










1 Department of Critical Care Medicine, Renmin Hospital of Wuhan University, 430060 Wuhan, Hubei, China
2 Central Laboratory, Renmin Hospital of Wuhan University, 430060 Wuhan, Hubei, China
Abstract
Cardiac arrest (CA) is a widespread public health problem with high mortality, severe neurological sequelae, and limited pharmacological therapies. We investigated the neuroprotective effect of a novel drug, FPS-ZM1 (FPS), on CA and explored its potential mechanism.
A potassium chloride–induced CA was induced for 9.5 min in mice, with i.p. injections of FPS or vehicle administered 24 and 1 h before induction. Postoperative assessments included survival rate, body weight change, neurological scores, and neuronal pathological damage. The expression levels of the high mobility group box 1 (HMGB1)/receptor for advanced glycation end products (RAGE) axis, pyroptosis-related molecules, oxidative stress markers, and the nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) axis were evaluated.
Post-CA brain injury (PCABI) activated the HMGB1/RAGE axis, triggering intensified oxidative stress and aggravated pyroptosis. In contrast, pretreatment with FPS attenuated CA-induced injuries. FPS pretreatment was found to suppress the activation of the HMGB1/RAGE axis, alleviate pyroptosis and the release of associated inflammatory mediators, and enhance the Nrf2/HO-1 antioxidant axis after PCABI.
FPS pretreatment mitigated PCABI by concurrently modulating the HMGB1/RAGE inflammatory axis and the Nrf2/HO-1 antioxidant pathway, suggesting that RAGE antagonism represents a promising therapeutic strategy for PCABI.
Graphical Abstract

Keywords
- cardiac arrest
- FPS-ZM1
- HMGB1 protein
- oxidative stress
- pyroptosis
- receptor for advanced glycation end products
Cardiac arrest (CA) is characterized by high morbidity, low survival rates
(typically
High mobility group box 1 (HMGB1) is an evolutionarily conserved protein found in multiple species, including both humans and rodents. It is widely expressed throughout the body, with significant expression in brain tissue [10]. Under normal physiological conditions, HMGB1 is essential for the formation of neuronal synapses, damage repair, and cellular homeostasis [10, 11]. In response to cerebral injuries, such as hypoxia and ischemia, HMGB1 is released into the extracellular space, where it acts as a damage-associated molecular pattern (DAMP). Once released into the extracellular space, HMGB1 binds to pattern-recognition receptors, including the receptor for advanced glycation end products (RAGE) and toll-like receptor 4, potentially triggering sterile inflammation [12, 13]. RAGE, the first identified high-affinity receptor for HMGB1 [14], exhibits increased expression that is primarily associated with exacerbated neuroinflammation after brain injury [15]. Research involving both human clinical trials and animal models has demonstrated increased concentrations of HMGB1 in the blood and cerebrospinal fluid of individuals undergoing CA. These elevated levels correlated significantly with adverse neurological outcomes, suggesting that HMGB1 acts as a potential prognostic biomarker [16, 17, 18]. Furthermore, increased HMGB1 expression in serum and brain tissue in post-CA rats modulates the inflammatory response [18, 19]. Given that RAGE is persistently activated after hypoxic-ischemic brain injury, thereby inducing neuroinflammation [20, 21], inhibiting the HMGB1/RAGE axis theoretically represents a potentially effective therapeutic strategy for PCABI.
Pyroptosis is an inflammation-driven subtype of programmed cell death that
occurs through activation of the inflammasome complex. The assembly of
inflammasome complexes requires several key components, including
apoptosis-associated speck-like protein containing a caspase recruitment domain
(ASC) and pro-caspase 1. Upon activation, gasdermin D (GSDMD) is cleaved,
resulting in the generation of transmembrane pores that disrupt cellular
integrity. Consequently, inflammatory cytokines like interleukin-1 beta
(IL-1
Nuclear factor erythroid 2-related factor 2 (Nrf2) plays a pivotal role in mediating the cellular antioxidant stress response and can activate the expression of multiple downstream antioxidant enzymes [29, 30]. Heme oxygenase-1 (HO-1) is a classic downstream effector directly regulated by Nrf2; its expression is upregulated under various stress conditions and exerts pronounced antioxidant and anti-inflammatory effects [31]. Stimulation of the Nrf2/HO-1 signaling pathway has been shown to suppress brain IRI-associated oxidative stress and inflammatory responses, thereby exerting neuroprotection [32, 33]. FPS-ZM1 (FPS), a non-toxic, high-affinity RAGE inhibitor, prevents RAGE from interacting with its ligands, including HMGB1 [34]. Shen et al. [35] showed that FPS enhanced Nrf2 and HO-1 expression, reduced oxidative stress in microglia, and thereby offered protection [36]. However, the effects of FPS administration on PCABI have not yet been fully elucidated.
We designed a series of experiments to evaluate the effects of FPS pretreatment on neurological outcomes in a cardiac arrest/cardiopulmonary resuscitation (CA/CPR) mouse model. Furthermore, we investigated the regulatory effects of FPS pretreatment on pyroptosis and oxidative stress, as well as the underlying mechanisms in PCABI.
Adult male C57BL/6 mice (aged 8–10 weeks, weighing 22–25 g) were sourced from Hunan SJA Laboratory Animal Co., Ltd. (Changsha, Hunan, China). Before the experiment, all mice were given at least one week to habituate to the colony room. During the study, the mice were kept under standardized conditions, with constant temperature and humidity control, a 12–12 h alternating light–dark cycle, and free access to standard rodent chow and water.
Mice were randomly assigned to four experimental groups (n = 13/group) using a computer-generated randomization sequence to minimize selection bias: (1) Sham + vehicle; (2) Sham + FPS; (3) CA/CPR + vehicle; (4) CA/CPR + FPS. Randomization was performed by an investigator (QL) who was not involved in subsequent data analysis.
One designated investigator (QL) was responsible for drug preparation and administration. All other investigators involved in surgical procedures, neurological function assessments, data collection, and statistical analysis remained blind to the group allocation throughout the entire experiment to minimize potential bias.
The CA model was generated in accordance with the protocol described previously
[33, 37]. In brief (Fig. 1), mice were weighed to confirm adherence to standard
weight parameters. Anesthesia was induced using 5% isoflurane (v/v; vol%;
R510-22-10, RWD Life Science Co., Ltd., Shenzhen, Guangdong, China), followed by
tracheal intubation. Anesthesia was maintained by mechanical ventilation
delivering 1.0%–1.5% isoflurane (v/v; vol%); a gas evacuation apparatus
(V101, RWD Life Science Co., Ltd.) was used to remove anesthetic exhaust. Mice
were ventilated through the tracheal tube at a tidal volume ranging from 0.3 to
0.5 mL. Electrocardiogram (ECG) was continuously monitored using
subcutaneously placed needle electrodes and a PowerLab 26T system (PL26T04;
ADInstruments Pty Ltd., Bella Vista, New South Wales, Australia). Body
temperature was kept at 37.0
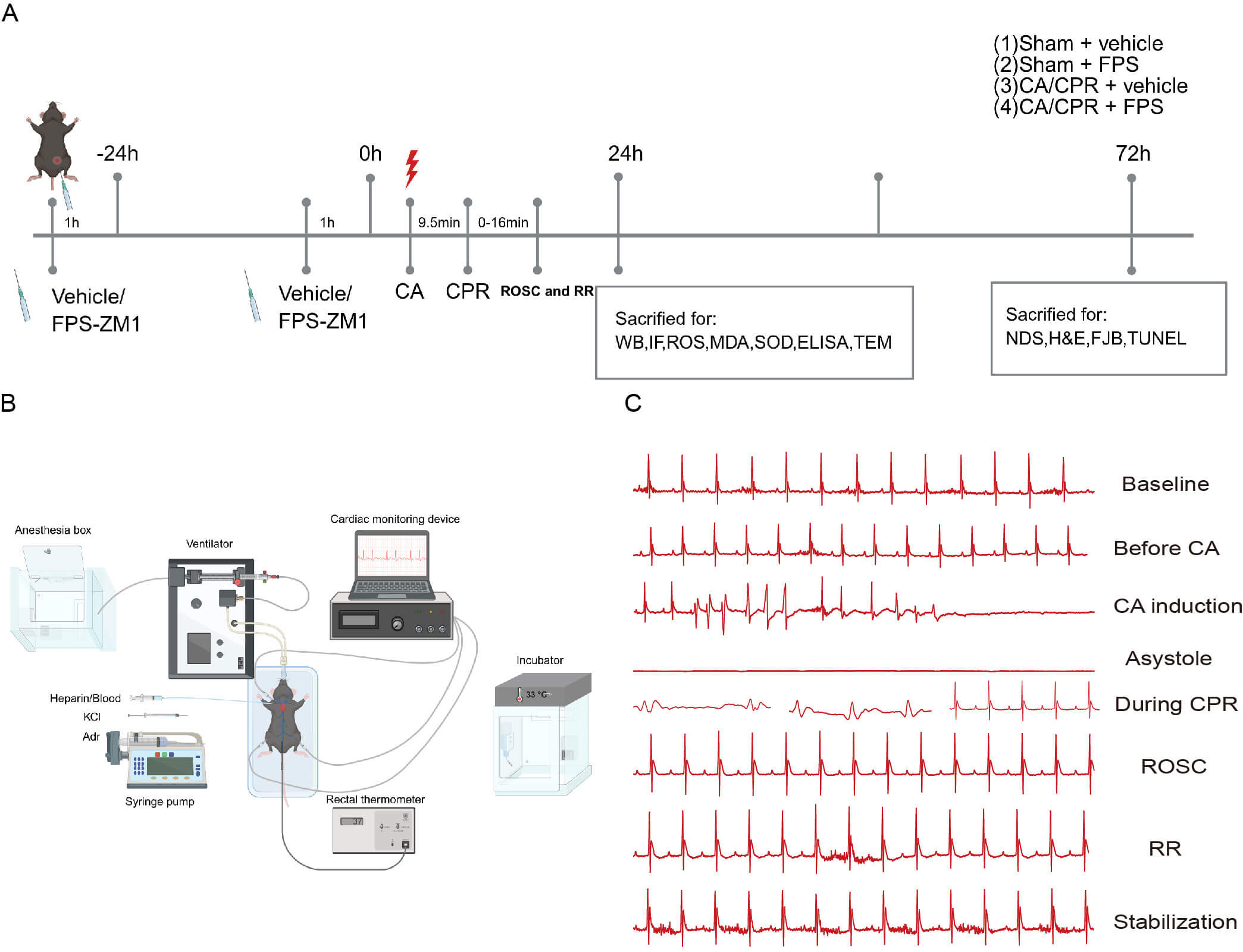 Fig. 1.
Fig. 1.
Mouse experimental procedure. (A) Mice received CPR after 9.5 min of CA. FPS (RAGE inhibitor, 10 mg/kg) or an equivalent dose of vehicle (saline) was injected intraperitoneally on the day before surgery, and another dose again one h before CA. Impact of FPS on HMGB1/RAGE pathway modulation, as well as oxidative stress and pyroptosis-related signaling pathways induced by CA/CPR, was assessed 1 day after CA/CPR. The neuroprotective impact of FPS-ZM1 (FPS) was assessed 3 days after CA/CPR. Groups: (1) Sham + vehicle; (2) Sham + FPS; (3) CA/CPR + vehicle; (4) CA/CPR + FPS (n = 13/group). (B) The process of establishing a KCl-induced CA model in mice included the following steps: anesthesia; tracheal intubation; jugular vein catheterization; temperature and electrocardiogram monitoring; drug infusion; and postoperative targeted temperature management (TTM). (C) ECG monitoring was performed throughout the procedure, capturing representative waveform patterns. CA, cardiac arrest; CPR, cardiopulmonary resuscitation; ROSC, return of spontaneous circulation; RR, respiratory recovery; WB, Western Blot; IF, immunofluorescence; ROS, reactive oxygen species; MDA, malondialdehyde; SOD, superoxide dismutase; ELISA, enzyme-linked immunosorbent assay; NDS, neurological deficit score; H&E, Hematoxylin-eosin; FJB, Fluoro-Jade B; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; KCl, potassium chloride; Adr, adrenaline; ECG, electrocardiogram; HMGB1, high mobility group box 1; RAGE, receptor for advanced glycation end products; TEM, transmission electron microscopy.
Compound FPS (RAGE inhibitor, 10 mg/kg, HY-19370, MedChemExpress LLC, Monmouth Junction, NJ, USA) was administered via i.p. injection 24 h pre-surgery, and again 1 h before the surgical procedure. The dosing protocol for the administration was based on previous research findings [38]. Fig. 1A displays the experimental protocol.
Neurological function was comprehensively assessed using two distinct scoring
systems (Supplementary Tables 1,2). First, a 12-point scale
[39] was used to assess the mice across six critical domains: responsiveness to
stimuli; corneal reflex; respiration; righting reflex; coordination; and
movement. Each domain was rated using a scale from 0 to 2, with 0 signifying
severe impairment and 2 signifying normal function. Second, a 9-point grading
scale [40] was used to evaluate neurological deficits based on motor performance.
Mice were tested for their ability to climb a vertical grid, balance on a
horizontal bar, and hang from a rope. Each task was scored from 0 to 3, where 0
represented severe disability and 3 represented normal function. Assessments were
performed under standardized conditions (quiet room, consistent illumination, and
minimal handling). Scoring was conducted independently by two investigators who
were blind to group allocation and study hypotheses. When the discrepancy was
Abnormal neuronal morphology was evaluated through H&E staining. Mice were anesthetized by induction in an anesthesia box with 5% isoflurane with the inspired gas mixture at a 7:3 oxygen-nitrogen ratio. After the mice were anesthetized, intracardiac perfusion with 0.9% NaCl was performed, followed by fixation in 4% paraformaldehyde (PFA, G1101, Wuhan Servicebio Technology Co., Ltd., Wuhan, Hubei, China). The brains were fixed overnight at 4 °C in 4% PFA, then dehydrated and embedded in paraffin (Wuhan Servicebio Technology Co., Ltd.). Serial coronal sections (4 µm) were sliced, deparaffinized, stained using H&E, and examined under an Olympus microscope (Olympus Corporation, Hachioji-shi, Tokyo, Japan). Image analysis was performed using ImageJ2 (version 2.16.0, Laboratory for Optical and Computational Instrumentation, University of Wisconsin–Madison, Madison, WI, USA).
Nissl staining was used in order to quantify the density of cortical neurons. Brain tissue was extracted from anesthetized mice after decapitation and fixed in 4% PFA. After fixation, the specimens were embedded in paraffin and sectioned coronally at a thickness of 4 µm. The slices were then dewaxed, rehydrated, and stained by immersing them in a 0.2% cresyl violet solution (G1086, Wuhan Servicebio Technology Co., Ltd.) for 10 min. Images of the brain sections were obtained with an Olympus microscope. Nissl body counting was performed using ImageJ software.
The TUNEL assay was used to identify apoptotic neurons, according to the protocol included with the In Situ Cell-Death Detection Kit (PN0017, Wuhan Pinuofei Biotechnology Co., Ltd., Wuhan, Hubei, China). Paraffin-embedded brain-tissue sections were deparaffinized and subsequently exposed to proteinase K (Wuhan Pinuofei Biotechnology Co., Ltd.) to increase cellular membrane permeability. Thereafter, the sections were incubated with the TUNEL reaction solution at room temperature for 1 h to facilitate the detection process. After DAPI (Wuhan Pinuofei Biotechnology Co., Ltd.) staining to label the nuclei, the slides were then coverslipped using an anti-fade mounting medium (Wuhan Pinuofei Biotechnology Co., Ltd.). The tissue sections were examined under an Olympus microscope, and images were captured for quantification of cortical apoptotic cells per slide.
FJB staining was used to evaluate neuronal degeneration and apoptosis. Brain sections were initially immersed in 0.06% potassium permanganate for 10 min, followed by incubation at room temperature for 20 min in 0.004% FJB solution (Merck Millipore, AG310, Merck Millipore, Darmstadt, Germany). After thorough rinsing and drying, the samples were coverslipped and analyzed using an Olympus microscope. The FJB-positive cell count within the cortical region was manually quantified using ImageJ software.
After deparaffinization, rehydration, and antigen retrieval, tissue sections were delineated and then treated with 3% bovine serum albumin (GC305010, Wuhan Servicebio Technology Co., Ltd.). Carefully prepared primary antibodies, sourced from the following suppliers, were then applied to the samples: HMGB1 (GB11103, Wuhan Servicebio Technology Co., Ltd.,1:500); RAGE (TC52809S, Abmart, Shanghai, China, 1:500); Nrf2 (#12721, Cell Signaling Technology, Danvers, MA, USA, 1:500); and caspase-1 (341030, Chengdu Zen Bioscience Co., Ltd., Chengdu, Sichuan, China, 1:500). The tissue slices were kept in a humidified chamber and incubated at 4 °C overnight to ensure adequate antibody binding. Next, the samples were exposed to the corresponding secondary antibody (#4412 and #4408, Cell Signaling Technology, 1:1000) at room temperature for 1 h. Fluorescence intensity was analyzed with ImageJ software. HMGB1/RAGE colocalization analysis was as follows. Briefly, double immunofluorescence images were analyzed using ImageJ. For each group, three mice were included; for each mouse, three sections were analyzed, and one scanning field covering the cortical region per section was quantified. Within predefined cortical regions of interest (ROIs), HMGB1 and RAGE-positive cells were identified using a consistent thresholding strategy, and HMGB1+RAGE+ double-positive cells were confirmed on individual channels and merged images. Colocalization was quantified as the percentage of HMGB1+RAGE+double-positive cells among total cells, with total cell number defined as the number of DAPI-positive nuclei within the ROI.
After cardiac perfusion with physiological saline, we immediately performed perfusion fixation using pre-chilled 2.5% glutaraldehyde fixative (G1102, Wuhan Servicebio Technology Co., Ltd.). The target cortical region was rapidly excised and sectioned into approximately 1 mm3 cubes using a sharp blade. These were transferred into EP tubes containing the aforementioned fixative solution for post-fixation at 4 °C for 4–6 h. Ultrastructural alterations in mouse cortical neurons were imaged using a TEM (HT7700, Hitachi High-Tech Corporation, Tokyo, Japan). Neurons were identified by systematic scanning from low to high magnification based on ultrastructural characteristics.
After rapid cryopreservation in liquid nitrogen, brain tissue was embedded in OCT (G6059, Wuhan Servicebio Technology Co., Ltd.) and sliced coronally. To visualize ROS, sections were treated with dihydroethidium stain (D7008, Sigma-Aldrich, St. Louis, MO, USA) for 30 min at room temperature in darkness. Images were acquired with an Olympus microscope, and the fluorescence signals were analyzed and quantified using ImageJ software.
The level of lipid peroxidation was quantified using the MDA assay, and SOD activity was measured as an indicator of cellular capacity to neutralize ROS. Brain cortex samples were carefully dissected, homogenized in saline, and then subjected to centrifugation to obtain the supernatant. The activities of SOD and MDA were then assessed using commercial assay kits (A001-3-2 and A003-1-2, Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China) following the guidelines provided by the manufacturer.
To extract proteins, mouse brain cortex samples were extensively homogenized in
a lysis buffer (G2002, Wuhan Servicebio Technology Co., Ltd.) using an ultrasonic
cell processor (Ningbo Scientz Biotechnology Co., Ltd., Ningbo, Zhejiang, China).
The extracted protein samples were combined with 5
Brain samples and peripheral blood were harvested in sterile tubes and stored at
–80 °C. Levels of cytokine (IL-1
All collected experimental results were subjected to statistical analysis using
GraphPad Prism (version 9.0.0.121, GraphPad Software, LLC, Boston, MA, USA), and
the findings are presented as mean
To investigate alterations in RAGE and HMGB1 expression in mice after CA/CPR, cortical brain tissue and serum samples were analyzed using immunofluorescence and ELISA 24 h post-CA. Immunofluorescence results revealed a marked increase in RAGE and HMGB1 expression within the cerebral cortex of CA/CPR mice (Fig. 2A–D). Subsequent ELISA assays further confirmed that serum HMGB1 levels were significantly higher in the CA/CPR group than in the sham group (Fig. 2E,F). Similarly, serum RAGE levels were markedly higher in CA mice than in the sham group (Fig. 2G,H). These results indicated that both RAGE and HMGB1 were substantially upregulated during PCABI, suggesting activation of the HMGB1/RAGE axis and its potential involvement in the pathogenesis of PCABI.
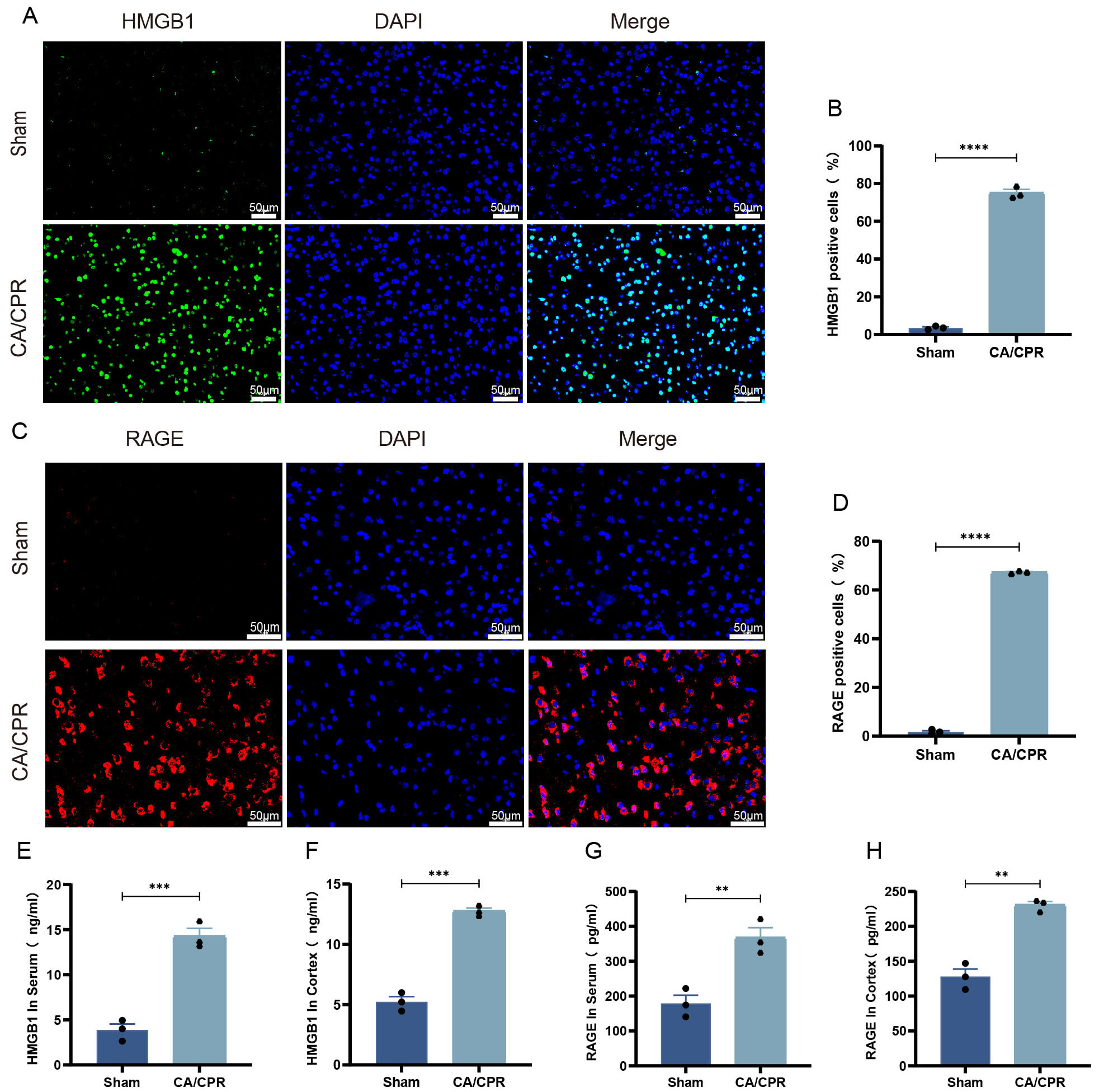 Fig. 2.
Fig. 2.
The HMGB1/RAGE axis is activated by CA/CPR. (A) Typical
immunofluorescence pictures of HMGB1. (B) Statistics of the proportion of
HMGB1-positive cells across different groups. (C) Typical immunofluorescence
pictures of RAGE. (D) Statistics of the proportion of RAGE-positive cells across
different groups. (E,F) ELISA quantification of HMGB1 levels in serum and cortex
in different groups. (G,H) ELISA quantification of RAGE levels in serum and
cortex in different groups. Data are depicted as mean
Although FPS is considered a specific RAGE inhibitor, the mechanistic basis for its suppression of RAGE activity in mouse brain tissue remains unclear. In the present study, immunofluorescence analysis revealed that, under physiological conditions, HMGB1 and RAGE exhibited only partial overlap in the mouse cerebral cortex, indicating a relatively low degree of colocalization (Fig. 3A). The CA/CPR + FPS group showed markedly less HMGB1/RAGE colocalization than did the CA/CPR + vehicle group (p = 0.0009, Fig. 3B). Consistent with the above observations, Western Blot analysis (Fig. 3C, the original Western blot image can be seen in the Supplementary Material-Western Blot) demonstrated significantly lower HMGB1 protein levels in cortex of the CA/CPR + FPS-pretreated group than in the CA/CPR + vehicle group (p = 0.0299, Fig. 3D), accompanied by a substantial decrease in RAGE protein levels (Fig. 3E). Furthermore, ELISA analysis revealed that HMGB1 and RAGE levels in both serum and cortex were significantly lower in the FPS–pretreated group than in the CA/CPR + vehicle group (Fig. 3F–I). Taken together, these findings suggested that FPS pretreatment exerted its effects, at least in part, by inhibiting CA/CPR-induced activation of the HMGB1/RAGE signaling pathway.
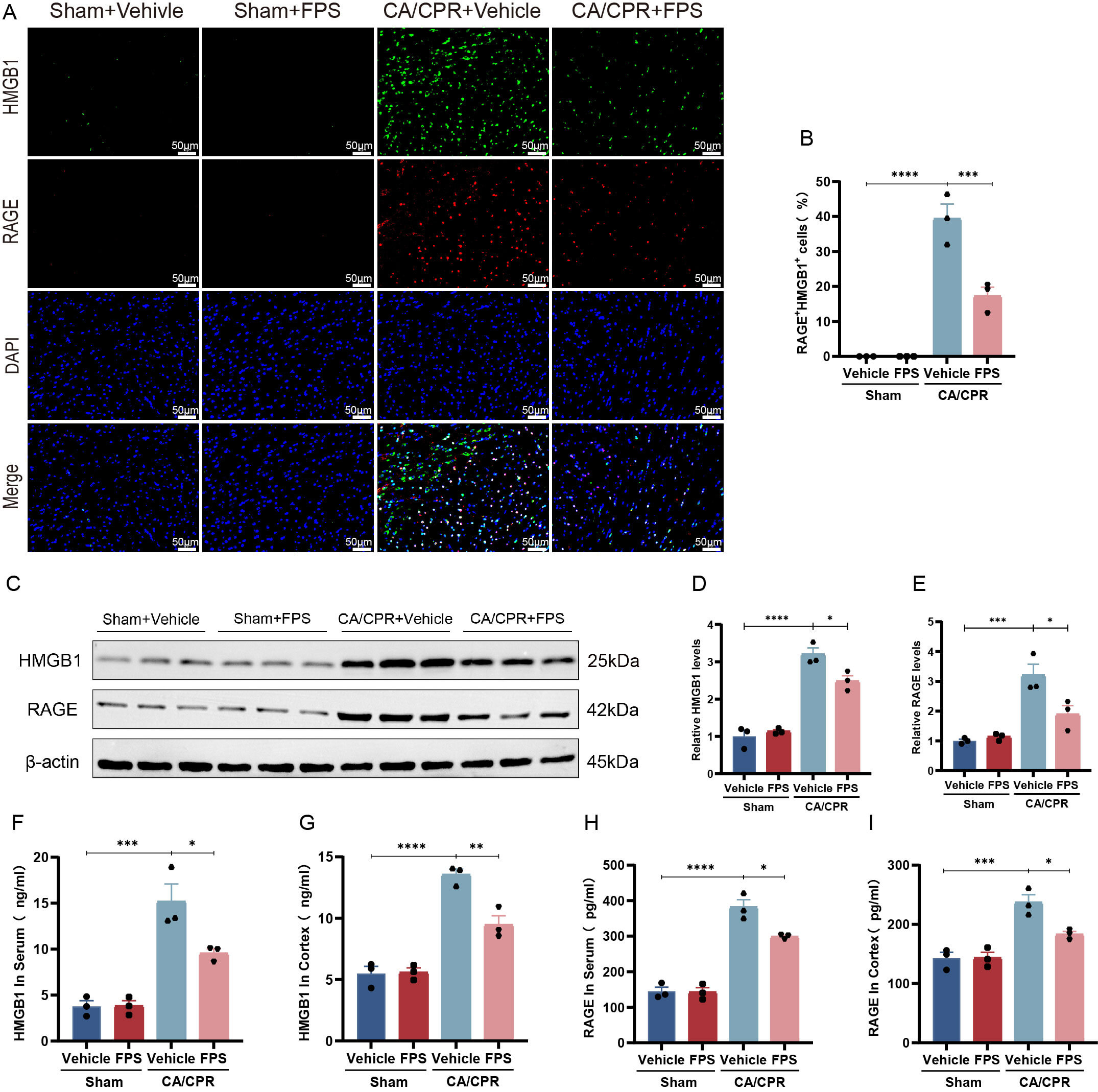 Fig. 3.
Fig. 3.
FPS pretreatment prevented activation of the HMGB1/RAGE axis
induced by CA/CPR. (A) Typical pictures of HMGB1 and RAGE double
immunofluorescence staining. (B) Statistics of the proportion of HMGB1 and RAGE
double-positive cells across different groups. (C) Typical protein immunoblotting
bands of HMGB1 and RAGE. (D,E) Relative protein expression semiquantitative
analysis of HMGB1 and RAGE across different groups. (F,G) ELISA quantification of
HMGB1 levels in serum and cortex in different groups. (H,I) ELISA quantification
of RAGE levels in serum and cortex in different groups. Data are depicted as mean
Recent research has demonstrated that the HMGB1/RAGE axis is critically involved
in mediating pyroptosis in neonatal hypoxic-ischemic brain injury and Kawasaki
disease. Pretreatment with FPS has been shown to inhibit pyroptosis under these
pathological conditions [27, 28]. Building on these findings, the present study
investigated whether FPS pretreatment could similarly attenuate CA/CPR-induced
pyroptosis. Immunofluorescence (Fig. 4A–D) analysis revealed that, after CA/CPR,
levels of activated caspase-1 and the proportion of TUNEL-positive cells in the
mouse cerebral cortex were significantly increased. By contrast, FPS pretreatment
markedly reduced activated caspase-1 levels and the number of TUNEL-positive
cells. Notably, we used TUNEL as an indicator of DNA fragmentation and overall
cell-death burden. To further explore pyroptosis, TEM revealed prominent
ultrastructural cortical neuronal damage after CA/CPR, including membrane
disruption, nuclear condensation, and severe organelle injury, which were
alleviated by FPS pretreatment (Fig. 4E). As expected, ELISA (Fig. 4F,G) assays
demonstrated that the concentrations of the cytokines IL-1
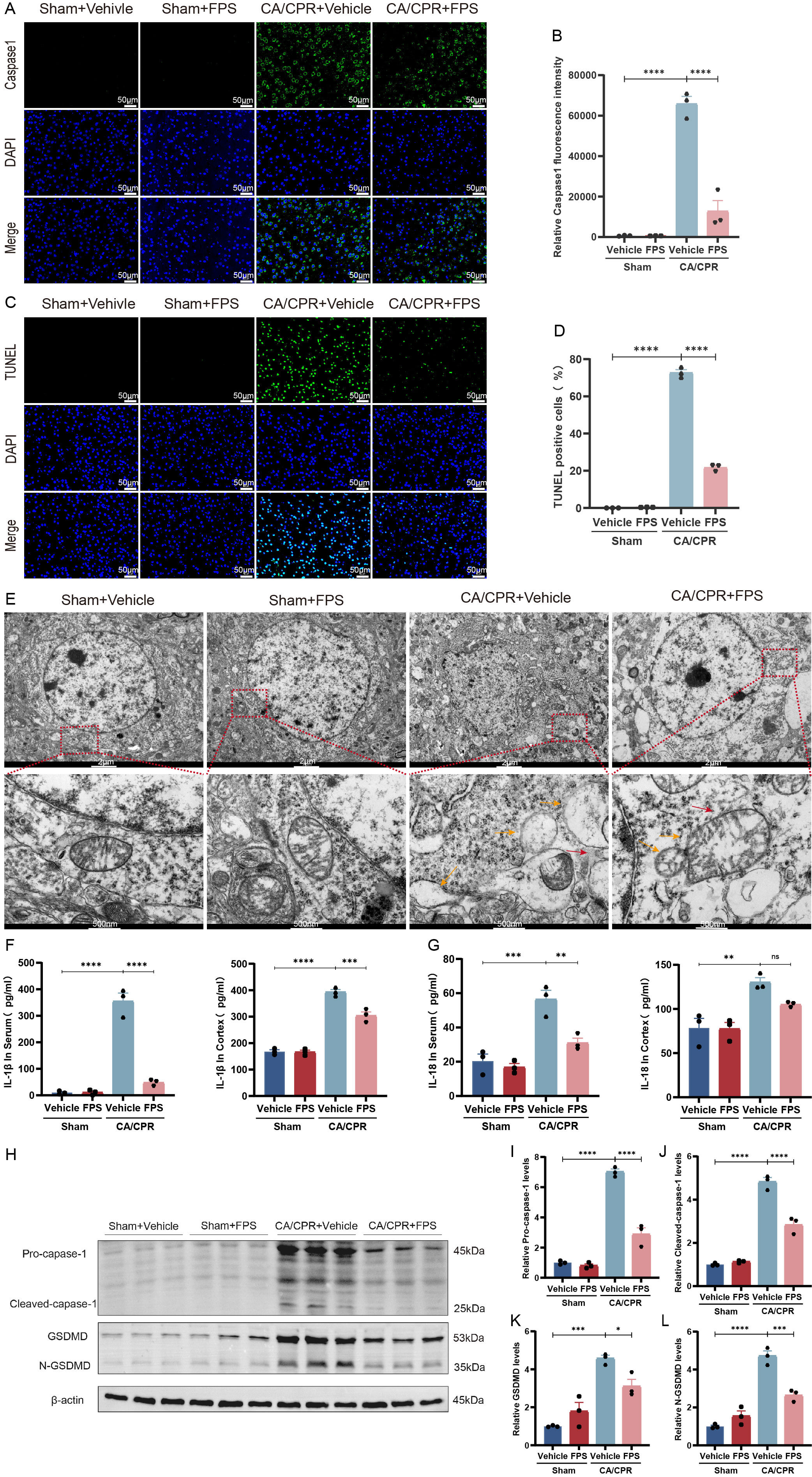 Fig. 4.
Fig. 4.
FPS pretreatment attenuated the level of pyroptosis after
CA/CPR. (A) Typical pictures of caspase-1 immunofluorescence staining. (B)
Quantitative assessment of caspase-1 fluorescence intensity in different groups.
(C) Typical pictures of TUNEL immunofluorescence staining. (D) Statistics of the
proportion of TUNEL-positive cells in different groups. (E) Typical TEM pictures
of cortical tissue (n = 3/group). Scale bar = 2 µm (low
magnification) and 500 nm (high magnification). Arrows indicate pyroptotic
neurons exhibiting cell membrane rupture (red) or organelle damage (orange).
(F,G) ELISA quantification of the inflammatory cytokine IL-1
To further elucidate the molecular mechanisms through which FPS mitigates PCABI, we measured oxidative stress levels induced by CA/CPR. As illustrated in Fig. 5A,B, immunofluorescence analysis demonstrated that CA/CPR significantly elevated ROS production in the cerebral cortex; whereas this effect was diminished by pretreatment with FPS. In addition, cortical SOD activity was markedly higher in the FPS-pretreated group than the CA/CPR + vehicle group (p = 0.0495, Fig. 5C), and accompanied by a substantial reduction in MDA levels (p = 0.0001, Fig. 5D). Collectively, these findings indicated that FPS pretreatment significantly alleviated CA/CPR-induced oxidative stress.
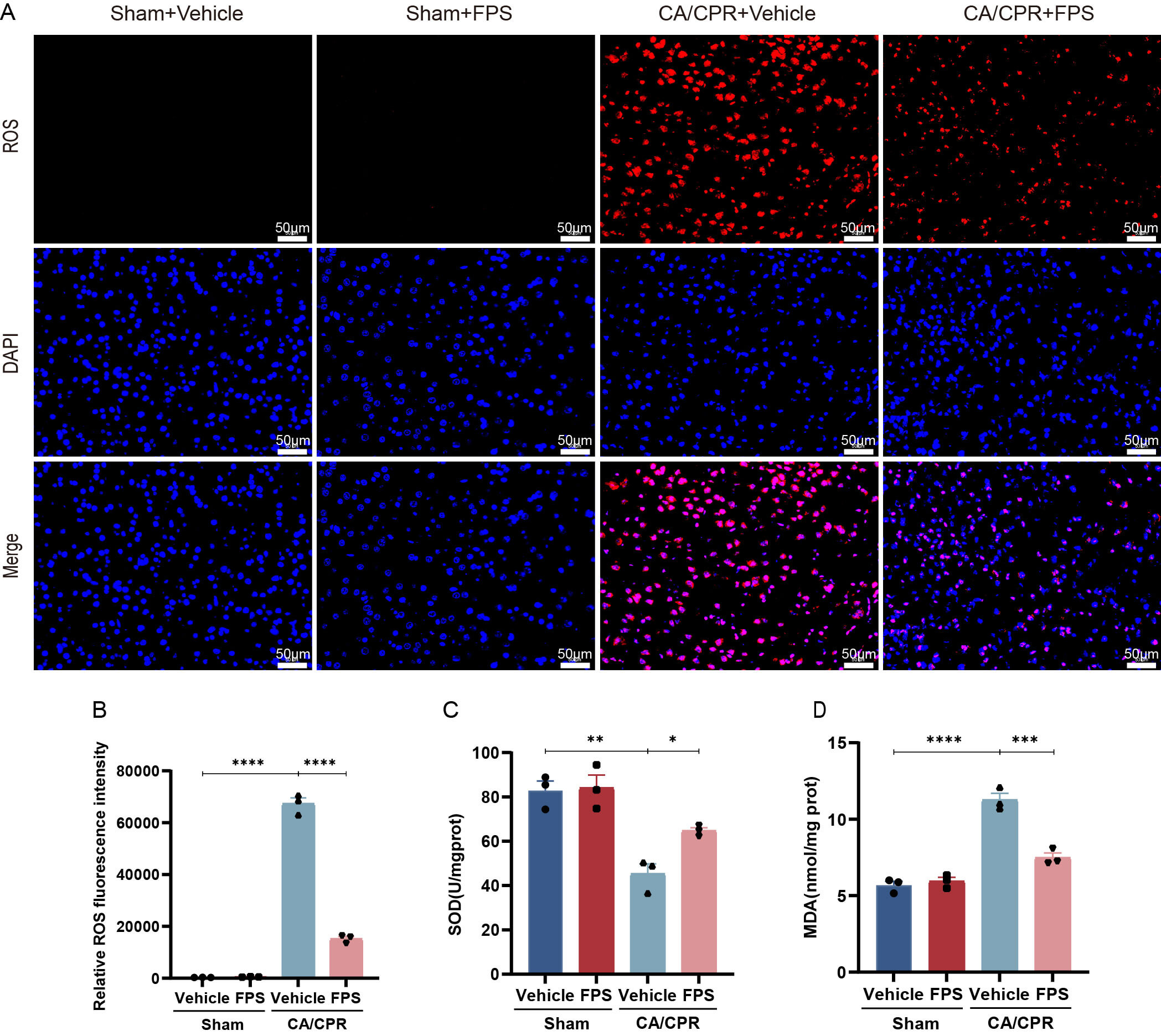 Fig. 5.
Fig. 5.
FPS pretreatment reduced oxidative stress after CA/CPR. (A)
Typical pictures of ROS immunofluorescence staining. (B) Quantitative assessment
of ROS fluorescence intensity in different groups. (C,D) Evaluation of SOD
activity and MDA levels. Data are depicted as mean
Previous research demonstrated that FPS possesses antioxidant properties and can upregulate Nrf2 and HO-1 levels in a dose-dependent fashion, thereby providing protective effects [35]. Based on those findings, we hypothesized that FPS pretreatment attenuates CA/CPR-induced oxidative injury by modulating the Nrf2-HO1 axis. To examine changes in Nrf2 and its downstream antioxidant signaling, we performed immunofluorescence staining (Fig. 6A,B) and Western Blot analysis (Fig. 6C–E, the original Western Blot image can be seen in the Supplementary Material-Western Blot). Immunofluorescence examination revealed that Nrf2 expression exhibited a slight elevation after CA/CPR. This suggested that CA/CPR manipulation stimulated Nrf2 expression to a modest increase, although it did not produce a sufficient protective effect. Notably, FPS pretreatment markedly increased the overall Nrf2 protein signal after CA/CPR. Consistent with these findings, Western Blot analysis further confirmed that FPS pretreatment significantly upregulated total Nrf2 protein levels as well as the key downstream effector HO-1 after CA/CPR. Collectively, these results indicated that FPS pretreatment strongly enhanced Nrf2 protein expression after CA/CPR, accompanied by upregulation of the antioxidant protein HO-1. These results are consistent with the effect of FPS pretreatment in reducing oxidative stress, suggesting that the Nrf2/HO-1 signaling axis may be involved in its protective effect against oxidative stress after CA/CPR.
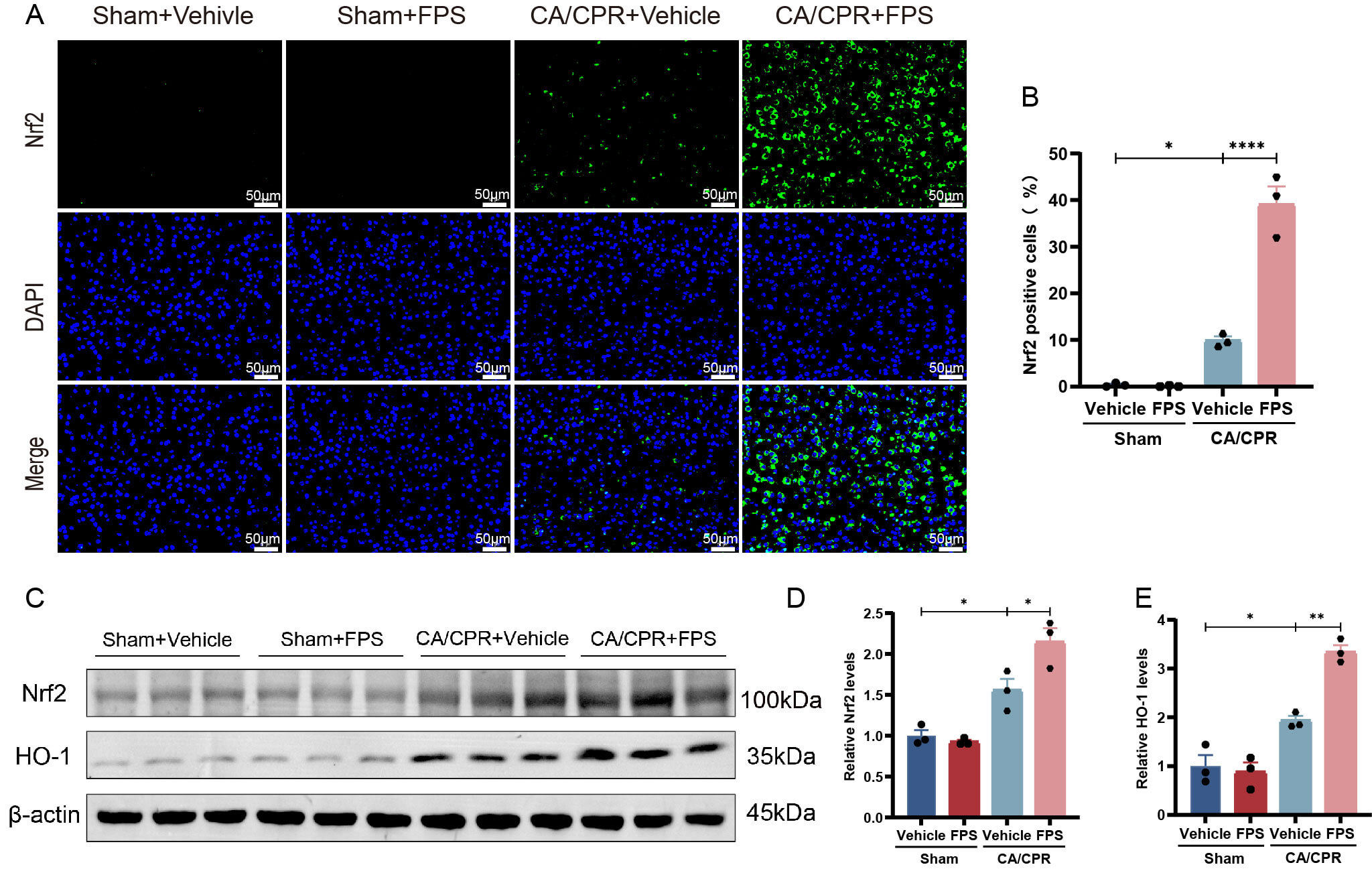 Fig. 6.
Fig. 6.
FPS pretreatment upregulated Nrf2 and HO-1 expression after
CA/CPR. (A) Typical pictures of Nrf2 immunofluorescence staining. (B) Statistics
of the proportion of Nrf2-positive cells in different groups. (C) Typical protein
immunoblotting bands of Nrf2 and HO-1. (D,E) Relative protein expression
semiquantitative analysis of Nrf2 and HO-1 in different groups. Data are depicted
as mean
To further characterize the neuropathological alterations in the brain after
CA/CPR, brain samples were harvested on postoperative day 3 and subjected to
H&E, Nissl, and FJB staining. The H&E (Fig. 7A,B) results showed that neurons
in the sham group displayed clear, intact morphology. In contrast, the CA/CPR +
vehicle group neurons were atrophied, with a reduced cytoplasmic volume, and
nuclei and nucleoli were blurred. In the CA/CPR + FPS group, the majority of
neurons retained relatively normal cytoplasmic and nuclear architecture, and only
a small proportion of cells showed mild shrinkage, characterized by decreased
cytoplasm and atypical nuclei. Nissl staining further demonstrated pronounced
neuronal damage in the CA/CPR + vehicle group, characterized by pyknosis,
karyorrhexis, and karyolysis, a notable loss of Nissl bodies, and a reduction in
Nissl-positive neuronal counts compared with the sham group (p
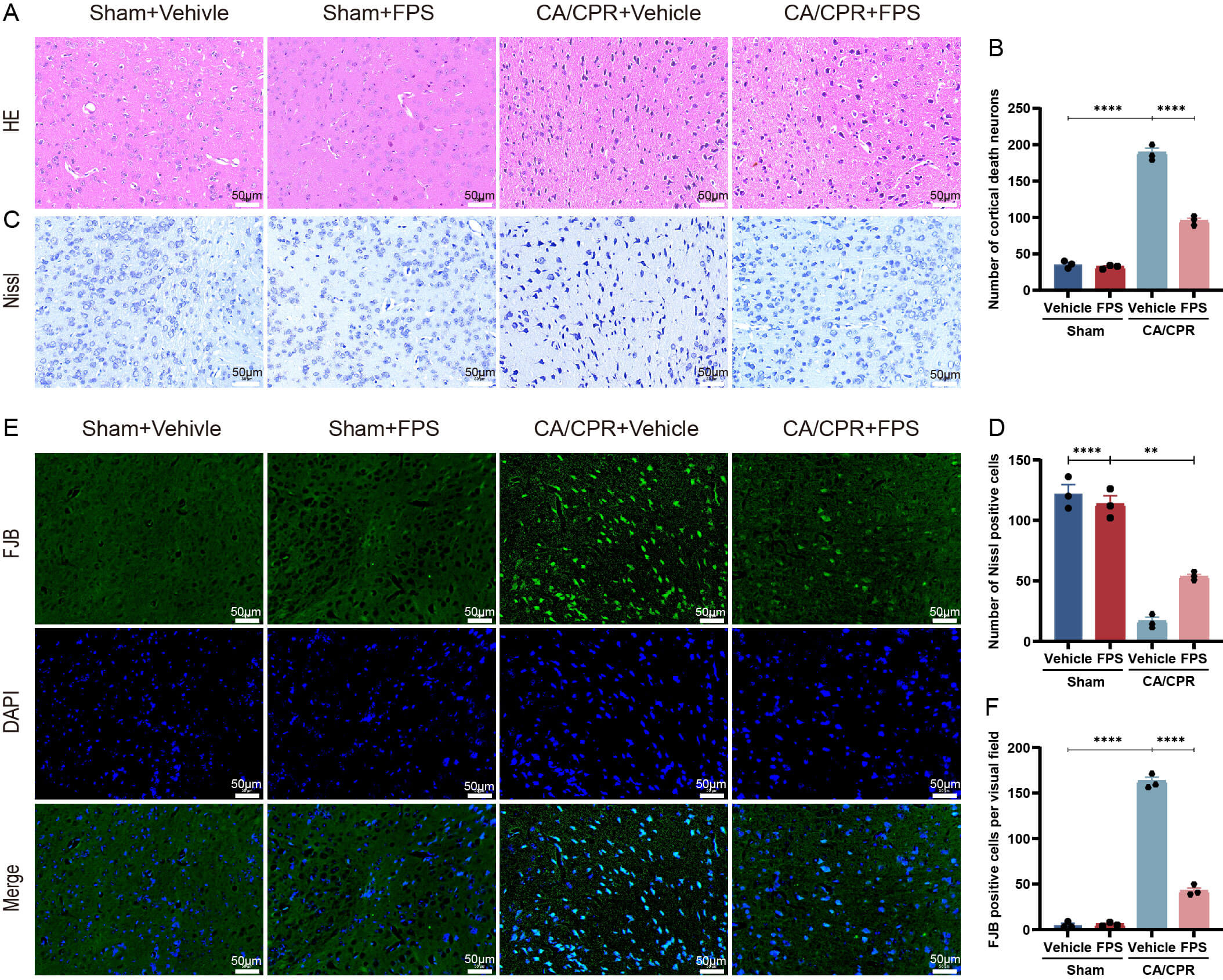 Fig. 7.
Fig. 7.
FPS pretreatment reduced neuron damage after CA/CPR. The mouse
brain cortex was stained with H&E, Nissl, and FJB 3 days after CA/CPR to assess
neuropathological damage. (A) Typical pictures of H&E staining. (B) Number of
dead cortical neurons. (C) Typical pictures of Nissl staining. (D) Quantification
of Nissl-positive cells. (E) Typical pictures of FJB staining. (F) Quantification
of FJB-positive cells. Data are depicted as mean
Neurological outcomes after CA/CPR were assessed using 9- and 12-point
neurological-function scoring systems. Body weight changes were monitored on days
1 and 3 after resuscitation, and survival rates were recorded daily. Survival
analysis (Fig. 8A) showed that the FPS pretreatment group showed markedly better
survival rates than did the CA/CPR + vehicle group, with higher survival rates at
1 day (69.23% vs. 46.15%), 2 days (69.23% vs. 30.77%), and 3 days (53.85% vs. 23.08%). Analysis of neurological-function scores indicated pronounced
functional impairment in the CA/CPR group relative to the sham group at both time
points. On the 9-point scale (Fig. 8B), the mean scores in the CA/CPR group were
1.00 on day 1 and 0.33 on day 3, whereas on the 12-point scale (Fig. 8C), the
corresponding scores were 6.17 on day 1 and 3.33 on day 3. In contrast, mice
receiving FPS pretreatment exhibited significantly better neurological
performance than did the CA/CPR + vehicle group: the 9-point scores increased to
2.22 on day 1 (p = 0.0079) and 1.86 on day 3 (p = 0.0128), and
the 12-point scores rose to 7.56 on day 1 (p = 0.0005) and 6.43 on day 3
(p
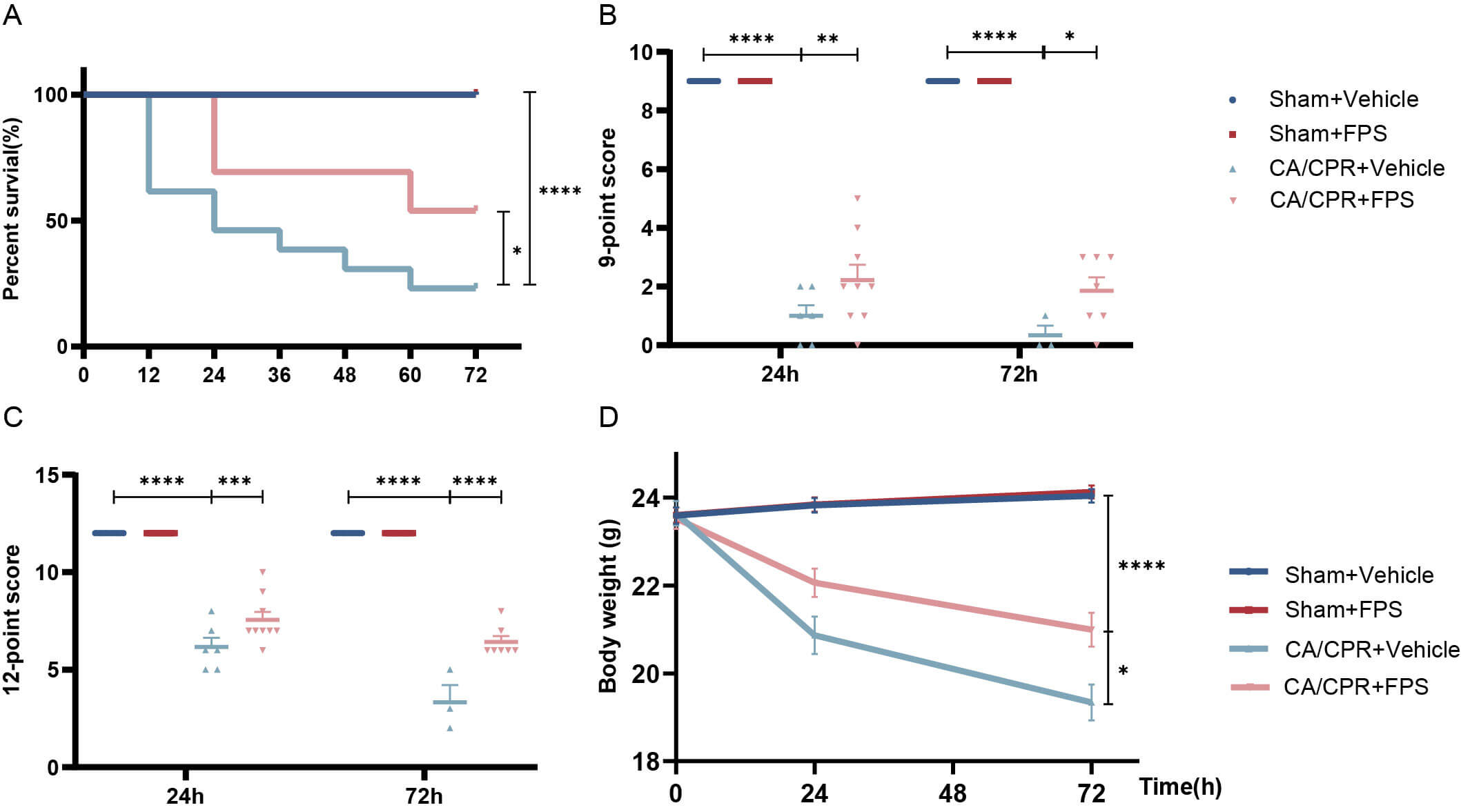 Fig. 8.
Fig. 8.
FPS pretreatment ameliorated neurological function and survival
after CA/CPR. (A) Survival percentage of mice was measured 3 days. Survival
curves were generated using the Kaplan–Meier method and compared by the log-rank
(Mantel–Cox) test. (B,C) 9-point and 12-point scoring systems at 1 and 3 days.
(D) Body weight reduction in mice was observed at 1 and 3 days. Data are depicted
as mean
In the present study, we demonstrated that the HMGB1/RAGE axis was activated after CA/CPR. Pharmacological inhibition of this axis with FPS pretreatment significantly reduced pyroptosis, ROS accumulation, and suppressed the release of inflammatory mediators. These findings demonstrated that FPS exerted both anti-inflammatory and antioxidant actions. Additional experiments further indicated that the antioxidant properties of FPS might be mediated, at least in part, by activation of the Nrf2/HO-1.
The pathophysiology of PCABI consists of two phases: global cerebral ischemia
and subsequent reperfusion after ROSC [41]. In the early phase of reperfusion,
levels of ROS rise sharply, and ischemic necrotic neurons release large amounts
of DAMPs. They are recognized by the innate immune system and trigger an
inflammatory cascade that exacerbates PCABI [42, 43]. Previous studies have shown
that the binding of HMGB1 to its receptor, RAGE, activates multiple downstream
signaling molecules, including nuclear factor kappa B (NF-
Pharmacological inhibition of RAGE can mitigate myocardial IRI in rats [47]. Singh and Agrawal [34] showed that FPS treatment reduced the interaction between RAGE and its ligand HMGB1, thereby reducing HMGB1 expression. Thus, it reduced neuroinflammation and exerted a neuroprotective effect in ischemic brain injury [48]. FPS is highly effective, well-tolerated, and readily crosses the blood-brain barrier; it is therefore considered a novel therapeutic candidate with substantial translational potential [49]. Our findings demonstrated that FPS pretreatment suppressed the upregulation of the HMGB1/RAGE axis after CA. In addition, FPS administration significantly improved overall outcomes after CA, including increased survival, enhanced neurological recovery, and reduced body weight loss. Histological analyses further provided morphological evidence for its neuroprotective effects: H&E, Nissl, and FJB staining showed that FPS pretreatment attenuated structural damage, increased the number of surviving neurons, and reduced apoptotic cell death, indicating a marked reduction in neuronal degeneration and loss. In summary, this study emphasized that FPS, through modulation of the HMGB1/RAGE axis, might represent a potential therapeutic agent in mitigating IRI induced by CA.
Pyroptosis is a key contributor to the initiation and development of PCABI [50, 51, 52]. Once released extracellularly, either actively or passively, HMGB1 binds
to RAGE receptors on macrophages, triggering a dynamic, receptor-mediated
endocytosis. That then leads to inflammasome activation, pyroptosis, and the
release of inflammatory mediators [26, 53]. Based on these findings, our study
investigated whether FPS pretreatment alleviated neuronal pyroptosis after CA/CPR
by inhibiting the HMGB1/RAGE axis. Our results demonstrated that CA/CPR indeed
induced significant neuronal pyroptosis in mice, as indicated by a marked
upregulation of key pyroptosis-related proteins (cleaved caspase-1, GSDMD,
N-GSDMD) and downstream inflammatory cytokines (IL-1
At present, ROS-mediated oxidative stress is essential in PCABI [54, 55]. The Nrf2 pathway has been shown to mitigate ROS-driven oxidative stress and improve neurological recovery after CA resuscitation [56, 57, 58]. RAGE activation may be closely associated with the accumulation of ROS in the nervous system [59]. FPS has been reported to inhibit NADPH oxidase activation and subsequent ROS production [60]. Consistent with previous findings, the present study showed that FPS notably upregulated the Nrf2/HO-1 antioxidant, reduced ROS accumulation, and alleviated oxidative stress after CA. However, this study did not include causal validation of the involved signaling pathways through in vivo gene-knockout experiments. Therefore, additional investigations are necessary to clarify the precise protective mechanism of FPS in PCABI.
This study had several limitations: (1) We evaluated the relationship between
the action of FPS and pyroptosis after CA. However, we could not exclude the
possibility that the HMGB1/RAGE pathway regulated by FPS may contribute to the
modulation of other types of cell death. For instance, studies have demonstrated
HMGB1/RAGE axis exacerbates myocardial IRI in mice by regulating apoptosis and
autophagy [53]. (2) We only investigated short-term changes to the HMGB1/RAGE
pathway and the effect of FPS pretreatment after CA/CPR and did not assess
long-term outcomes. Future studies should include additional long-term assessment
time points (e.g., 7 and 14 days after CA) to systematically evaluate the dynamic
changes associated with persistent activation of the HMGB1/RAGE axis and the
sustained effects of FPS. Previous research has demonstrated that HMGB1 protein
levels in rat hippocampus are significantly increased at 1 day after CA and
continue to rise over the next 7 days [61], suggesting that this pathway may
contribute to brain injury over a longer time. (3) The assessment of inflammation
in the present study was not exhaustive. We only analyzed IL-18 and IL-1
In summary, pretreatment with FPS in a CA model was found to exert neuroprotective function by regulating the HMGB1/RAGE axis. It attenuated oxidative stress and pyroptosis associated with PCABI, thereby reducing cerebral damage and improving neurological outcomes.
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
YZ and ZY conceived and designed the study. YZ, YW, and JH conducted the behavioral experiments and acquired the data. YZ, QL, LL, SX, and LPL participated in data acquisition and contributed to data analysis and interpretation. YZ drafted the manuscript. ZY, QL, LL, SX, and LPL critically revised the manuscript for important intellectual content. LPL and ZY supervised the study. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animals received approval from the Laboratory Animal Centre of Renmin Hospital Wuhan University Committee (No. March 4, 2025 [20250304]) and were performed in accordance with the institutional guidelines for the care and use of laboratory animals.
We appreciate Jianfei Sun for providing guidance on animal experiments and the support of BioRender.
This work received financial support from the National Natural Science Foundation of China (No. 81772039), the Knowledge Innovation Program of the Wuhan Municipal Science and Technology Bureau (No. 2022020801010474), and the “Chutian Talent Program” for healthcare professionals in Hubei Province (No. CZ2024020001).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN50023.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.