



 , Lingzhen Tan 1,2,3,†, Lin Lao 1,2,3,†, Xiaomeng Huang 2, Sheng Zhang 4,5,6,*
, Lingzhen Tan 1,2,3,†, Lin Lao 1,2,3,†, Xiaomeng Huang 2, Sheng Zhang 4,5,6,* , Haitao Sun 1,3,6,*
, Haitao Sun 1,3,6,*1 Clinical Biobank Center, Guangdong Provincial Clinical Research Center for Laboratory Medicine, Department of Laboratory Medicine, Zhujiang Hospital, Southern Medical University, 510280 Guangzhou, Guangdong, China
2 The Second School of Clinical Medicine, Southern Medical University, 510280 Guangzhou, Guangdong, China
3 Neurosurgery Center, The National Key Clinical Specialty, The Engineering Technology Research Center of Education Ministry of China on Diagnosis and Treatment of Cerebrovascular Disease, Guangdong Provincial Key Laboratory on Brain Function Repair and Regeneration, The Neurosurgery Institute of Guangdong Province, Zhujiang Hospital, Southern Medical University, 510280 Guangzhou, Guangdong, China
4 Department of Neurobiology, School of Basic Medical Sciences, Southern Medical University, 510515 Guangzhou, Guangdong, China
5 Department of Neurology, The Eighth Affiliated Hospital, The First People’s Hospital of Shunde, Southern Medical University, 528300 Foshan, Guangdong, China
6 Key Laboratory of Mental Health of the Ministry of Education, Guangdong-Hong Kong-Macao Greater Bay Area Center for Brain Science and Brain-Inspired Intelligence, Southern Medical University, 510515 Guangzhou, Guangdong, China
†These authors contributed equally.
Abstract
Both myelination and remyelination in the central nervous system (CNS) rely on the differentiation of oligodendrocyte precursor cells (OPCs) into mature oligodendrocytes (OLs). However, the microenvironment and underlying mechanisms of these two processes are significantly different. Myelination is driven by intrinsic developmental programs, whereas remyelination is triggered as a response to demyelinating injury. These distinct origins shape unique microenvironmental conditions that critically influence the respective processes. The microenvironment comprises, the extracellular matrix (ECM) and cellular components. Neurons, microglia, and astrocytes play stage-specific roles, ensuring proper myelin turnover under physiological conditions and regeneration after demyelination. Dysregulation of the microenvironment represents a critical driver of aberrant myelination and remyelination failure, as well as a key limitation of current pro-regenerative strategies. In this review, we discuss the cellular and molecular basis of microenvironmental regulation, recent advances in pathological microenvironmental alterations across demyelinating diseases (multiple sclerosis and neuromyelitis optica spectrum disorder) and myelin-associated disorders (Alzheimer’s disease and ischemic stroke), and emerging pro-remyelination strategies targeting the microenvironment, such as Bruton’s tyrosine kinase (BTK) inhibitors and microglia replacement strategies. We provide a non-cell autonomous perspective to advance the understanding of OLs differentiation and CNS remyelination.
Graphical Abstract
Keywords
- myelination
- remyelination
- microenvironment
- neuron
- microglia
- astrocyte
- multiple sclerosis
- Alzheimer’s disease
- ischemic stroke
Myelin, a fundamental structural component in the central nervous system (CNS), facilitates saltatory conduction of action potentials and provides metabolic support of in the CNS [1]. Oligodendrocyte precursor cells (OPCs) constitute a progenitor population that differentiates into mature oligodendrocytes (OLs) to form myelin sheaths [2]. Physiological myelination begins in infancy and involves continuous structural remodelling [2]. The microenvironment, composed of cellular components and the extracellular matrix (ECM), is essential for myelin formation and maintenance [3]. Neurons, microglia, and astrocytes interact with OPCs/OLs to maintain myelination homeostasis [4]. Following pathological demyelination, remyelination emerges as a critical neuroprotective mechanism to mitigate disease progression and neurodegeneration [5]. When the CNS encounters external stress such as inflammation or disrupted blood-brain barrier (BBB) integrity, the brain enables peripheral immune cells to infiltrate the CNS, and the microenvironment exhibits significant challenge [6]. These interactions alter microenvironmental conditions, ultimately determining the success or failure of remyelination [6]. Impaired remyelination represents a pivotal driver of neuronal damage and a central therapeutic challenge in regenerative therapy [5].
CNS myelinopathies include neurodevelopmental disorders (e.g., X-linked adrenoleukodystrophy (X-ALD), metachromatic leukodystrophy (MLD)) and demyelinating diseases (e.g., multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD)) [1]. Emerging studies have identified myelin defects as critical engineers of neurodegenerative diseases—including Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Parkinson’s disease (PD)—as well as neurovascular pathologies such as ischemic stroke [7, 8]. These findings expand the spectrum of demyelination-associated pathologies and provide novel therapeutic perspectives for incurable CNS disorders through myelin-targeting strategies. Current therapeutic strategies are evolving from inflammatory modulation towards remyelination-promoting approaches, particularly to address progressive disability in chronic MS [9]. However, many pro-remyelination therapies are unable to overcome the detrimental effects of pathological microenvironmental factors and fail to achieve therapeutic outcomes [10]. Thus, understanding microenvironmental alterations is crucial for advancing knowledge of CNS disease mechanisms and developing novel therapies.
While myelination and remyelination share fundamental cellular developmental pathways, they are regulated by distinct microenvironmental signals and mechanisms. Previous reviews have predominantly characterized the static components of myelination and remyelination microenvironments, focusing largely on inflammatory demyelinating diseases. To broaden understanding, this review examines the cellular and molecular mechanisms of microenvironmental modulation in both processes and investigates their pathogenic roles across various CNS disorders. Notably, we highlight emerging insights into myelinopathies beyond inflammatory demyelination, including Alzheimer’s disease and ischemic stroke. Finally, we evaluate innovative strategies targeting microenvironmental modulation to promote myelin regeneration.
Myelin is a multilayered membrane structure formed by OLs processes wrapping
around axonal segments [1]. It consists primarily of lipids (
Myelin sheaths segmentally insulate axons, creating distinct electrophysiological domains along the neural fiber [11]. The exposed nodal regions, known as the Node of Ranvier, are enriched with voltage-gated sodium channels, enabling efficient action potential propagation through saltatory conduction [12]. Conversely, the myelinated internodal regions exhibit reduced membrane potential fluctuations below the excitation threshold [12]. The conduction velocity in myelinated fibers is influenced by factors such as myelin thickness, internode length, and axon diameter, which are represented by the g-ratio (the ratio of axon diameter to fiber diameter) [11]. An optimal g-ratio (approximately 0.6–0.7) maximizes saltatory conduction efficiency [13]. Additionally, axons face energetic challenges due to their long distance from the cell body and high metabolic demand [14]. Myelin supports these needs by enabling metabolic coupling through monocarboxylate transporters (MCTs), which transfer lactate and pyruvate from OLs to axons for oxidative phosphorylation [15]. Apart from the effects on conduction and metabolic support, potassium buffering mechanisms, involving Kir4.1 channels, paranodal gap junctions, and Na+/K+-ATPase on myelin, help maintain extracellular K+ balance and prevent neuronal hyperexcitability [16, 17].
The CNS contains an abundant population of long-lived, proliferative OPCs that continuously differentiate into mature oligodendrocytes. During this process, OLs undergo an immense expansion of membrane surface area—up to several thousandfold—enabling them to myelinate multiple axons. This remarkable growth is achieved with relatively high metabolic demands on the vascular supply [2, 18, 19]. The progress involves a series of precisely regulated myelination stages [2]. Initially, OPCs are recruited to axonal domains, where they extend processes to establish selective axonal contact [2]. Once stable contact is made, polarized redistribution of proteins and reorganization of the cytoskeleton drive the morphological transformation into pre-myelinating OLs [2]. Finally, terminal maturation culminates in the concentric wrapping of axons and compaction of the myelin sheath [2]. Specific transcriptional regulators govern each stage, with sequential expression of Sox2 (promoting OPC proliferation), Olig2/Sox10 (lineage commitment), Sox2/Tcf7l2/Nkx2.2 (OLs maturation), and Myrf (terminal differentiation) [20, 21, 22, 23, 24]. Moreover, myelination is bidirectionally influenced by microenvironmental factors: Neuronal electrical activity and gliogenic growth factors promote myelination, while axonal surface, such as Jagged1, and inhibitory ECM components like chondroitin sulfate proteoglycans (CSPGs) help prevent excessive myelination [3, 25, 26].
Human myelination follows distinct spatiotemporal patterns. It progresses rapidly in early life and adolescence, stabilizes in adulthood, and declines with aging [27]. Spatially, myelination occurs in a specific sequence: projection fibers complete myelination first, followed by commissural fibers, and ultimately association fibers [27]. Notably, even after myelination is complete, structural remodeling continues throughout life, involving dynamic adjustments to myelin sheath quantity, length, thickness, and internode properties [27]. This ongoing plasticity plays a key role in neural circuit refinement and supports advanced neurological functions, including motor skill acquisition and memory consolidation [28, 29, 30].
The formation and maintenance of CNS myelin depend on both the intrinsic properties of OLs and the balance of microenvironmental factors [3]. Recent research has shown that disrupting meningeal lymphatic vessels in adult mice alters the CNS microenvironment, affecting glial cell function and cytokine profiles, leading to reduced populations of mature OLs and impaired myelination [31]. The microenvironment comprises cellular elements and ECM, with key contributors including neurons, astrocytes, and microglia (Fig. 1) [32, 33, 34]. The ECM contains inhibitory molecules such as CSPGs and hyaluronic acid, which can also influence myelination. While this review emphasizes cellular interactions, a detailed examination of ECM components can be found in Su et al.’s [26] comprehensive review.
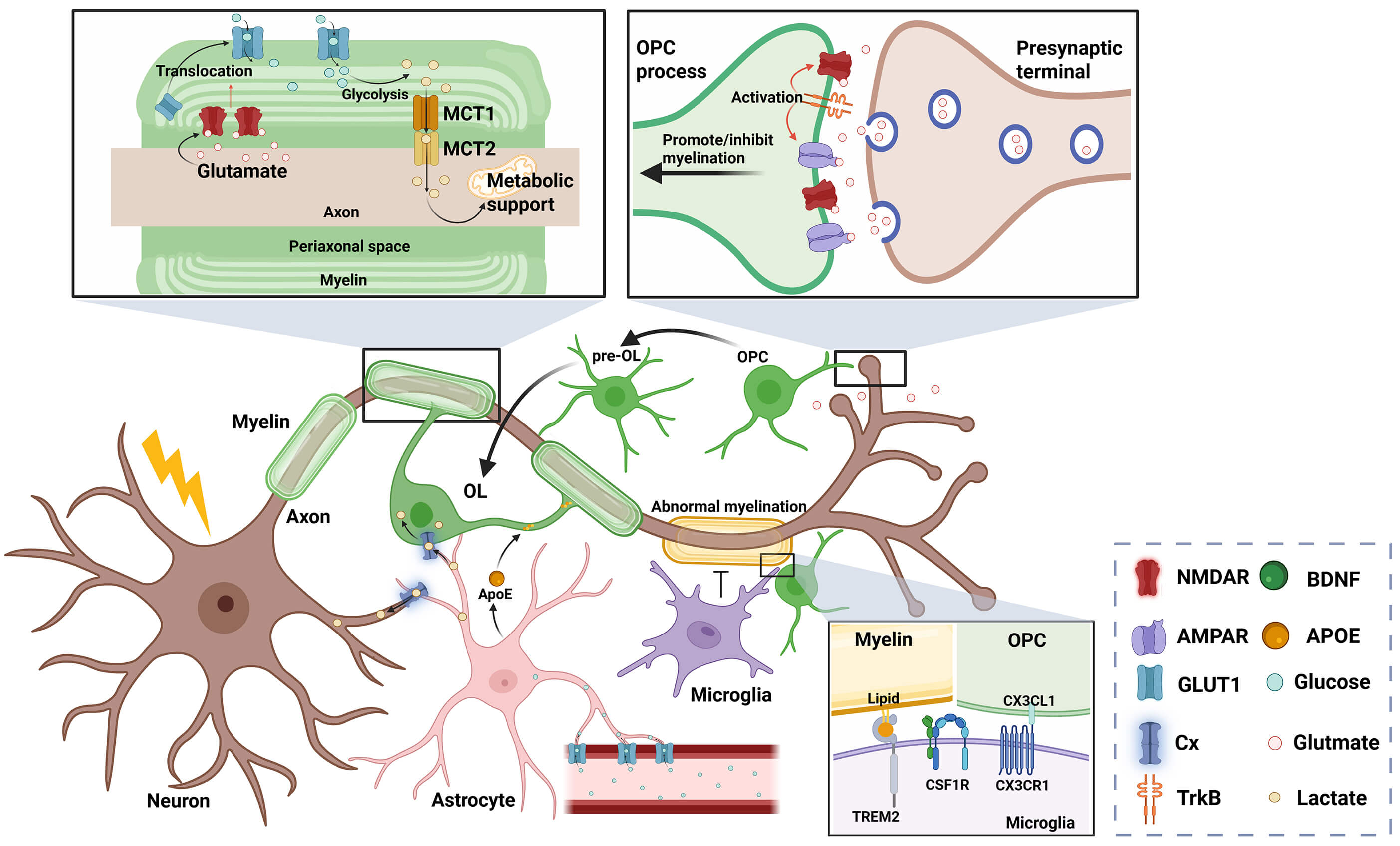 Fig. 1.
Fig. 1.
Physiological myelination microenvironment. Neurons, microglia,
and astrocytes regulate myelin structural stability and normal turnover. Neurons
mediate neurotransmitter and BDNF release through electrical activity to initiate
and regulate myelination. Glutamatergic signaling exerts differential effects at
distinct sites: Axonal terminals release glutamate onto OPC processes to modulate
their proliferation/differentiation, while glutamate from the periaxonal space
acts on NMDARs to regulate myelin-mediated metabolic support for axons. Microglia
phagocytose excessive/misplaced myelin sheaths and OPCs through TREM2 or CX3CR1
to prevent aberrant myelination. Astrocytes uptake glucose from the vasculature,
supply energy substrates to neurons and OLs via connexin-mediated metabolic
coupling, and provide cholesterol for myelination through APOE-mediated lipid
transport. Cx, connexin; BDNF, brain-derived neurotrophic factor; OPC, oligodendrocyte
precursor cell; NMDAR, N-methyl-D-aspartate receptor; TREM2, triggering receptor
expressed on myeloid cells 2; CX3CR1, CX3C chemokine receptor 1; OLs,
oligodendrocytes; APOE, Apolipoprotein E; MCT, monocarboxylate transporter;
AMPAR,
Neuronal activity plays a crucial role in regulating OPCs proliferation, axonal selection, and myelin structural dynamics through adaptive myelination, a process fundamental to CNS development and circuit remodeling [32, 35]. Mechanistically, experience-driven neuronal depolarization triggers patterned action potentials that release signaling molecules, which in turn modulate OPCs lineage progression [35].
Neurotransmitters, particularly glutamate, mediate axoglial communication by
binding to receptors on both OPCs and myelin, with emerging evidence highlighting
the roles of
Neuronal activity also induces the release of neurotrophic factors such as brain-derived neurotrophic factor (BDNF) [46, 47]. Released via synaptic vesicles, BDNF binds to TrkB receptors on OPCs, driving their differentiation [48]. Additionally, BDNF enhances glutamatergic signaling by simultaneously activating NMDARs and AMPARs on OPCs. [49]. Geraghty et al. [50] reveal that OPC-specific TrkB deletion in mice exhibits impaired cortical projection myelination and leads to cognitive deficit. In contrast, axonal membrane proteins exert inhibitory control over myelination, including polysialylated neural cell adhesion molecule (PSA-NCAM), Jagged1, transient axonal glycoprotein-1 (TAG-1), and leucine-rich repeat and immunoglobulin domain-containing protein 1 (LINGO-1) [51, 52, 53, 54]. Their structural features and inhibitory mechanisms are detailed in Almeida’s review [55].
Interestingly, Fang et al. [56] reveal that OPCs regulate neuronal function through mechanisms independent of myelination. OPC processes establish activity-dependent contacts with neuronal somata, promoting lysosomal exocytosis in neurons, which protects against neuronal senescence and degeneration. However, further studies are needed to clarify the temporal sequence between cellular contact and lysosomal fusion [56].
The CNS harbors two main macrophage populations: parenchymal microglia and non-parenchymal border-associated macrophages (BAMs) [57]. BAMs reside in perivascular spaces, meninges, and choroid plexus [57, 58]. Under physiological conditions, microglia secrete neurotrophic factors and regulate synaptic pruning, refining neural circuits during development [59]. Perivascular macrophages maintain BBB integrity by restricting peripheral immune cell entry [60]. Meningeal macrophages may regulate meningeal lymphatic vessel dynamics and act as antigen-presenting cells (APC) [31]. Choroid plexus macrophages, located near microvilli, are thought to contribute to cerebrospinal fluid (CSF) homeostasis [61]. Collectively, BAM functions require further validation.
A specialized “microglial fountain” subset emerges postnatally in regions of active myelination, such as the corpus callosum and cerebellum. These cells express activation-associated markers, including CD11c, and are critical for early oligodendrogenesis and the maintenance of adult OPC pools [62]. Their lifespan coincides with the period of extensive myelination in healthy murine brains, and they represent a major source of insulin-like growth factor-1 (IGF-1), a potent pro-myelination signal [63, 64]. Selective depletion of this subset impairs myelination, confirming their essential role in developmental myelinogenesis [63].
Microglia regulate OPC lineage development and myelination through phagocytosis, lipid metabolism regulation, and neurotrophic factor secretion [65]. Pioneering work by Schafer et al. [66] reveals their role in activity-dependent synaptic pruning in the developing retina. Subsequent studies revealed their ability to phagocytose excess OPCs and aberrant myelin in an activity-dependent manner, preventing hypermyelination [67, 68]. The neuron-derived chemokine CX3CL1 plays a critical role by binding to the microglial receptor CX3C chemokine receptor 1 (CX3CR1), which triggers activity-dependent synaptic remodeling [69]. Beyond synaptic modulation, CX3CR1 signaling also enables microglia to recognize and phagocytose OPCs [68]. Loss of CX3CR1 not only disrupts synaptic pruning but also reduces myelin density in the corpus callosum, linking microglial-dependent oligodendrogenesis to neuronal circuit refinement [68].
Microglia express triggering receptor expressed on myeloid cells 2 (TREM2), a member of the immunoglobulin superfamily. TREM2 recognizes anionic myelin components, including sphingomyelin, phosphatidylcholine, and fatty acids, which regulate diverse microglial functions encompassing survival, migration, phagocytosis, and metabolic regulation [70, 71]. It is activated in response to mild myelin injury or aging, coordinating microglial responses that stabilize neural networks [70]. Homozygous TREM2 mutations cause Nasu-Hakola disease, characterized by subcortical demyelination and early-onset dementia [72]. TREM2 also plays an important role in regulating microglial lipid metabolism [73, 74, 75]. Microglial TREM2 deficiency impairs cholesterol efflux pathways and induces cholesterol ester (CE) accumulation during myelin clearance [75].
Microglia also support OPC development by releasing IGF-1, IGF-2, tumor necrosis
factor-alpha (TNF-
Colony-stimulating factor 1 receptor (CSF1R), a cell-surface receptor, regulates
the survival of microglia and monocyte-derived macrophages (MDMs) in the CNS
[77]. A landmark clinical case of a patient with homozygous CSF1R mutation showed
complete absence of microglia, accompanied by hypoplasia of the corpus callosum
and progressive white matter atrophy, which provides direct evidence of the
indispensable role of microglia in myelin development and white matter
homeostasis [78]. Methodologically, CSF1R knockout animal models are used to
investigate microglial functions in the CNS [79]. However, traditional CSF1R
knockout models deplete both microglia and MDMs, confusing cell-specific analyses
[80]. To overcome this, McNamara et al. [33] developed the FIRE-deleted
mouse model, which selectively eliminates microglia by deleting the CSF1R Fms
intronic regulatory element [81]. Interestingly, this model revealed that
microglia particularly maintain adult myelin integrity through transforming
growth factor
Astrocytes contribute to myelination through both structural and metabolic interactions [83]. Their processes wrap around cerebral microvessels to support BBB formation and regulate cerebral blood flow [83]. White matter astrocytes exhibit distinct features with compact somata and elevated glial fibrillary acidic protein (GFAP) expression [83, 84]. GFAP-deletion mice show delayed optic nerve and spinal cord myelination, cerebellar white matter disorganization, and BBB leakage [85].
Metabolically, astrocytes supply energy substrates to both neurons and oligodendrocytes [86]. They transport glucose from blood vessels via GLUT1 and provide lactate for neurons and OLs [87]. This metabolic coupling is mediated by gap junctions and hemichannels formed by connexin (Cx), which transfer glucose, lactate, and Ca2+ across glial networks [88]. Mice lacking Cx47 and Cx30 exhibit OLs degeneration characterized by white matter vacuolization and myelination defects [89]. Astrocytes are also essential for lipid homeostasis, particularly cholesterol metabolism [90]. The availability of cholesterol is a crucial limitation on myelination [91]. While OLs initiate cholesterol synthesis during initial myelination, astrocytes become the predominant cholesterol source during myelin maintenance through APOE-mediated transport [92, 93]. To mention, the APOE4 genotype is the strongest genetic risk factor for Alzheimer’s disease. Carriers of APOE4 exhibit abnormal cholesterol accumulation, oligodendrocyte dysfunction, and reduced myelin content, which links astrocytic lipid metabolism to both myelin biology and neurodegenerative vulnerability [94].
Remyelination is the regeneration of myelin sheaths around intact axons following demyelination, a process crucial for restoring axonal conduction and metabolic support [5]. Although it recapitulates key stages of developmental myelination, including OPCs migration, proliferation, and differentiation into myelinating OLs, remyelination exhibits distinct morphological and cellular features [20]. Regenerated myelins show thinner and shorter sheaths with an elevated g-ratio compared to developmental myelin, which may compromise conduction velocity [95]. Lineage-tracing studies using radiocarbon (14C) birth-dating demonstrated that mature OLs surviving from demyelination can contribute to myelin regeneration in adult MS lesions through proliferation-independent mechanisms [96]. However, zebrafish models indicate that while residual OLs exhibit remyelination capacity, their regenerative efficiency and structural precision are markedly lower than that of newly generated OLs derived from OPCs [97]. Moreover, Schwann cells, normally peripheral myelinating glia, can also participate in CNS remyelination via OPCs transdifferentiation, particularly in astrocyte-deficient lesions [98, 99, 100].
A critical distinction between myelination and remyelination is that controlled acute inflammation is indispensable for initiating remyelination [6]. In the early phase of inflammation, chemokines and semaphorins released by microglia and astrocytes recruit OPCs to lesion sites [101]. Optimal OPCs recruitment density activates mechanosensitive Piezo1 channels and elevates Netrin-1 levels, promoting their differentiation [102, 103]. However, a recent study have shown that the mechanotransduction channel TMEM63A, rather than Piezo1, is expressed in OLs and promotes their differentiation. Additionally, activated microglia clear inhibitory myelin debris and release trophic signals necessary for OPCs proliferation and differentiation [104]. The limited period in which OPCs migrate and interact with pro-remyelination signals in the microenvironment is called the “remyelination-permissive window” [104, 105]. If exceeding this period, chronic inflammation establishes a hostile microenvironment characterized by inhibitory axonal molecules (e.g., PSA-NCAM), astrogliotic scarring, and inhibitory ECM deposition (e.g., CSPG), all of which hinder OPCs migration and differentiation [106, 107]. Collectively, remyelination requires collaboration among diverse cellular populations. Transcriptomic studies highlight dynamic functional states of microglia, MDMs, astrocytes, and neurons across distinct phases of regeneration, and have linked these states to regenerative outcomes [108, 109]. MS lesions with robust remyelination are characterized by reduced accumulation of foamy microglia and enrichment for epithelial–mesenchymal transition (EMT) pathways, along with pro-regenerative factors such as CXCL12, EGF, and HGF [108]. In the cuprizone (CPZ) model, during remyelination, astrocytes upregulate genes associated with detoxifying metabolic pathways and exhibit distinct transcriptional subpopulations, suggesting functional diversity [109]. Neurons, however, exhibit relatively limited transcriptional changes during remyelination, which needs further validation [109].
In demyelinating conditions, circulating MDMs infiltrate through the compromised BBB and collaborate with resident microglia to mediate antigen presentation and myelin debris clearance [110, 111]. Despite overlapping functions, MDMs and microglia diverge in their roles across disease stages. Comparative studies reveal that MDMs exhibit higher expression of MHC II molecules and co-stimulatory markers compared to microglia, positioning them as primary drivers in demyelination pathogenesis [110]. In lysolecithin (LPC)-induced lesions, both cell types initially display comparable densities and phagocytic capacities for myelin debris [112]. As demyelination progresses, however, microglia undergo robust activation and proliferation, becoming the dominant population, whereas MDMs are largely restricted to the acute demyelinating phase [113]. Microglia also restrain excessive macrophage infiltration by remodeling the extracellular matrix, modulating secretory profiles, and facilitating BBB repair [113]. Experimental microglial depletion exacerbates axonal loss and disrupts OPCs recruitment and differentiation, underscoring their indispensable neuroprotective and pro-regenerative roles [112, 113].
During remyelination, microglia shift from a pro-inflammatory to a
pro-regenerative phenotype through necroptosis-mediated elimination of
pro-inflammatory subsets. This transition is tightly coupled to the stage of OPC
differentiation [114, 115]. Additionally, microglia P2X4R activation can also
promote pro-regenerative polarization and remyelination [116]. Pro-regenerative
microglia enhance remyelination by secreting a diverse repertoire of trophic and
immunomodulatory factors, including activin A, IL-4, IGF1, TNF-
Notably, the oversimplified in vitro classification of microglia into “resting”, “M1” (pro-inflammatory), or “M2” (pro-regenerative) phenotypes fails to capture their functional complexity in vivo, and has been abandoned by current researchers [113]. Single-cell transcriptomic profiling now provides a new paradigm for precisely distinguishing the functional states of microglia [117]. RNA sequencing in mouse models across developmental, aged, and post-demyelination has identified at least nine transcriptionally distinct microglial phenotypes [117]. A subset characterized by chemokine ligand 4 (Ccl4) expression is specifically associated with demyelinating pathology [117]. In another research, microglial subsets expressing high levels of the transcription factor MAFB are crucial for remyelination [109]. These findings suggest that future therapies may need to target specific microglial subsets to maximize repair while minimizing adverse effects.
Efficient myelin debris clearance is a prerequisite for remyelination.
Accumulated debris not only damages axons but also inhibits OPC recruitment and
differentiation [118, 119]. Microglia recognize debris through receptors such as
TREM2, complement receptor 3 (CR3), and signal-regulatory protein-
Debris clearance imposes high lipid-processing demands on microglia, especially for cholesterol and sphingolipids [126]. Proper lipid metabolism is critical for cellular homeostasis and inflammation resolution [126]. While the autophagy-lysosomal pathway enables lipid degradation, its hyperactivation can cause lysosomal overload, perpetuate pro-inflammatory phenotypes, and impair remyelination [127]. In LPC models, suppressing excessive autophagy-lysosomal activity or supplementing conjugated linoleic acid (CLA) during acute demyelination mitigates inflammation and promotes remyelination [127].
Microglia also recycle cholesterol from degraded myelin and transfer it to OPCs to support remyelination [93]. Liver X receptors (LXRs) regulate cholesterol efflux and have anti-inflammatory effects in acute demyelination [128, 129]. However, in chronic lesions, impaired LXRs activation leads to toxic intracellular cholesterol accumulation, which triggers endoplasmic reticulum stress and apoptosis in microglia [130]. To buffer lipid toxicity, microglia esterify excess cholesterol into lipid droplets, a protective response that promotes foam cell formation [130]. Notably, TREM2 deficiency disrupts lipid droplet biogenesis, aggravates ER stress, and further impairs remyelination [131].
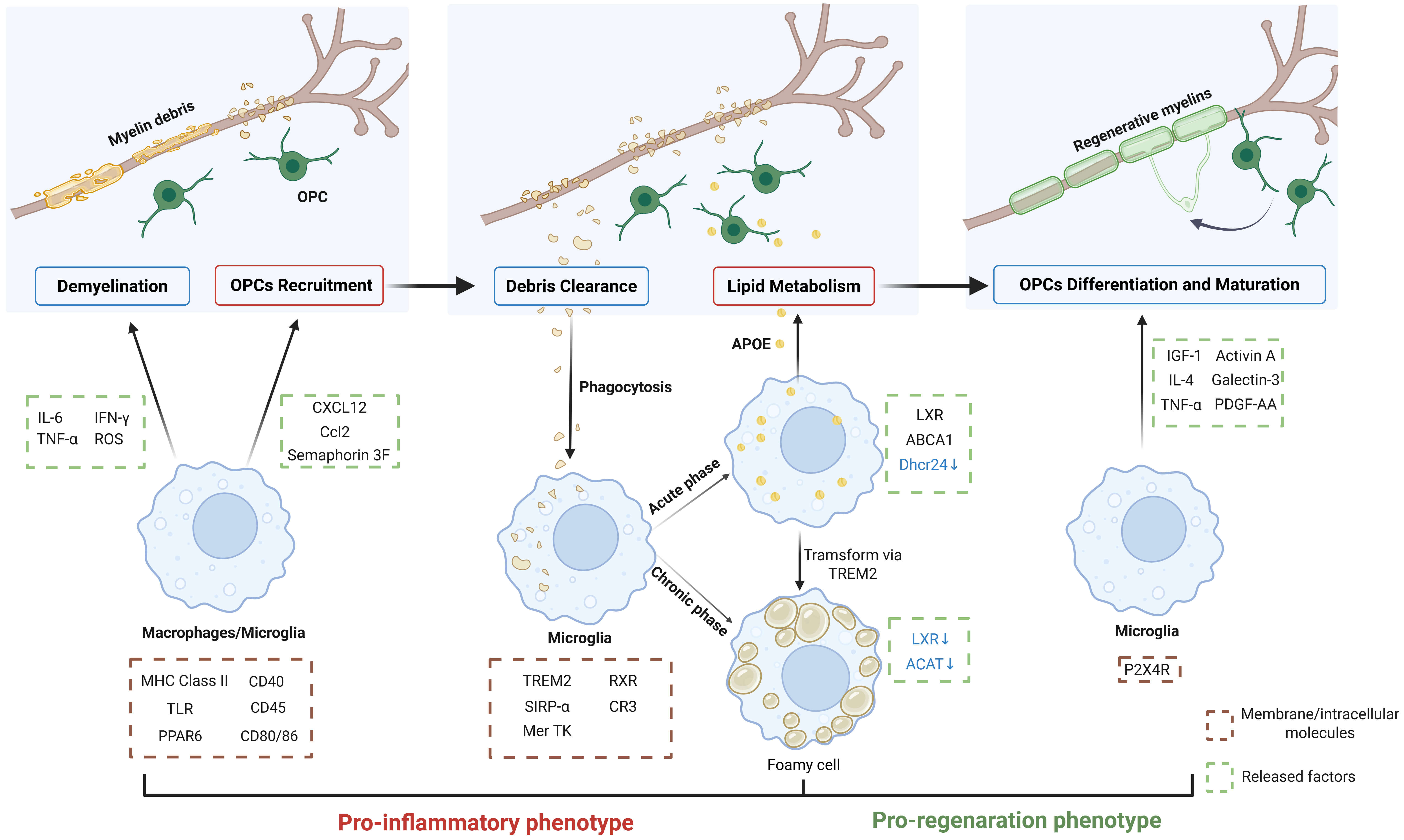
Collectively, microglia play crucial roles in different phases of demyelinating disease (Fig. 2).
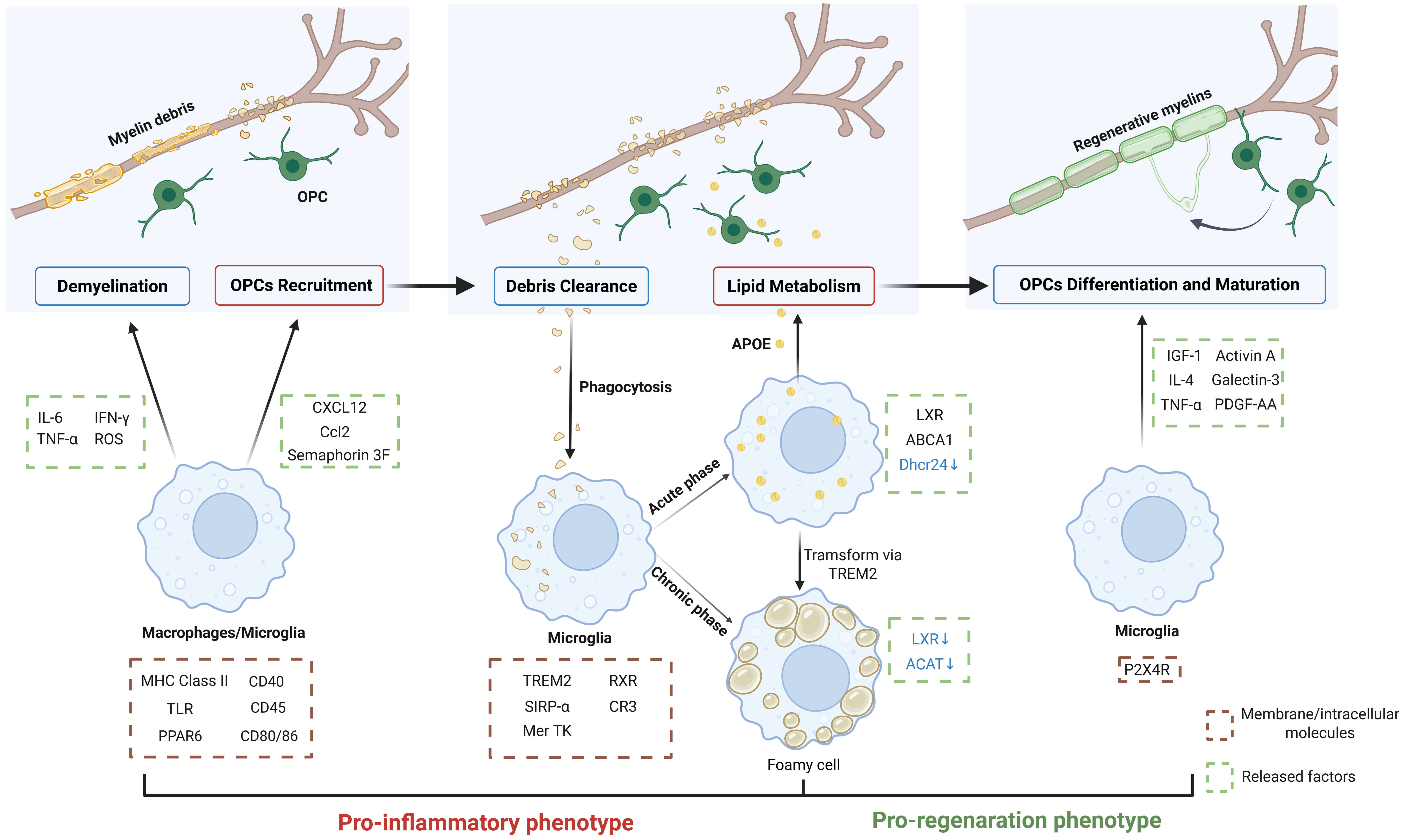 Fig. 2.
Fig. 2.
Microglia in remyelination. Microglia and monocyte-derived
macrophages serve as the primary immune mediators of demyelination, releasing
cytotoxic factors to damage myelin sheaths. While amplifying inflammatory
responses, they simultaneously secrete chemokines and semaphorins to recruit OPCs
to lesion sites. During the acute inflammatory phase, microglia efficiently clear
myelin debris, subsequently transferring processed lipids to OPC lineages to
support remyelination. If inflammation resolution is impaired, chronic
inflammation triggers microglia transformation into lipid-overloaded foam cells
with compromised lipid-processing capacity. As inflammation subsides, microglia
progressively transform to the pro-regenerative phenotype, releasing factors that
promote OPC differentiation and maturation. Successful remyelination requires
precisely coordinated regulation of these cellular functions across pathological
stages. IL-6, interleukin-6; IFN-
Although not traditionally classified as immune cells, astrocytes play pivotal
immunomodulatory roles in both demyelination and remyelination [6].
Interestingly, in EAE and MS lesions, astrocytes acquire epigenetic immune memory
similar to classical immune cells. This process is mediated by ATP-citrate lyase
(ACLY), which enhances transcription of NF-
During demyelination, astrocytes detect damage and become reactive. They release cytokines and chemokines that recruit immune cells and promote debris clearance [6]. Astrocyte-derived signals exert dual effects on remyelination: leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), and IGF-1 enhance OPCs proliferation; platelet-derived growth factor AA (PDGF-AA) and fibroblast growth factor 2 (FGF-2) drive OPCs differentiation; while endothelin-1 (ET-1) and FGF-9 inhibit remyelination [34]. Additionally, reactive astrocytes secrete ECM components that can limit lesion expansion but also hinder OPC migration and differentiation [34].
Astrocytes also regulate lipid metabolism during remyelination by supplying cholesterol through the ABCA1-mediated efflux pathway [133]. This process is linked to nuclear factor erythroid-2-related factor-2 (Nrf2) downregulation in astrocytes [133]. Interestingly, chronic MS lesions exhibit sustained Nrf2 activation, which disrupts cholesterol metabolism and reduces OLs survival [133]. While constitutive Nrf2 activation impairs regeneration, it remains essential for antioxidant defense [134, 135]. Nrf2 activation induces transcription of cytoprotective genes, including NAD(P)H quinone and oxidoreductase 1 (Nqo1), which mitigate oxidative stress via reactive oxygen species (ROS) scavenging, damage repair, and anti-inflammatory actions [136]. In EAE models, astrocytes display a distinct molecular profile marked by Nrf2 downregulation and MAFG upregulation. MAFG, a member of the sMAF (small musculoaponeurotic fibrosarcoma) transcription factor family, exerts gene expression control through combining with partner proteins. Accumulated MAFG represses Nrf2-driven transcriptional programs in astrocytes, consequently amplifying oxidative stress and neuroinflammation [137]. Consistently, Nrf2-knockout mice show cerebellar astrogliosis and myelin pathology, highlighting the need for balanced Nrf2 activity in maintaining myelin integrity [134].
Increasing evidences further highlight the indispensability of astrocytes for remyelination. In ethidium bromide (EB)-induced demyelination, OPCs fail to differentiate in astrocyte-depleted lesions [138]. However, the heterogeneous functional states of astrocytes remain poorly understood due to the lack of definitive surface markers. To address this, Clark et al. [139] developed nucleic acid detection and sequencing (FIND-seq) technology, a platform that tags astrocyte subsets using DNA/RNA signatures. Using this approach, they identified the role of nuclear receptor NR3C2 in suppressing transcriptional programs of pro-demyelinating astrocyte subsets [139]. However, conclusions regarding astroglial remyelination in MS drawn from rodent models require further validation, given the evolutionary divergence in genomic and transcriptomic profiles between mice and humans.
Although less studied, neuronal activity may play a key role in regulating remyelination. The underlying mechanisms likely involve both direct neurotransmitter effects and indirect modulation of the microenvironment [140, 141]. Gautier et al. [140] reveal that in EB-induced focal demyelination, demyelinated axons in the corpus callosum form nascent synapses with OPCs, which promotes remyelination via AMPAR activation. Conversely, blocking neuronal activity impairs OPCs differentiation [140]. Moreover, neuronal activity induces axonal ATP release, which acts on astrocytes in a paracrine manner and stimulates their LIF secretion, enhancing myelin formation by mature OLs [141].
Repeated optogenetic neuronal stimulation enhances remyelination in callosal
fibers and restores action potential conduction in demyelinated lesions [142].
However, outcomes vary depending on stimulation parameters, methods, and animal
age [142]. While optogenetic tools enable precise neuronal control, their
invasiveness limits clinical application [143]. Non-invasive brain stimulation
(NIBS) broadly modulates the microenvironment, which activates microglia,
astrocytes, and neural stem cells to promote anti-inflammatory microglial
polarization and neurogenesis [144, 145]. In rodent spinal cord demyelination
models, NIBS increases remyelination, upregulates neurotrophic factors (BDNF,
NGF), and suppresses pro-inflammatory cytokines (TNF-
Experimental, histopathological, and neuroimaging studies demonstrate a progressive aging-related decline in remyelination efficiency [147, 148, 149]. In MS, aging accelerates the accumulation of chronic lesions, with many patients transitioning to a progressive disease phase between 40 and 45 years. This shift markedly increases the risk of irreversible axonal degeneration and permanent disability [150, 151, 152]. Age-related remyelination failure is largely driven by OPCs dysfunctions, which involve two interconnected mechanisms: Intrinsic OPCs senescence and microenvironmental deterioration [153, 154, 155, 156]. Transplantation experiments highlight the dominant influence of the microenvironment. Aged OPCs regain a youthful regenerative capacity and transcriptional profile when placed in a young CNS matrix, whereas young OPCs adopt impaired function in an aged environment [102]. This dysfunction may primarily arise from age-dependent increases in ECM stiffness, which excessively activates the mechanosensitive ion channel Piezo1 on OPCs, reducing their regenerative potential [102]. Furthermore, fibroblast growth factor 17 (FGF17), which supports OPC proliferation and differentiation, declines with age in both human cerebrospinal fluid (CSF) and mouse neurons [157]. Remarkably, infusing young CSF or FGF17 into aged mice enhances hippocampal myelination and rescues memory deficits, underscoring the potential of CSF-based therapies [157]. However, clinical translation remains limited by incomplete knowledge of FGF17 dynamics in human CSF and the unknown safety and efficacy of CSF infusion in patients.
The aged microenvironment is characterized by chronic, low-grade inflammation
[6]. Interestingly, demyelinating injury promotes the accumulation of senescent
microglia, which upregulate components of the senescence-associated secretory
phenotype (SASP). These changes exacerbate inflammation and suppress
remyelination [112]. Senescent microglia and macrophages exhibit diminished
migration and phagocytic capacity, leading to myelin debris accumulation [130, 158, 159]. This dysfunction is associated with downregulation of the retinoid X
receptor (RXR) pathway [160]. RXR is both a ligand-activated receptor and a
transcription factor of genes related to macrophage immune regulation function
and cholesterol transport, which is a positive regulator of remyelination [161].
Deletion of microglial RXR
In addition, senescent microglia display impaired cholesterol processing, with insufficient activation of LXR-dependent efflux pathways. This defect leads to the accumulation of crystalline cholesterol within foam cells, triggering lysosomal rupture and NLRP3 inflammasome activation [130]. Moreover, astrocytes in the aged CNS also exhibit senescent features, including hypertrophic reactive morphology and autophagic vacuole accumulation [163]. These cells reduce cholesterol biosynthesis due to downregulation of key enzymes, limiting the lipid supply available for oligodendrocytes [164, 165].
Autoimmune disorders of the CNS arise from failures in immune tolerance, leading to aberrant responses against self-antigens in neural tissues [166]. Major examples include demyelinating diseases such as multiple sclerosis (MS), acute disseminated encephalomyelitis, neuromyelitis optica spectrum disorder (NMOSD), glial fibrillary acidic protein astrocytopathy (GFAP-A), and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). Other autoimmune encephalitides include anti-NMDAR encephalitis, anti-leucine glioma inactivated 1 (LGI-1) encephalitis, and anti-gamma aminobutyric acid-B (GABA-B) receptor encephalitis, etc. [166]. Among these, MS serves as the prototypical model and is widely studied using experimental autoimmune encephalomyelitis (EAE), lysolecithin (LPC)-induced, ethidium bromide (EB)-induced, cuprizone (CPZ)-induced, and viral infection models [167]. Of these, only EAE and viral models recapitulate autoimmune pathophysiology, with EAE remaining the most widely used platform to dissect mechanisms of autoimmune demyelination [168, 169]. Insights from both EAE and MS patients indicate that disease progression occurs through three major phases [170].
Activation of the peripheral immune system. Central thymic tolerance eliminates most reactive T cells, though some escape to the periphery [170]. Under physiological conditions, peripheral tolerance mechanisms, including regulatory T cells and intrinsic lymphocyte inhibition, constrain their activation [170]. However, tolerance breach of peripheral tolerance via molecular mimicry or epitope spreading activates these escaped T cells. Once activated, they acquire the ability to disseminate in circulation and compromise BBB integrity [171, 172, 173].
Compromise of the blood-brain barrier. The CNS immune privilege relies on multiple protective barriers, including the BBB, blood-CSF barrier, and meningeal-lymphatic isolation [174]. Reactive T cells trigger the expression of adhesion molecules on endothelial cells, such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin, facilitating their migration across the BBB [173, 175]. Recent studies reveal additional entry routes: meningeal lymphatic vessels that permit direct access of immune cells into the CNS [176]. Additionally, through the CSF, blood-derived innate immune cells located in the meningeal, perivascular, and ventricular spaces present antigen to T cells patrolling in CSF, which may also cause CNS inflammation [171].
Demyelination in brain parenchyma. Within the CNS, reactive T cells recognize
antigen-presenting cells (APCs) such as microglia, macrophages, and astrocytes,
which trigger their reactivation [170]. Reactivated T cells drive clonal
expansion of CD4+ T helper (Th) subsets, particularly Th1 and Th17 cells,
which secrete pro-inflammatory cytokines and chemokines that amplify inflammation
and recruit additional peripheral immune cells [170]. Myelin loss and axonal
injury are then mediated by neurotoxic molecules, including ROS, nitrogen species
(RNS), matrix metalloproteinases (MMPs), and TNF-
Multiple sclerosis (MS) is characterized by focal inflammation, demyelination,
and eventual neurodegeneration [169]. In addition to T cell-driven pathology,
infiltrating peripheral immune cells such as monocyte-derived macrophages (MDMs)
and neutrophils amplify neuroinflammation [169]. The mechanisms driving their
generation remain incompletely understood. Recent work by Shi et al.
[178] reveals that reactive T cells home to bone marrow in a CXCR4-dependent
manner, where they stimulate hematopoietic expansion through CCL5 secretion. This
finding not only deepens the understanding of reactive T cells as disease
initiators but also highlights the therapeutic potential for hematopoietic stem
cell transplantation (HSCT) in MS [178]. In a clinical trial, HSCT has shown
superior efficacy to disease-modifying therapies (DMTs) in patients with
relapsing-remitting MS, significantly delaying disease progression and improving
neurological function [179]. Autologous HSCT has now emerged as a promising
treatment option for patients with active RR-MS who show inadequate response to
DMTs. However, several challenges remain, including precise patient selection,
optimal timing of treatment, and integration into existing therapeutic sequences,
which warrant further investigation to fully realize its clinical potential
[180]. Dendritic cells (DCs) also shape disease course by suppressing reactive T
cell activation and pro-inflammatory responses through lactate-mediated
activation of the HIF-1
Beyond immune-mediated mechanisms, progressive MS is driven by neuronal and
axonal injury. Oxidative stress, iron toxicity, and glutamate-induced calcium
dysregulation contribute to cumulative neurodegeneration [177]. Neurodegeneration
in MS remains an unresolved challenge [177]. Aberrant activation of the
stimulator of interferon genes (STING) pathway in neurons may further accelerate
neuronal damage [182]. In this process, IFN-
Astrocytes also play critical roles in MS pathogenesis. They are recognized as
mediators of environmental MS risk (e.g., herbicide exposure) and drivers of
neurodegenerative processes in chronic MS [183, 184]. In progressive MS stages,
astrocytes exhibit disrupted sphingomyelin metabolism, which leads to elevated
cytosolic phospholipase A2 (cPLA2) activity. This shift activates mitochondrial
antiviral-signaling protein (MAVS) activation, which amplifies
NF-
Pathological and neuroimaging studies reveal that MS lesions evolve dynamically across disease stages, which is reviewed by Matthews et al. [186]. Remyelination capacity correlates with lesion chronology [187]. Acute lesions exhibit robust OPCs recruitment and increased density of cells, showing “shadow plaque” in pathological sections, whereas chronic lesions show OPC depletion and arrested pre-myelinating oligodendrocytes [187, 188, 189]. Mapping this lesion heterogeneity remains a challenge. Lin et al. [190] developed a multimodal pipeline integrating MRI images with spatial transcriptomics and tissue staining, identifying 28 lesion microenvironments in the course of the EAE marmoset model and identifying perivascular and periventricular aSERPINE1+ astrocyte subsets as early lesion initiators. Notably, they suggest that the ratio of proton density-weighted signal to T1 relaxation time is an important MRI biomarker, which can predict lesion formation 30 days before conventional MRI examination, offering a potential breakthrough in early MS diagnosis [190].
Current MS therapies mainly target peripheral inflammation, which provides symptom relief but fails to prevent progressive neurodegeneration [177, 189]. Enhancing endogenous remyelination has emerged as a promising therapeutic paradigm to halt disease progression [9]. Early efforts focused on promoting OPC differentiation, yet efficacy depends on OPCs availability, which varies among patients [191, 192, 193]. Acute active lesions often contain adequate OPCs that fail to differentiate, whereas chronic progressive lesions are characterized by OPCs depletion and microenvironmental inhibition of remyelination [101, 187, 194]. These differences underscore the necessity for stage-specific therapeutic strategies tailored to the evolving lesion microenvironment.
Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune demyelinating disease marked by recurrent episodes of visual, motor, or sensory dysfunction, often leading to neurological disability [195]. The core pathogenic mechanism involves Aquaporin 4 (AQP4)-mediated inflammatory demyelination, accompanied by retinal ganglion cell (RGC) degeneration through microglial activation [196]. Notably, both demyelination and remyelination in NMOSD rely heavily on the regulation of astrocytes [197].
NMOSD is now recognized as a primary astrocytopathy [197]. Cerebrospinal fluid GFAP levels serve as biomarkers for astrocytic injury and clinical severity in NMOSD [198]. Acute phase NMOSD exhibits significantly higher GFAP concentrations compared to MS, with rapid normalization during chronic phases [198]. AQP4 water channels, expressed on astrocyte end-feet, are the principal targets of pathogenic AQP4-IgG autoantibodies [199]. The binding of these antibodies during acute relapses activates the classical complement cascade, driving infiltration of granulocytes and lymphocytes [200]. Then, escalating inflammation initially damages astrocytes, subsequently extending to OLs and neurons [201]. Astrocytic dysfunction also impairs remyelination [202]. Notably, AQP4-IgG-positive patients demonstrate reduced CXCL12 expression, a key astrocyte-derived chemokine required for OPCs activation and remyelination [202]. Although astrocytes occupy a central role in NMOSD pathogenesis, the disorder arises from broader dysregulation involving oligodendrocytes, microglia, neurons, and infiltrating immune cells, a complexity that requires further validation [203].
Neurodegenerative and neurovascular disorders have long been described as neuronal loss and injury. However, growing evidence highlights myelin degeneration and OL dysfunction as shared features across these conditions [7, 204]. Traditionally, myelin breakdown was regarded as secondary to neuronal pathology. However, recent work suggests that primary myelin defects in Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and ischemic stroke may actively drive disease onset and progression [8, 205, 206, 207]. Here, we focus on AD and ischemic stroke, where demyelination-related mechanisms are increasingly recognized, and pro-remyelination strategies show therapeutic promise [208, 209, 210].
The prevalence of AD is rising sharply with population aging, projected to
exceed 139 million cases by 2050, imposing substantial individual and societal
burdens [211]. Classically, AD is defined by extracellular amyloid-
AD was long considered a gray matter disorder, but white matter pathology has
gained increasing recognition [214, 215]. White matter hyperintensities often
appear in presymptomatic stages, predicting cognitive decline independently of
A
Importantly, myelin abnormalities may occur before A
Mitochondrial dysfunction, a shared pathological hallmark of aging and AD,
triggers oxidative stress that creates a microenvironment detrimental to myelin
maintenance [224, 225]. A
APOE4 and TREM2 variants constitute major genetic risk factors for AD [230, 231]. The APOE4 variant disrupts the lipid-binding region of APOE, impairing cholesterol transport [94]. Single-cell transcriptomic analyses of postmortem human brains further link APOE4 to myelin pathology, showing reduced expression of myelination-related genes and altered cholesterol homeostasis in oligodendrocytes of APOE4 carriers [94]. In co-culture experiments, APOE4 impairs intercellular lipid transport between astrocytes and OL [232]. Variants in TREM2, another AD risk gene, compromise microglial lipid sensing and myelin debris clearance [233]. Conversely, TREM2 overexpression restores phagocytic capacity, ameliorating both neuropathology and behavioral deficits in AD models [233].
Notably, remyelination-based interventions improve cognition in animal models, underscoring the therapeutic relevance of myelin protection and repair in AD [205].
Ischemic stroke (cerebral infarction) is caused by stenosis or occlusion of cerebral blood supply arteries, resulting in neurological dysfunction in affected brain regions [234]. White matter injury strongly correlates with post-stroke dementia risk [234]. In middle cerebral artery occlusion (MCAO) mouse models, acute OLs death causes myelin loss within two weeks of stroke, while insufficient remyelination in later weeks impairs neuronal survival and functional recovery [8]. These findings highlight myelin integrity as both a prognostic marker and a therapeutic target in ischemic stroke. Notably, Pro-remyelination approaches, including muscarinic receptor 1 deletion or treatment with the pro-OPC differentiation drug clemastine, maintain neuronal survival and enhance functional recovery in MCAO models [8].
Multiple mechanisms converge to damage myelin after ischemia, including metabolic failure, excitotoxicity, and inflammation [235]. Energy depletion impairs ion channel functions, such as Kir4.1 on OPCs, disrupting myelination [236, 237]. Excitotoxicity is driven by extracellular glutamate accumulation, resulting from the reversed operation of the glutamate transporter GLT-1 under ischemic conditions, which triggers harmful calcium overload in neurons and OLs via AMPAR overactivation [238, 239]. Furthermore, abnormal ATP signaling activates P2X7 receptors on glia, inducing oxidative stress and exacerbating OL injury [240, 241].
Post-ischemic myelin debris accumulation and local inflammatory cascades impede endogenous white matter repair [242]. HDAC3, a histone deacetylase implicated in various neurological disorders, is selectively upregulated in microglia after ischemic stroke [243, 244]. The HDAC3/PU.1 axis drives pro-inflammatory microglia subset proliferation, exacerbating demyelination and neuronal injury [244]. Moreover, perivascular microglia respond to BBB breakdown by engulfing leaked low-density lipoprotein (LDL), but excessive lipid ingestion overwhelms their clearance capacity, impairing debris degradation and worsening white matter injury [245]. Interestingly, astrocytes also acquire phagocytic activity in chronic ischemic stages, mediated by LCN2-LRP1 signaling, but their debris uptake worsens demyelination [246]. Moreover, astrocyte-derived CXCL5 can inhibit microglial debris clearance, aggravating white matter damage and cognitive decline in carotid stenosis mouse models [247].
Extracellular vesicles (EVs), the nanoscale lipid bilayer particles containing proteins, lipids, and microRNAs, add another layer of regulation [248]. Astrocyte-derived EVs deliver pro-remyelinating factors such as FGF-2 and vascular endothelial growth factor (VEGF) to enhance OPCs migration and differentiation under ischemic conditions [249, 250]. Interestingly, a recent study reveals that macrophage-derived foam cells within atherosclerotic plaques communicate with CNS microglia via EVs [251]. These EVs deliver miR-101-3p to microglia, which suppresses Nrf2 activity, triggering microglial metabolic dysfunction and exacerbating ischemic demyelination [251]. This pathway is independent of traditional hypoperfusion-mediated white matter injury, suggesting that atherosclerotic plaques may initiate myelin damage through early microenvironmental interference before overt clinical manifestations [251].
Pathological overlap between AD and stroke further supports a microenvironmental
perspective. Approximately 80% of AD patients exhibit cerebral small vessel
disease from vascular A
The development of high-throughput screening platforms has accelerated the
discovery of compounds that promote OPCs differentiation, with several advancing
to clinical trials [208, 254, 255]. Notable candidates include muscarinic
receptor antagonist clemastine, histamine receptor antagonist GSK239512, and
RXR-
| Drug | Mechanism | Result |
| Ac-4,4-diF-GlcNAc [259] | Inhibits astrocytic CSPG synthesis and reduces monocyte and lymphocyte infiltration. | Mitigated clinical severity in EAE mice and reduced immune cell infiltration. |
| S3 [260] | A modified flavonoid. Suppresses hyaluronidase activity, reduces high-MW HA degradation products, and reverses HA-mediated inhibition of OPC maturation. | Enhanced remyelination in LPC-induced demyelination models and improved conduction velocity in lesions. |
| Opicinumab [261, 262] | A human monoclonal antibody against axonal inhibitor LINGO-1. Blocks LINGO-1-mediated OPC differentiation inhibition. | Improved optic nerve conduction in optic neuritis patients (phase 2 trials). |
| No efficacy observed in relapsing MS (phase 2 trials). | ||
| Evobrutinib [263] | A highly selective BTK inhibitor. Suppresses CNS immune infiltration, enhances phagocytosis, and promotes remyelination. | Reduced MRI-enhancing lesions in relapsing MS patients (phase 2b trial). |
| CC-292 [264] | A BTK inhibitor. Inhibits PLC |
Modulates phagocyte function in 5 |
| Ibudilast [265, 266] | A non-selective PDE inhibitor. Antagonizes TLR4, inhibits pro-inflammatory cytokines, and promotes neurotrophic factor release. | Slowed brain atrophy in MS patients (phase 2 trials). |
| Reduced AD pathology in the hippocampal dentate gyrus of 344-AD rat models and enhanced spatial learning/memory. | ||
| Masitinib [267, 268] | A selective tyrosine kinase inhibitor. Suppresses microglia, macrophages, and mast cell activity. | Slowed disability progression in MS patients (phase 3 trials). |
| Improved cognition in mild-to-moderate AD patients (phase 3 trials). | ||
| AL002 [123, 269] | A TREM2-agonistic monoclonal antibody. Enhances myelin debris clearance, OPC maturation, and modulates inflammation. | Enhanced remyelination and axonal integrity in CPZ-induced demyelination models. |
| Reduced amyloid plaque formation and behavioral deficits in 5 | ||
| Metformin [258] | Activates AMPK pathway, improves cellular senescence and mitochondrial function, and restores OPC responsiveness to differentiation signals. | Enhanced remyelination in EB-induced demyelination models. |
Note: This table summarizes the agents that have the potential to promote
remyelination through targeted modulation of the microenvironment in recent
years, as well as their mechanisms of action and results. CSPG, chondroitin
sulfate proteoglycan; EAE, experimental autoimmune encephalomyelitis; LINGO-1,
immunoglobulin domain-containing protein 1; BTK, Bruton’s tyrosine kinase;
PLC
Pathological deposition of ECM components and axonal surface molecules critically impair remyelination [25]. CSPGs restrict cell migration and repair in MS and traumatic injuries [271, 272]. Inhibiting CSPG biosynthesis with the glucosamine derivative Ac-4,4-diF-GlcNAc reduces neuroinflammation and improves clinical outcomes in EAE models [259]. Hyaluronic acid, which accumulates in MS and EAE lesions, halts OPC differentiation at premyelinating stages [273]. The flavonoid derivative S3, by blocking hyaluronidase, restores OPC maturation in lysolecithin-induced demyelinating mice [260]. In addition, neurite outgrowth inhibitor receptor co-receptor (LINGO-1) inhibits OPCs differentiation and axonal regeneration [274]. Clinical trials of the LINGO-1 antibody opicinumab showed modest benefit in optic neuritis but no efficacy in relapsing MS [261, 262].
Bruton’s tyrosine kinase (BTK), a regulator of B cell and myeloid cell
activation [275]. It is an important target for the treatment of mantle cell
lymphoma and chronic lymphocytic leukemia and has emerged as a therapeutic target
in MS [275]. In EAE, the oral BTK inhibitor evobrutinib suppresses peripheral
immune activation, shifts microglia away from a pro-inflammatory phenotype, and
enhances phagocytosis, collectively promoting remyelination [276]. In clinical
trials, evobrutinib lowered MRI-enhancing lesion counts in a dose-dependent
manner in relapsing MS patients [263]. However, comparative studies revealed that
its therapeutic efficacy did not surpass its active comparator, teriflunomide
[277]. Broader immunomodulators are also under investigation, including ibudilast
and masitinib. Ibudilast, a multi-target agent inhibiting cyclic nucleotide
phosphodiesterases (PDEs) and TLR4, showed efficacy in slowing cerebral atrophy
progression in phase 2 MS trials [265]. Masitinib, a selective tyrosine kinase
inhibitor, significantly delayed disability progression in phase 3 MS studies
[267]. Notably, these agents are also being tested in AD [267]. BTK upregulation
observed in AD patient brains and 5
Several molecular targets have been identified to enhance phagocytosis,
including TREM2, CD36, and Mer tyrosine kinase (MerTK) [120]. In CPZ-induced
models, the TREM2 agonist AL002a enhances myelin clearance, increases OPC
density, and boosts remyelination [123]. TREM2 activation also shows promise in
AD, where the agonist AL002c reduces A
Heterochronic parabiosis study shows that a youthful systemic environment can restore remyelination in aged mice [279]. Pharmacological advances have identified promising anti-aging agents, including RXR agonists, nicotinamide, and metformin [161, 258, 280]. Combining agents may be particularly effective: pairing the pro-differentiation agent clemastine with metformin, for example, can both stimulate OPCs differentiation and overcome age-related resistance [281]. In aged demyelination models, metformin reduces SASP markers, reverses regenerative deficits in OPCs, and restores OPCs responsiveness to pro-regenerative agents [258].
Mesenchymal stem cell (MSC) therapy has shown potential in treating MS through its immunomodulatory and neurorestorative properties [282]. MSCs secrete anti-inflammatory cytokines, including indoleamine 2,3-dioxygenase (IDO) and prostaglandin E2 (PGE2), which effectively suppress T-cell proliferation, rebalance T-cell subsets, and attenuate autoimmune responses [283]. They also regulate B-cell and dendritic cell activity through direct contact and paracrine signaling, lowering autoantibody production and weakening antigen presentation [284]. Beyond immunomodulation, MSCs release neurotrophic factors, including BDNF and NGF, that support OPCs survival and differentiation [285]. In EAE models, MSC administration decreases demyelinated lesions and ameliorates neurological deficits [282]. Preclinical studies and early-phase clinical trials indicate favorable safety profiles and tolerability of MSC therapy, with some trials reporting symptom improvement and reduced disease activity [286]. However, challenges, including optimal dosing, delivery routes, and treatment timing, require further investigation to advance clinical translation [287].
Another strategy aims to restore CNS homeostasis by replacing dysfunctional microglia. This can be achieved through bone marrow transplantation (BMT), hematopoietic stem cell transplantation (HSCT), or direct transplantation of microglia [288]. BMT and HSCT have been used for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), X-ALD, MLD, and lysosomal storage disorders (LSDs), where therapeutic benefits partially stem from bone marrow-derived monocyte-macrophages compensating for impaired microglial functions [289, 290, 291, 292, 293]. Strikingly, a recent study reveals that BMT effectively replaces genetically defective microglia in patients with ALSP, leading to halted disease progression [294]. Cell replacement therapy also shows promise in globoid cell leukodystrophy (GLD/Krabbe disease), a severe leukodystrophy caused by galactocerebrosidase (GALC) mutations, where macrophages transform into neurotoxic globoid cells [295]. Transplantation of GALC-expressing monocytes into GLD mouse models reduces pathological hallmarks and extends survival [295].
Despite these advances, conventional transplantation approaches have limitations. Chemotherapy and radiotherapy have the side effects of exacerbating CNS injury, and bone marrow-derived cells are unable to completely replicate microglial and BAM functions [293, 296]. To overcome these barriers, emerging strategies combine resident macrophage depletion with targeted microglial transplantation [293]. In LSD and AD mouse models, the CSF1R inhibitor PLX3397 enables efficient microglial replacement, leading to improved neurological outcomes [296]. Moreover, another study reveals that PLX3397 pretreatment enhances monocyte-derived macrophage replacement of BAMs in mice [293]. Given that PLX3397 is already FDA-approved for tenosynovial giant cell tumors and under investigation for glioblastoma, its use in transplantation strategies underscores translational potential [297]. These synergistic approaches suggest that integrating microglial depletion with cell transplantation could provide a viable therapeutic avenue for neurodegenerative and demyelinating diseases.
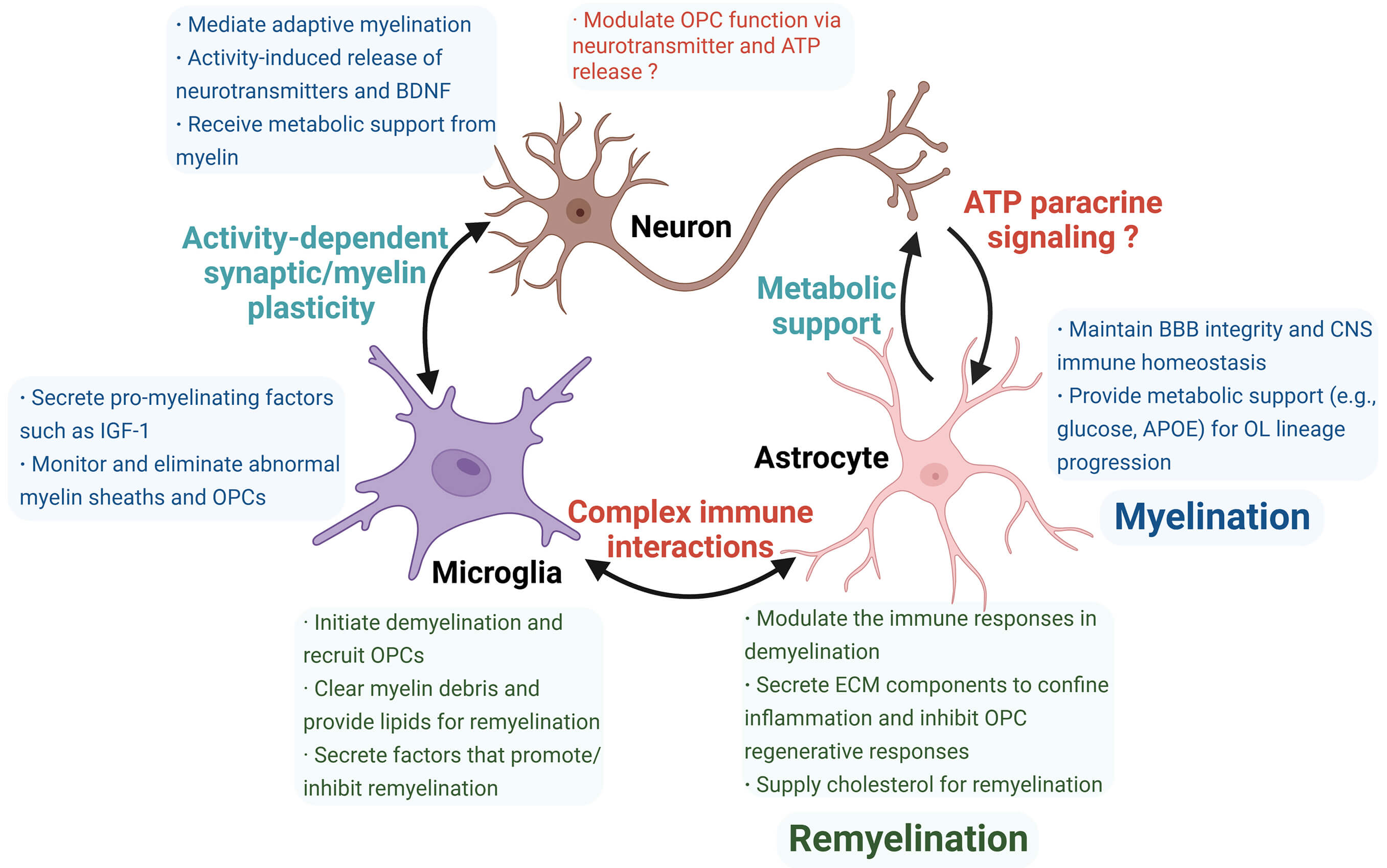
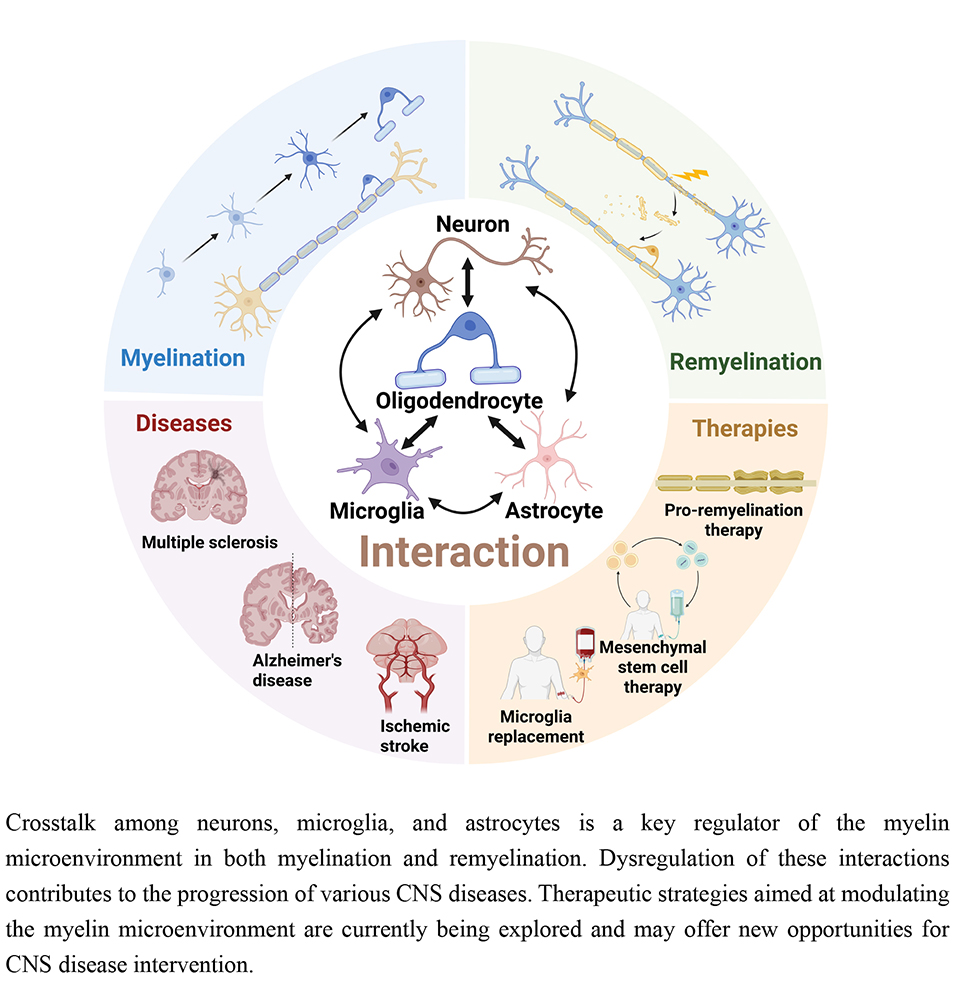
The microenvironment regulates dynamic myelin remodeling by coordinating OPCs proliferation, recruitment, differentiation, and myelin formation. Microenvironmental homeostasis is essential for protecting myelin against immunological, degenerative, and ischemic challenges, as well as for regeneration after injury. This review synthesizes current knowledge on microenvironmental regulation in demyelination and remyelination, especially the roles and interactions between neurons, microglia, and astrocytes (Fig. 3). These interactions form a highly complex network, and disentangling the dominant regulatory roles remains a central research challenge. Future progress will depend on the development of biomarkers and analytical platforms capable of resolving microenvironmental composition with spatiotemporal precision. Such tools would allow the identification of critical cellular and molecular determinants of myelin injury and repair. Recent breakthroughs, such as the discovery of injury-initiating aSERPINE1+ astrocyte subsets through integrated neuroimaging-transcriptomic approaches, exemplify this paradigm [190]. Microenvironmental targeting medicines and cell transplantation strategies hold significant promise for developing interventions tailored to lesion-specific pathological states (Fig. 4). Successful clinical translation of remyelination strategies could transform the treatment of diverse CNS disorders and bring the field closer to personalized medicine.
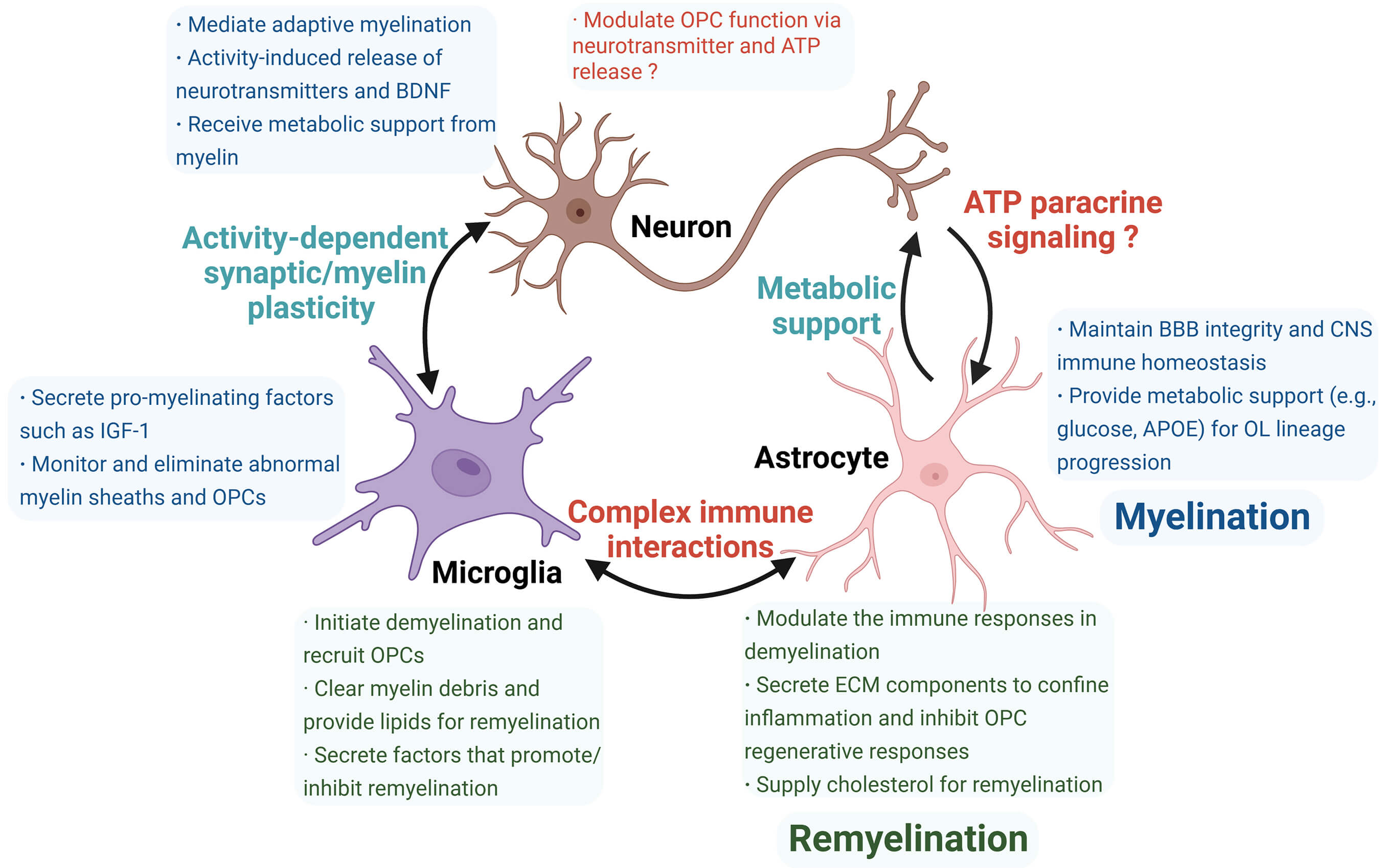 Fig. 3.
Fig. 3.
Summary of roles and interactions of neurons, microglia, and astrocytes in myelination (blue box) and remyelination (green box). Neurons, microglia, and astrocytes perform distinct functions during myelination and remyelination, contributing to microenvironmental differences between these two processes. Cellular crosstalk represents an emerging research area with unresolved questions (red highlights), including the incompletely understood role of neurons in remyelination and their interactions with astrocytes, which require further validation. Additionally, complex immune communication between microglia and astrocytes remains unclear. ATP, adenosine triphosphate; ECM, extracellular matrix. The figure was drawn using Biorender (https://www.biorender.com).
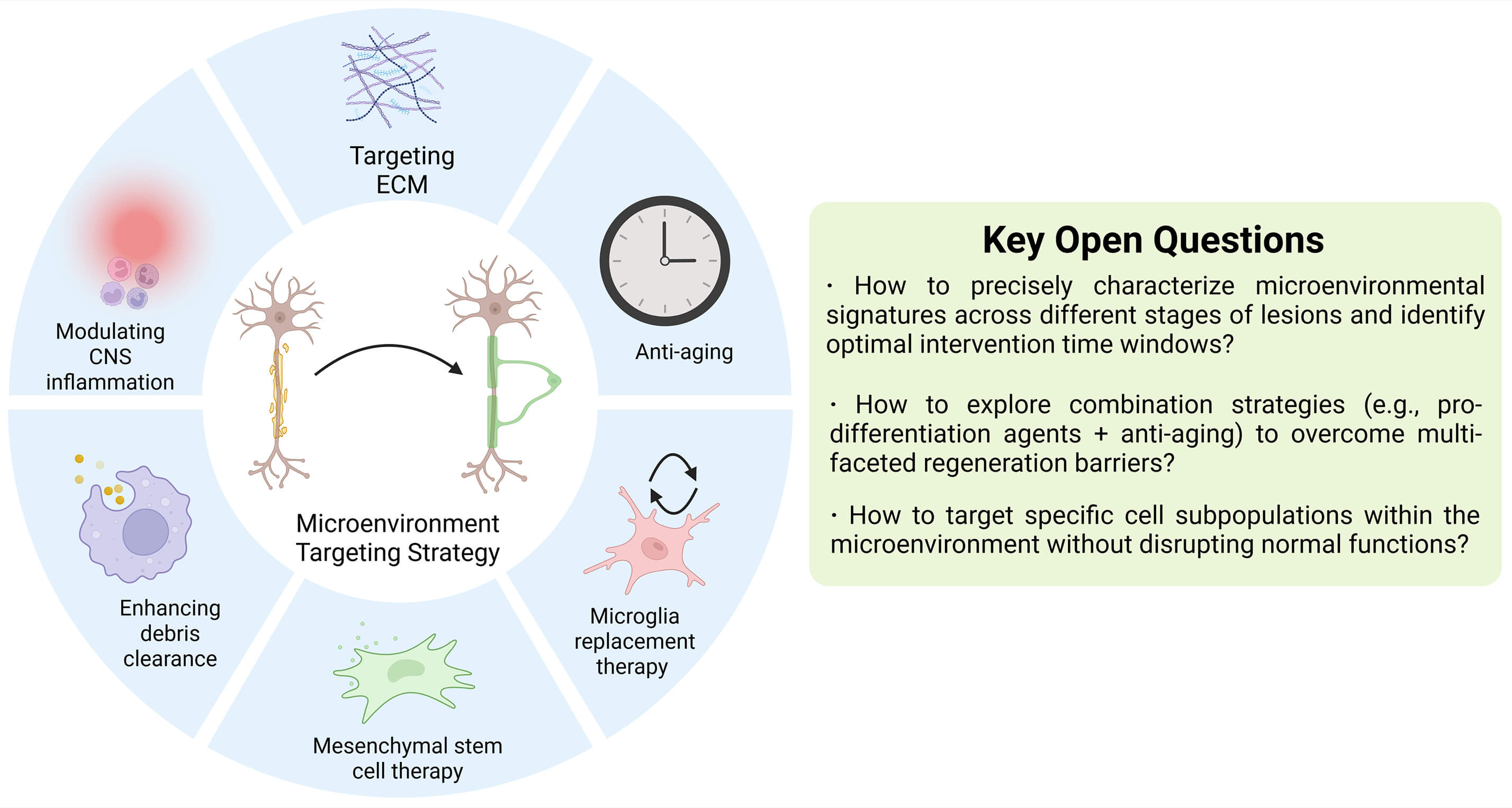 Fig. 4.
Fig. 4.
Summary and outlook for microenvironment-targeting therapeutic strategies. To date, most strategies remain at the preclinical stage. A notable exception is microglia replacement therapy, which has achieved clinical success in ALSP patients by halting disease progression. However, the complexity and high heterogeneity of microenvironmental alterations in autoimmune demyelinating diseases and neurodegenerative disorders pose significant challenges for therapeutic development. Future research should prioritize addressing these core issues to facilitate translation. ALSP, adult-onset leukoencephalopathy with axonal spheroids and pigmented glia; CNS, central nervous system. The figure was drawn using Biorender (https://www.biorender.com).
YW, LT, LL, XH, SZ, HS contributed to the study conception and design. YW, LL, and LT collected and sorted the references and designed and prepared the figures and tables. The first draft of the manuscript was written by YW, LL and LT. The content editing was performed by SZ, HS and YW. The format editing was in the charge of XH. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to express gratitude to Shilan Chen and Tianying Hu for the methodological guidance in writing and language polishing. In addition, we would like to thank BioRender for assistance with figures preparation.
This work is supported by the Guangdong Basic and Applied Basic Research Foundation, No. [2023A1515030045; 2025A1515010528] (to H.S.); Guangdong Provincial Clinical Research Center for Laboratory Medicine No. [2023B110008] (to H.S.); Presidential Foundation of Zhujiang Hospital of Southern Medical University, No. [yzjj2022ms4] (to H.S.); Special Funds for the Cultivation of Guangdong College Students’ Scientific and Technological Innovation No. [pdjh2024a088] (to H.S.); Guangdong Students’ Platform for Innovation and Entrepreneurship Training Program No. [202512121005] (to H.S.); National Natural Science Foundation of China, No. [82201497] (to S.Z.); Guangdong Basic Applied Basic Research Foundation, No. [2025A1515010744] (to S.Z.) and the Pearl River Talent Program, No. [2023QN10Y775] (to S.Z.).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.