dietary interventions exert substantially greater effects on the brain and liver than on skeletal muscle, indicating tissue-specific adaptations according to their respective energy demands [130]. This tissue specificity appears conserved across species, as RNA sequencing of various water buffalo tissues reveals distinct mitochondrial gene expression patterns in high-energy-demand organs like the heart and brain, particularly showing apparent tissue-specific differences in nuclear-encoded mitochondrial genes related to amino acid metabolism [131]. More importantly, this specificity is particularly pronounced between different brain regions: neurons in the hippocampal Cornu Ammonis 2 (CA2) subregion not only exhibit unique enrichment of mitochondrial-related pathways but also possess more than twice the mtDNA copy number per cell compared to Cornu Ammonis 1 (CA1) and dentate gyrus (DG) neurons, indicating significant mitochondrial functional differentiation even within the same brain structure [132]. These tissue-specific differences show close genetic associations with type 2 diabetes. Genome-wide analyses reveal that the expression of mitochondrial-related genes, such as Tu translation elongation factor, mitochondrial (TUFM), correlates not only with the volume of specific brain regions like the caudate nucleus and putamen but also significantly intersects with type 2 diabetes risk. This finding provides an important molecular basis for understanding regional selective vulnerability in diabetic cognitive impairment [133].
At the cellular level, mitochondrial functional heterogeneity demonstrates highly refined regulatory characteristics. Neuronal mitochondria show distinct regional and subcellular distribution differences: in CA1 and DG regions, axonal mitochondria exhibit simpler morphology than dendritic mitochondria, while somatic mitochondria display intermediate complexity between axons and dendrites. Notably, this cell compartment-specific mitochondrial morphology distribution remains relatively stable during aging, even with significant neuronal atrophy in the hippocampal CA1 region [134]. Astrocytes in the adult human neocortex and hippocampus express different key mitochondrial enzymes that directly constrain their respective oxidative phosphorylation capacities, establishing fundamental metabolic differences between cells across brain regions [135]. Under the pathological conditions of diabetic cognitive impairment, specific microglial subsets (such as disease-associated microglia) develop metabolic imbalance through heightened expression of mitochondrial complex I-related genes, increasing ROS production and driving neuroinflammation and neurotoxicity via mechanisms like reverse electron transfer [136]; meanwhile, cerebrovascular pericytes demonstrate increased mitochondrial superoxide production, impaired respiratory function and ATP generation, consequently disrupting BBB integrity [137]. These findings collectively reveal how different cell types, through their specific mitochondrial regulatory mechanisms, form complex interaction networks in DCI that ultimately collectively drive disease progression.
The molecular basis underlying this specificity reveals that mitochondrial gene expression and function follow distinct rules across different tissues rather than a unified whole-body program. Recent research transcending the nervous system demonstrates that mitochondria in different brain regions—and even within subregions—may possess unique molecular signatures of “adaptability” or “vulnerability”. This insight provides crucial direction for DCI precision treatment: successful intervention strategies must account for the complex cellular architecture within the brain and target mitochondrial characteristics specific to vulnerable brain regions (e.g., hippocampus) and cell types (e.g., neurons), enabling development of tailored therapeutic approaches that address the multi-level specificity of mitochondrial regulation in DCI.
Beyond mitochondrial dysfunction, DCI progression is driven by a complex, self-reinforcing pathological network comprising systemic metabolic disturbances, cerebrovascular damage, and chronic neuroinflammation. These elements do not operate in isolation but engage in extensive crosstalk, creating a vicious cycle that accelerates neurological decline.
First, metabolic disturbances serve as the initiating factor in DCI. Persistent hyperglycemia promotes the formation of AGEs. AGEs are a heterogeneous group of molecules generated via the non-enzymatic glycation of proteins, lipids, and nucleic acids. The accumulation of AGEs can induce amyloid plaque formation, tau hyperphosphorylation, and disrupt cellular signalling—such as by downregulating SIRT1—thereby exacerbating IR and neurodegeneration [138]. Preclinical study indicates that sodium-glucose cotransporter 2 (SGLT2) inhibitors, by normalizing blood glucose, can reverse cerebrovascular dysfunction and cognitive impairment in rats with chronic hyperglycemia, potentially through mitigating oxidative stress-induced vascular damage [139].
Second, neuroinflammation plays a critical amplifying role in this process. As resident immune cells in the CNS, microglia respond to various exogenous and endogenous stimuli, including lipopolysaccharide (LPS), ROS, excess glucose, and fatty acids. Excessive inflammatory cytokines not only increase oxidative stress but also exacerbate neuroinflammation through specific mechanisms [140]. Study has found that under high-glucose conditions, accumulated lipid droplets co-localize with the microglia-specific inflammatory amplifier TREM1, aggravating lipotoxic injury and promoting a neuroinflammatory cascade via the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome. In db/db and high-fat diet (HFD)/STZ mouse models, pharmacological blockade of TREM1 with LP17 inhibited the accumulation of lipid droplets and TREM1, reduced inflammatory damage to hippocampal neurons, and consequently improved cognitive function [141].
Third, cerebrovascular dysfunction acts as a key nexus in this vicious cycle. Type 2 diabetes induces endothelial cell dysfunction through transcriptional and post-transcriptional regulatory mechanisms, increasing vascular permeability and negatively impacting cognitive function [142]. Factors such as hyperglycemia lead to increased ROS, reduced nitric oxide bioavailability, impaired vasodilation, and initiation of inflammation [143]. Furthermore, impaired cerebrovascular endothelial function disrupts BBB integrity, allowing neurotoxic substances and inflammatory cytokines from the bloodstream to more easily enter the brain. This forms a positive feedback loop that further exacerbates neuroinflammation and metabolic disturbances [143].
In summary, the pathogenesis of DCI is a symphony of interacting pathologies. Metabolic disorders ignite inflammation and vascular injury; inflammation, in turn, amplifies vascular damage and directly assaults neurons and their mitochondria; and vascular dysfunction creates a leaky BBB that further intensifies both metabolic and inflammatory insults. This tightly intertwined network underscores that the development of DCI is not the result of a single linear pathway but rather the outcome of a complex, synergistic interplay among vascular, inflammatory, and metabolic mechanisms. Therefore, future therapeutic strategies must move beyond single-target approaches and aim to disrupt this interconnected network at multiple points simultaneously.
DCI currently lacks FDA-approved targeted therapies. Existing treatment
strategies face limitations: while some novel glucose-lowering medications show
potential for improving cognitive scores—for instance, nine systematic reviews and meta-analyses have demonstrated that glucagon-like peptide-1 (GLP-1) receptor agonists reduce the decline in global cognitive function, specifically in overall learning (p = 0.039; p
The important pathological mechanism of DCI stems from the direct damage inflicted by metabolic disturbances on neuronal mitochondria. The combined effects of persistent hyperglycemia, hypoglycemic fluctuations, and consequent oxidative stress disrupt the structural and functional homeostasis of mitochondria within neurons, thereby triggering hippocampal damage and progressive cognitive decline. This mechanism is clearly validated in animal studies, which demonstrate that hypoglycemia can induce significant hippocampal neuronal injury and cognitive impairment, with this damage being markedly more severe in a diabetic context [34]. At the mechanistic level, work by Tang et al. [148] in vivo experiments further reveals that the Cav-1 protein alleviates neuronal damage and ultimately improves DCI by precisely regulating the critical mitochondrial fission-mitophagy axis—specifically by suppressing excessive fission and promoting the clearance of defective mitochondria. This evidence chain clearly demonstrates that neuronal mitochondrial dysfunction is a pathological event throughout the development and progression of DCI, providing a solid theoretical foundation for establishing the restoration of mitochondrial function as a fundamental therapeutic target.
Mitochondrial transfer represents a pivotal strategy for restoring the function of damaged cells. To achieve this therapeutic objective, the EV-mediated delivery pathway demonstrates multiple unique advantages over alternative methods. First, EVs provide natural lipid bilayer protection for the mitochondria they carry, significantly enhancing their stability in the circulatory system and effectively avoiding the innate immune response that may be triggered by direct injection of free mitochondria. Second, EVs possess inherent targeting capability and can be artificially modified by scientists, making precise delivery to impaired neurons possible with far greater efficiency than the nondirectional, random connections of TNTs. Furthermore, unlike gap junctions, which are primarily limited to the exchange of small molecules between adjacent cells, EVs can achieve long-distance, barrier-crossing transport of intact, functional mitochondria. Collectively, these characteristics establish EV-mediated mitochondrial transplantation not only as an efficient intercellular rescue mechanism but also as a highly promising therapeutic strategy for clinical translation, offering novel solutions to overcome the challenges of immunogenicity, targeting, and applicability associated with other delivery methods.
The therapeutic advantages of EVs have been empirically validated in numerous
studies. For instance, Islam et al. [129] demonstrated that BMSCs
release mitochondrial-containing microvesicles that are engulfed by epithelial
cells, and intranasal administration of BMSC-MVs in mice elevated alveolar ATP
concentrations (p
Multiple foundational studies have indicated that compared with traditional long-term treatment methods, EV-mediated mitochondrial transplantation has certain advantages in improving cognitive function, yet the clinical translation of these research findings still faces numerous challenges. Although the lipid bilayer of EVs confers a degree of immune-privileged properties, reducing the risk of strong innate immune reactions that may be triggered by the direct injection of free mitochondria, this is not absolute. Studies indicate that the immunogenicity of EVs is influenced by multiple factors, including cellular origin [151], particle size [152], surface composition [153], internal cargo [154, 155], production [151] and storage [156] methods, dosage [157], infusion rate [158], and the potential formation of a biomolecular corona [159]. Furthermore, there are bottlenecks in isolation technology. Traditional ultracentrifugation has low recovery rates (approximately 30%) and is prone to lipoprotein contamination, whereas emerging techniques such as size-exclusion chromatography and polymer-based precipitation improve yield but exhibit insufficient purity. In terms of heterogeneity control, single-cell analysis revealed that in ovarian tumour cells (SKOV3), up to 58.6% of a specific subpopulation functions as high-efficiency secretion units for epithelial cell adhesion molecule-positive (EPCAM+) exosomes. This demonstrates a significantly greater degree of specialization than that in normal epithelial cells, as shown in the human telomerase-immortalized fallopian tube epithelial cell line FTE187 (24.4%). Notably, this research overturns the conventional belief that “high expression implies high secretion”, demonstrating that even within the same cell population, the subpopulation with the strongest surface markers is not necessarily the primary functional executor [160]. This profound functional heterogeneity poses a fundamental challenge to traditional quality control strategies that rely on population-averaged analysis. Additionally, the International Society for Extracellular Vesicles has not yet established standardized culture conditions, hindering scalable production. Finally, engineering modification represents a key strategy for enhancing the prophylactic efficacy of exosomes. Through genetic engineering or electroporation techniques, therapeutic molecules can be precisely loaded into EVs, including surface modifications and cargo loading strategies. More critically, the therapeutic strategy itself carries an inherent immunogenic risk that constitutes another major challenge. A simple “mitochondrial supplementation” strategy requires extreme caution, as exogenous mitochondria or their components may trigger cyclic GMP-AMP synthase (cGAS) recognition during delivery, leading to type I interferon responses and neuroinflammation, which could exacerbate the pathological progression of DCI [161]. This risk has been validated in multiple experimental models. For instance, Zeng et al. [162] directly demonstrated in vitro that IL-6 induces mtDNA leakage and activates the cGAS–STING pathway, leading to the production of EVs enriched in mtDNA and the immune checkpoint molecule PD-L1, which ultimately drives immune escape. This finding clearly reveals the specific detrimental effects of cGAS–STING activation in pathological contexts. Furthermore, Fan et al. [163] confirmed in vivo that EVs derived from damaged hepatocytes promote skeletal muscle inflammation via the mtDNA–cGAS/STING axis, whereas inhibiting liver EV secretion or STING signalling effectively alleviates cirrhosis-associated muscle atrophy. Based on these mechanisms, we explicitly propose that future strategies employing engineered EVs for mitochondrial transplantation must prioritize “avoiding cGAS–STING pathway activation” as a core design principle, achievable through measures such as ensuring mitochondrial membrane integrity or combining anti-inflammatory agents to systematically mitigate immunogenic risks. Collectively, these multifaceted challenges—spanning immunogenicity, production efficiency, functional heterogeneity, and manufacturing standardization—significantly hinder the clinical translation of EV-based mitochondrial transplantation therapies.
To systematically mitigate these risks, several targeted strategies have been developed: for instance, selecting less differentiated cells (such as MSCs) as the source of EVs [151]; prioritizing small EVs to enhance delivery efficiency [152]; modifying the surface with “don’t eat me” signals to avoid clearance by immune cells [153]; optimizing endogenous loading to evade immune recognition [154, 155]; improving production processes to avoid ultracentrifugation and dead-end filtration [151]; adding cryoprotectants to enhance storage stability [156]; avoiding repeated administration to reduce cumulative exposure [157]; implementing premedication and low infusion rates to improve tolerance [158]; and incorporating complement regulatory factors to suppress complement system activation [164]. By systematically outlining both risk factors and corresponding mitigation strategies, we aim to provide a more comprehensive risk management perspective for future research in this field. Notably, to address the low yield of EV-Mito, a recent in vitro and in vivo study revealed that the release of EV-Mito from MSCs is regulated by the CD38/IP3R/Ca2+ signalling pathway. Activation of this pathway via nonviral genetic engineering enabled the “super donor MSCs”, which yielded three times more EV-Mito than normal MSCs did. In a proof-of-concept model of Leber hereditary optic neuropathy, Super EV-Mito successfully rescued mtDNA defects and alleviated related symptoms [165]. Addressing the aforementioned challenges in the engineering modification breakthrough direction, researchers have developed multiple surface engineering strategies to enhance EV functions. Researchers have developed multiple surface engineering strategies to enhance EV functions. For instance, one approach incorporates polyethylene glycol (PEG)-conjugated nanobodies into EVs, which can confer targeting capability to the isolated vesicles [166]. In parallel, another study developed a novel exosome-drug conjugate system termed CAR-equipped exosome-drug conjugate (CAR-EDC), which effectively integrates chemotherapy and immunotherapy [167]. This system utilizes exosomes derived from genetically engineered chimeric antigen receptor macrophages (CAR-M) cells as carriers. The surface CAR molecules ensure targeting specificity, while endogenously high levels of C-X-C motif chemokine ligand 10 (CXCL10) provide immune activation functionality. Furthermore, the covalent loading of the chemotherapeutic drug SN-38 introduces cytotoxic activity. This multi-mechanism synergy enables CAR-EDC to achieve therapeutic effects superior to those of traditional antibody-drug conjugates (ADCs) [167]. This confirms that the engineering objective of achieving targeting specificity is supported by validated technical pathways, and that the proposed direction is built upon a solid and diverse technological foundation. In summary, independent studies across various neurological disease models provide robust cross-validation for the broad applicability of EV-mediated mitochondrial transplantation. Whether in models of stroke, multiple sclerosis, or hereditary optic neuropathy, EVs and engineered vesicles derived from diverse cellular sources consistently improve neurological function and pathological markers through mitochondrial transfer. Collectively, this evidence establishes the strategy as a promising therapeutic paradigm that transcends individual disease models, laying a solid logical foundation for its potential application in DCI.
We recognize that while in vitro models offer mechanistic clarity, they
cannot fully replicate the complex in vivo microenvironment; similarly,
animal models, although reflective of overall physiological responses, face
significant obstacles in translation to humans because of species differences.
Encouragingly, emerging clinical data are beginning to bridge this gap. A
triple-blind clinical trial revealed that MSC-derived exosome eye drops
(NCT04213248) effectively improved corneal damage and severe dry eye disease
through anti-inflammatory and tissue repair mechanisms (tear secretion: Con group
vs. treatment 2 week group, p = 0.010; IL-6: Con vs. treatment 2 week or
4 week, p = 0.020 or 0.011), with these improvements persisting for
three months after treatment discontinuation [168, 169]. In a Pseudomonas
aeruginosa-induced lung injury mouse model, a human adipose-derived MSC-EVs
(haMSC-EVs) nebulized formulation (NCT04313647) increased the 96-hour survival
rate to 80% by reducing pulmonary inflammation and histological severity (Con
group vs. haMSC-EVs group: p
Although there are currently no direct clinical studies on the use of EV-mediated mitochondrial transplantation for the treatment of cognitive dysfunction in diabetes, we constructed a rational extrapolation method on the basis of existing successful EV clinical trials. We highlight that translating this platform into cerebral mitochondrial supplementation therapy faces core challenges, including ensuring efficient and targeted delivery of functional mitochondria to specific neurons and maintaining mitochondrial viability during large-scale production. Future research should focus on developing novel engineered EVs to enhance their brain-targeting ability and consider designing an early-phase clinical trial incorporating neuroimaging assessments. This trial would evaluate, in patients with type 2 diabetes and mild cognitive impairment, the added value of mitochondria-loaded EVs (e.g., administered via innovative routes such as intranasal delivery) compared with conventional EVs in improving neuronal energy metabolism and cognitive function, thereby establishing a novel therapeutic paradigm for EV-mediated mitochondrial transplantation.
Current evidence indicates that EV-mediated mitochondrial transplantation holds clear potential for improving neurological function, yet the field remains marked by critical mechanistic gaps and conflicting research outcomes that demand further investigation. A central unresolved question concerns the precise mechanisms linking peripheral mitochondrial dysfunction to central neuronal damage. While preliminary evidence suggests the existence of neuro-peripheral mitochondrial stress signalling in aging and disease states, the specific molecules responsible for transmitting these signals (such as certain mitochondrial stress secretory factors) and their reception mechanisms in the CNS remain at the forefront of research [171, 172]. Simultaneously, contradictory experimental data highlight the complexity of biological systems: for instance, m/lEVs may play a more significant role in mediating mitochondrial transfer than small EVs, contrasting with conventional understanding [78]; more notably, Li et al. [173] found in vivo experiments that mitochondrial transplantation produced significant therapeutic benefits in juvenile mouse models but yielded limited effects in adult models. This age-dependent efficacy disparity underscores the profound influence of the host microenvironment on treatment outcomes. These gaps and contradictions demonstrate that we still have considerable ground to cover in fully understanding the mechanisms of EV-mediated mitochondrial transplantation, which constitutes a key direction for future research in this field.
Neuronal mitochondrial dysfunction is a potential trigger of age-related neurodegeneration and cognitive decline, making the targeting of mitochondrial homeostasis a critical breakthrough point for the prevention and treatment of CNS diseases. Extensive research has revealed that mitochondrial function is regulated by multimodal signalling, providing new perspectives and methods for elucidating interactions between neuronal mitochondria and the extracellular environment or other cells. These interactions may involve various physiological and pathological processes, such as tissue development, regeneration, aging, metabolism, immunity, and inflammation. By analysing material exchange and signal transduction between mitochondria and multimodal signalling, the molecular mechanisms and regulatory networks of these processes can be elucidated, providing a basis for the discovery of new biomarkers and therapeutic targets. Studies on multimodal signalling regulation of mitochondrial function also provide new ideas and platforms for developing diagnostic and therapeutic strategies [26, 78, 106, 108, 109, 129]. The detection of EVs containing mitochondrial components or products can reflect mitochondrial function and status, thereby assessing the health of neurons or brain tissue. Modification of the quantity or properties of EVs carrying mitochondrial components or products can influence neuronal or brain tissue mitochondrial function, enabling disease prevention or treatment. For example, injecting EVs containing normal mtDNA or proteins can repair damaged or defective mitochondria, thereby ameliorating mitochondrial-related neurodegenerative diseases. However, the translation of current animal model-based research findings into clinical practice faces significant challenges. First, species differences represent a major obstacle, as fundamental distinctions between humans and animal models like mice in immune system function, BBB permeability, and metabolic pathways limit the direct extrapolation of animal experimental results. Second, standardized protocols for large-scale production and quality control of EV therapies have not been established, hindering the reproducibility and standardization of their clinical application. Furthermore, controlling functional heterogeneity remains a substantial challenge, as even within the same cell population, significant differences exist among functional subpopulations secreting EVs, posing a fundamental challenge to traditional quality control strategies relying on population-averaged analysis. Although engineering modifications represent a key strategy for enhancing targeting and efficacy, systematically evaluating how to balance modification efficiency with immunogenicity risks and ensure long-term safety remains necessary. Finally, the immunogenicity risk in the clinical translation pathway cannot be overlooked, as exogenous mitochondria or their components may trigger innate immune responses through pathways such as cGAS-STING, requiring this to be a core consideration in carrier design. In summary, the regulation of neuronal mitochondrial function by multimodal signalling is an emerging and cutting-edge research field with significant biological implications and clinical application value. Future work should further explore the mechanisms, effects, regulatory factors, and influencing factors of mitochondria-multimodal signalling interactions, as well as their changes under various physiological and pathological conditions. These findings provide more evidence and theoretical support for understanding the role of mitochondrial function in maintaining neuronal homeostasis and its impact on the onset and progression of diabetes and its complications.
Neuronal mitochondrial dysfunction is one of the important pathological links in the occurrence and progression of DCI, making the regulation of neuronal mitochondrial homeostasis a key breakthrough point for the prevention and treatment of CNS diseases. Among these strategies, EV-mediated mitochondrial transplantation is the most promising strategy. It promotes therapeutic targeting from the traditional molecular level to the level of organelle functional repair and replacement while offering broad prospects for clinical translation. However, the clinical translation of this concept faces several challenges, primarily reflected in the suboptimal targeting and delivery efficiency of EVs, the low in vivo survival rate of exogenous mitochondria, and the unclear mechanisms underlying their functional integration with the host mitochondrial network. Therefore, future research should focus on enhancing the safety, targeting precision, and overall efficacy of mitochondrial transplantation while also delving deeper into the regulatory mechanisms of transplanted mitochondria within neurons.
YL conducted the investigation and wrote the original draft. PZ contributed to the conception and design of the study, analyzed and interpreted key findings regarding therapeutic mechanisms, and was responsible for funding acquisition and project administration. He also contributed critically to the writing through review and editing. Both authors contributed to critical revision of the manuscript for important intellectual content. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the Foundation of National Natural Science Foundation of China (82174112) and the Science and Technology Project of Haihe Laboratory of Modern Chinese Medicine (22HHZYSS00015).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.