





1 Department of Physiology, Faculty of Science, Charles University, 128 00 Prague, Czech Republic
Abstract
Astrocytes are increasingly recognized as central players in the pathogenesis of Alzheimer’s disease (AD), exhibiting both neuroprotective and neurotoxic functions, which complicates their role in disease progression. Under physiological conditions, astrocytes support neuronal homeostasis, facilitate synaptic function, and promote the clearance of Amyloid-β (Aβ), thereby contributing to neuroprotection. In the context of AD, however, reactive astrocytes can adopt detrimental phenotypes, releasing pro-inflammatory cytokines, generating oxidative stress, and disrupting neuronal networks, thereby exacerbating neurodegeneration. Consequently, the shift from a protective to a neurotoxic phenotype may not only drive neuronal loss but also accelerate AD progression. The dual roles of astrocytes and the dynamic changes in their functions—protecting neurons under normal conditions while promoting pathology when dysregulated—underscore their complex contribution to AD pathophysiology. Elucidating the mechanisms underlying astrocyte-mediated neuroprotection and neurotoxicity is essential for developing targeted therapeutic strategies aimed at modulating astrocyte activity to slow or prevent disease progression. This review aims to present and critically discuss recent advances and ongoing controversies concerning the involvement of astrocytes in AD.
Keywords
- Alzheimer’s disease
- Amyloid-β
- astrocytes
- astrogliosis
- calcium dyshomeostasis
- PI3K/Akt
- JAK/STAT
- NF-κB
- Nrf2
- neuroinflammation
Alzheimer’s disease (AD) is the most common form of dementia, accounting for at least two-thirds of cases in individuals over the age of 65. It is a slowly progressive neurodegenerative disorder that affects memory, cognition, and behavior, and is characterized by confusion, communication difficulties, and personality changes. Although the symptoms of AD have been extensively studied, there is currently no cure to halt or reverse its progression. However, certain medications are available that may help slow the course of the disease [1].
AD is a multifactorial neurodegenerative condition resulting from a combination
of factors, including protein aggregation, chronic neuroinflammation, and
neuronal loss. Although our understanding of AD pathogenesis remains incomplete,
several hallmark pathological features have been identified—namely,
extracellular neuritic plaques and intracellular neurofibrillary tangles. These
are composed of accumulated Amyloid-
In recent years, increasing attention has been directed toward the role of glial cells in both normal brain function and neurodegenerative diseases [4, 5, 6]. In this context, the present review focuses on the contribution of astrocytes to AD pathogenesis and progression, summarizing recent advances and outlining key priorities for future research.
Astrocytes are the most abundant glial cells in the adult human brain, with numbers estimated between 40 and 130 billion, resulting in a glia-to-neuron ratio of approximately 1:1 [7]. They play essential roles in neuronal development, activity, and homeostasis, emphasizing the importance of glia–neuron interactions [8]. First named for their star-like shape by Mihály Lenhossék in 1895 [9], astrocytes are structurally and functionally diverse, supporting central nervous system (CNS) function throughout life. They regulate synapses, neurotransmitters, ion and water balance, and maintain the extracellular matrix, while also contributing to metabolic support, synapse formation, and synaptic pruning [6, 10]. Additionally, astrocytes are critical for the blood–brain barrier (BBB), neuroprotection, waste clearance, and neurogenesis, acting alongside microglia as a frontline defense [11].
Under CNS injury or disease, astrocytes become reactive, undergoing morphological, transcriptional, and functional changes in a process called astrogliosis. Reactive astrocytes can adopt distinct phenotypes, including A1 pro-inflammatory, potentially neurotoxic cells, and A2 neuroprotective cells that support repair. The role of astrocytes varies across pathological contexts, and morphology alone does not always indicate function [12, 13, 14]. According to current perspectives, reactive astrocytes should not be classified solely within simplified frameworks such as neurotoxic versus neuroprotective or A1 versus A2 states. Instead, a more comprehensive approach that integrates multiple molecular and functional parameters, along with their impact on pathological hallmarks in relevant models, is preferred [12]. The goal is to move beyond rigid categorization and to identify the key variables that drive distinct reactive astrocyte states, phenotypes, and functions within specific pathological contexts. Achieving this requires the use of multidimensional datasets and co-clustering approaches to accurately define the diversity and distinctiveness of astrocyte phenotypes. Consequently, future classification systems should incorporate a range of criteria, including transcriptomic and proteomic profiles, morphological characteristics, and specific cellular functions.
Astrocytes play a fundamental role in maintaining CNS homeostasis by supporting key physiological processes, including neurotransmitter clearance, energy metabolism, ion buffering, and immunomodulation. Disruption of these functions is thought to occur progressively during aging and disease progression, thereby amplifying other pathological mechanisms in the brain. Under adverse conditions, astrocytes can adopt pathological phenotypes characterized by distinct morphological and molecular alterations. Neurodegenerative processes are commonly associated with dysregulated astrocyte reactivity, astrogliosis, functional impairment, and, in some cases, the induction of cellular senescence or cell death. One of the earliest astrocyte-driven mechanisms contributing to neurodegeneration involves changes in the astrocytic secretome. Astrocytes release a wide range of cytokines, chemokines, and interleukins that can amplify inflammatory signaling through multiple pathways, including autocrine activation, stimulation of microglia, and recruitment of peripheral immune cells [11]. Persistent activation of these pathways promotes a chronic inflammatory environment that ultimately leads to neuronal dysfunction, neurotoxicity, and cell death. Under pathological conditions, astrocytes not only lose their supportive roles but may actively contribute to disease progression through the secretion of toxic mediators, including pro-inflammatory cytokines and neurotoxic lipids [15]. Although altered astrocyte reactivity, functional impairment, and cytotoxicity often occur concurrently, the precise mechanistic relationships among these processes remain incompletely understood.
Accumulating evidence indicates that several essential astrocytic functions are compromised during both acute inflammation and chronic neurodegeneration. For instance, astrocytes promote synapse formation during development by secreting synaptogenic factors such as SPARC-like protein 1 (SPARCL1), thrombospondins (TSP1 and TSP2), and glypicans (GPC4 and GPC6) [16, 17, 18]. However, the expression of these molecules is markedly reduced in neurotoxic reactive astrocytes in both rodent models and human tissue, resulting in a diminished capacity to support synaptogenesis in neuron–astrocyte co-culture systems [19]. These findings underscore how the loss of astrocyte-mediated support directly contributes to synaptic dysfunction in neurodegenerative diseases.
Importantly, astrocyte dysfunction does not occur in isolation but is closely intertwined with neuroinflammatory processes that characterize many CNS disorders. A comprehensive understanding of astrocyte involvement in neurodegeneration therefore requires detailed investigation of their role in inflammatory signaling networks.
Neuroinflammation is a defining feature of many neurodegenerative disorders,
including AD, and astrocytes play a central role in this process. Inflammation is
typically initiated as a protective response to infection or injury, but in the
CNS it can also be triggered by endogenous molecules associated with
neurodegenerative pathology, including A
Astrogliosis represents a complex and highly context-dependent response characterized by several defining features: (1) it encompasses a broad range of molecular, cellular, and functional changes occurring in response to nearly all types of CNS insults; (2) these changes occur along a graded continuum depending on the severity of injury; (3) reactive responses are shaped by diverse signaling pathways and intercellular interactions; and (4) astrocyte reactivity can result in both gain- and loss-of-function effects [22].
The contribution of neuroinflammation and astrogliosis to neurodegeneration
remains an area of active debate. While some evidence suggests that these
processes arise as secondary responses to protein aggregation, other studies
indicate that immune signaling may actively drive disease progression. For
example, variants in the TREM2 gene—which encodes an innate immune receptor
highly expressed in microglia—can increase the risk of late-onset AD by two- to
four-fold, comparable to the risk associated with a single apolipoprotein E (APOE)
Astrocyte reactivity is often influenced by microglial activation states. Microglia themselves can be modulated by signals from other CNS cells as well as by systemic factors such as gut microbiome–derived metabolites that activate the aryl hydrocarbon receptor [25]. This highlights the complex, multicellular nature of neuroinflammatory signaling networks. Another important factor shaping astrocyte responses is cellular heterogeneity. Advances in single-cell and single-nucleus sequencing have revealed numerous astrocyte subpopulations that differ in morphology, gene expression, and functional properties. These subtypes can change dynamically during development, aging, and disease, thereby altering the overall composition of the astrocyte population [26]. In AD, pronounced transcriptomic changes in astrocytes occur along the spatiotemporal progression of the disease, reflecting shifts between homeostatic and reactive astrocyte states [27]. Importantly, individual astrocytes may display both protective and detrimental properties depending on environmental signals and the stage of the disease [19]. Recent spatial transcriptomics data mapping glial states and molecular events within plaque–glial niches in human AD brain tissue provide new insights into the cellular and molecular heterogeneity underlying neurodegenerative pathology [28]. Current single-cell sequencing techniques also enable the correlation of transcriptional changes in astrocytes from AD patients with specific clinical indicators [29]. While astrogliosis and inflammatory signaling represent major components of astrocyte pathology, they are accompanied by additional cellular changes that affect astrocyte physiology and survival.
Beyond inflammatory activation, astrocytes in neurodegenerative conditions
frequently exhibit profound functional impairments that further compromise
neuronal homeostasis. In both AD patients and experimental models, astrocytes
show disturbances in key processes such as calcium signaling and glutamate
buffering. For instance, astrocytes in AD models display abnormal intracellular
Ca2+ dynamics characterized by hyperactive signaling patterns that parallel
neuronal hyperexcitability observed during disease progression [30]. Elevated
astrocytic expression of the Ca2+/calmodulin-dependent phosphatase
calcineurin has also been detected in early-stage AD and may contribute to the
induction of inflammation-related genes [31]. Additionally, decreased levels of
inositol 1,4,5-trisphosphate receptor type 2 have been reported [32, 33]. Such
remodeling of calcium signaling can impair astrocyte reactivity and disrupt
homeostatic support mechanisms [34]. Alterations in glutamate metabolism
represent another important pathological feature. Reduced expression of the
glutamate transporter EAAT2 and diminished glutamate uptake activity have been
reported in the frontal cortex of AD patients [35]. Impaired glutamate transport
has also been associated with increased A
In addition to functional impairment, astrocytes may undergo cellular
senescence, particularly during aging and chronic stress. Senescent astrocytes
enter permanent cell-cycle arrest but remain metabolically active and develop a
senescence-associated secretory phenotype (SASP). This phenotype promotes chronic
inflammation and can negatively influence surrounding neural cells [39].
Accumulation of senescent astrocytes has been increasingly linked to
neurodegenerative disease progression [40, 41]. Metabolically, senescent
astrocytes undergo a shift from glycolytic metabolism to increased oxidative
phosphorylation, which reduces lactate supply to neurons [42, 43]. This shift is
associated with enhanced inflammatory signaling and a transition from
neurotrophic to neurotoxic phenotypes. In parallel, several essential astrocytic
functions—including glutamate uptake, cholesterol synthesis, and ATP
production—are significantly reduced [44, 45, 46]. These deficits may contribute to
BBB dysfunction, synaptic impairment, decreased A
Astrocytes may undergo cell death under pathological conditions. Both apoptotic and non-apoptotic mechanisms have been reported, including ferroptosis, an iron-dependent form of cell death associated with lipid peroxidation [50, 51]. Although astrocyte death can trigger immune responses that promote the clearance of aggregated proteins, chronic cell loss and sustained inflammation may exacerbate neurodegenerative processes [52]. Astrocytic death phenotypes have been observed in the brains of AD patients [50, 53].
Taken together, these findings highlight the multifaceted ways in which astrocyte dysfunction contributes to brain pathology. These mechanisms are particularly evident in AD, where astrocytic alterations influence multiple aspects of disease progression. The key features of normal and pathological astrocyte phenotypes potentially involved in AD development are schematically illustrated in Fig. 1.
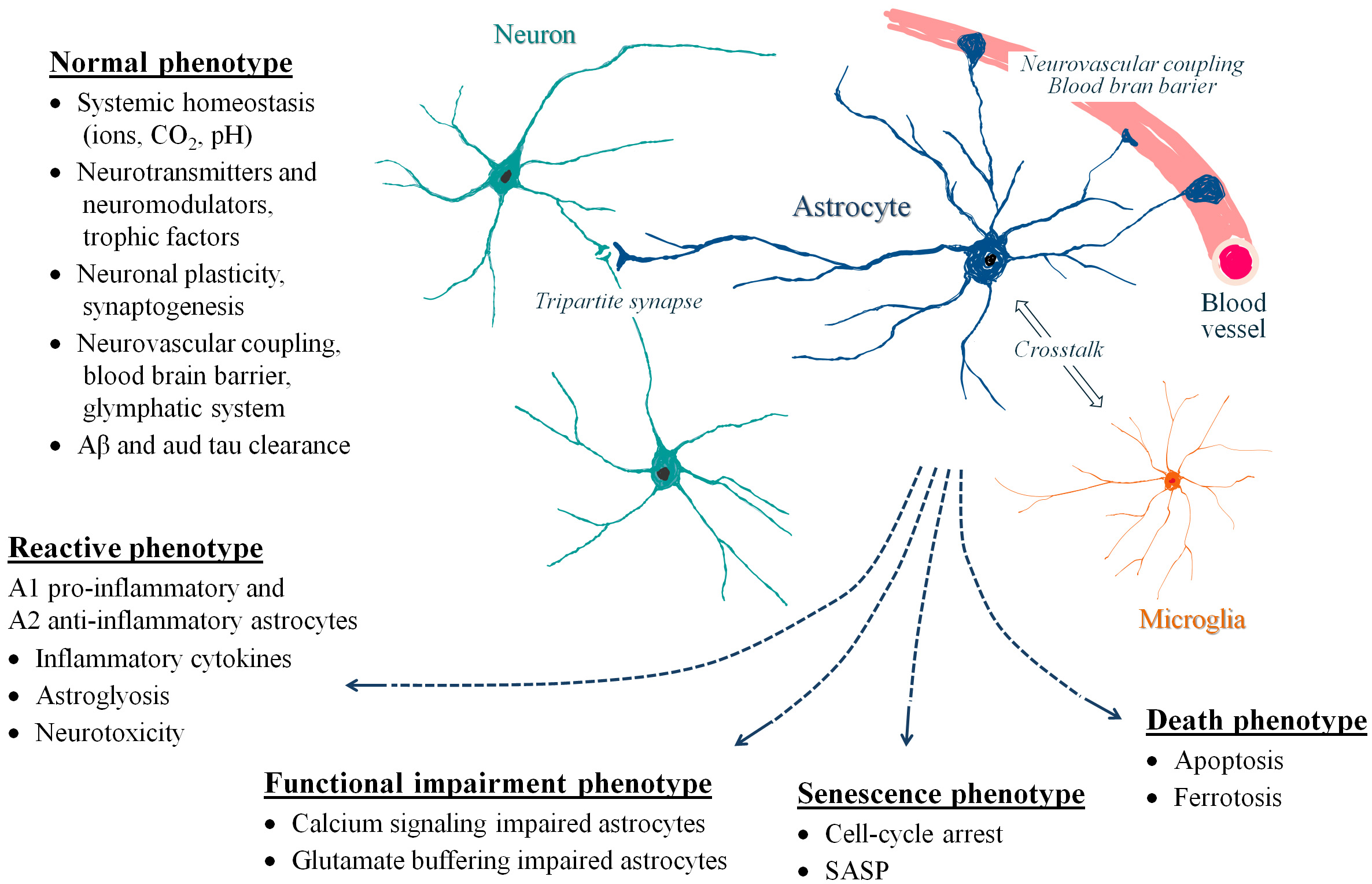 Fig. 1.
Fig. 1.
Schematic overview of characteristic features of normal
and pathological astrocyte phenotypes potentially involved in AD pathology. Normal, healthy astrocytes play a crucial role in maintaining CNS homeostasis,
including regulation of pH and ion balance (Na+, K+, Ca2+),
production of trophic factors (BDNF, GDNF, CNTF, NGF), neurotransmitter
recycling, modulation of neuronal plasticity, formation and maintenance of the
neurovascular unit and the BBB, support of glymphatic function, and clearance of
A
Astrocytes play a central role in the pathophysiology of AD, exerting both
protective and detrimental effects depending on disease stage and pathological
context. Under physiological conditions, they maintain neuronal homeostasis by
regulating metabolic pathways, neurotransmitter cycling, ion balance, and the
clearance of toxic metabolites. However, chronic exposure to pathological stimuli
such as A
Increased amounts of both A1 and A2 astrocytes identified in post-mortem brain
tissue from patients with AD suggest the roles for both these phenotypes in the
disease pathogenesis [56]. In the early stages of AD, astrocyte activation may
serve protective functions. Reactive astrocytes can enhance A
Several molecular mechanisms underlie astrocyte-mediated contributions to AD
pathology. Reactive or senescent astrocytes often exhibit altered cytokine
secretion and impaired glutamate uptake, increasing the risk of excitotoxic
neuronal injury [64, 65]. They may also promote tau hyperphosphorylation and
impair A
Metabolic dysregulation represents another key aspect of astrocyte involvement
in AD. Astrocytes play a central role in brain energy metabolism through the
astrocyte–neuron lactate shuttle, which supplies neurons with metabolic
substrates. Disturbances in cerebral glucose metabolism—commonly observed in
AD—may partly result from impaired astrocytic glucose uptake and metabolism
[73, 74]. Disruption of astrocyte-derived lactate production can compromise
neuronal energy supply and synaptic transmission [75, 76]. These metabolic
deficits likely arise from a combination of astrocytic dysfunction, vascular
abnormalities, and impaired glucose transport across the BBB. In addition,
growing evidence indicates that astrocytes play a key role in the regulation of
fatty acid and cholesterol metabolism, which is disrupted in astrocytes in AD
[77]. Alterations in astrocytic lipid metabolism may directly affect neuronal
health by disturbing cholesterol homeostasis, promoting neuroinflammation through
lipid peroxidation byproducts, and impairing energy metabolism. Furthermore,
these metabolic changes may contribute to AD pathophysiology by compromising the
astrocyte-mediated clearance of A
Genetic evidence further supports the involvement of astrocytes in AD.
Apolipoprotein E4 (ApoE4), the strongest genetic risk factor for sporadic AD, is
predominantly produced by astrocytes in the healthy brain. While
astrocyte-derived ApoE regulates A
Despite these pathological mechanisms, astrocytes retain important
neuroprotective capacities. They contribute to A
Emerging evidence suggests that in AD the initial pro-inflammatory stimulus may
arise from stressed or damaged neurons. This primary insult can then initiate a
secondary inflammatory cascade mediated by complex intercellular interactions
between astrocytes and microglia, ultimately driving disease progression [60, 85]. Astrocyte reactivity may gradually shift toward neurotoxic phenotypes
characterized by a transcriptional profile that impairs synaptic support and
promotes neuronal death [19]. A central driver of astrocyte subtype switching
during AD progression is A
Overall, astrocytes exhibit complex and context-dependent roles in AD. While
they can protect neuronal networks by maintaining metabolic and synaptic
homeostasis and facilitating protein clearance, persistent activation and
dysfunction may transform them into contributors to neurodegeneration. This
duality is particularly evident in AD, where astrocytes transition from initially
protective to progressively detrimental states in response to sustained
pathological stress. Understanding the molecular pathways that regulate astrocyte
subtype transitions is essential for developing therapeutic strategies targeting
their function in AD. In particular, signaling mechanisms such as astrocytic
calcium dynamics and the JAK/STAT, PI3K/Akt, NF-
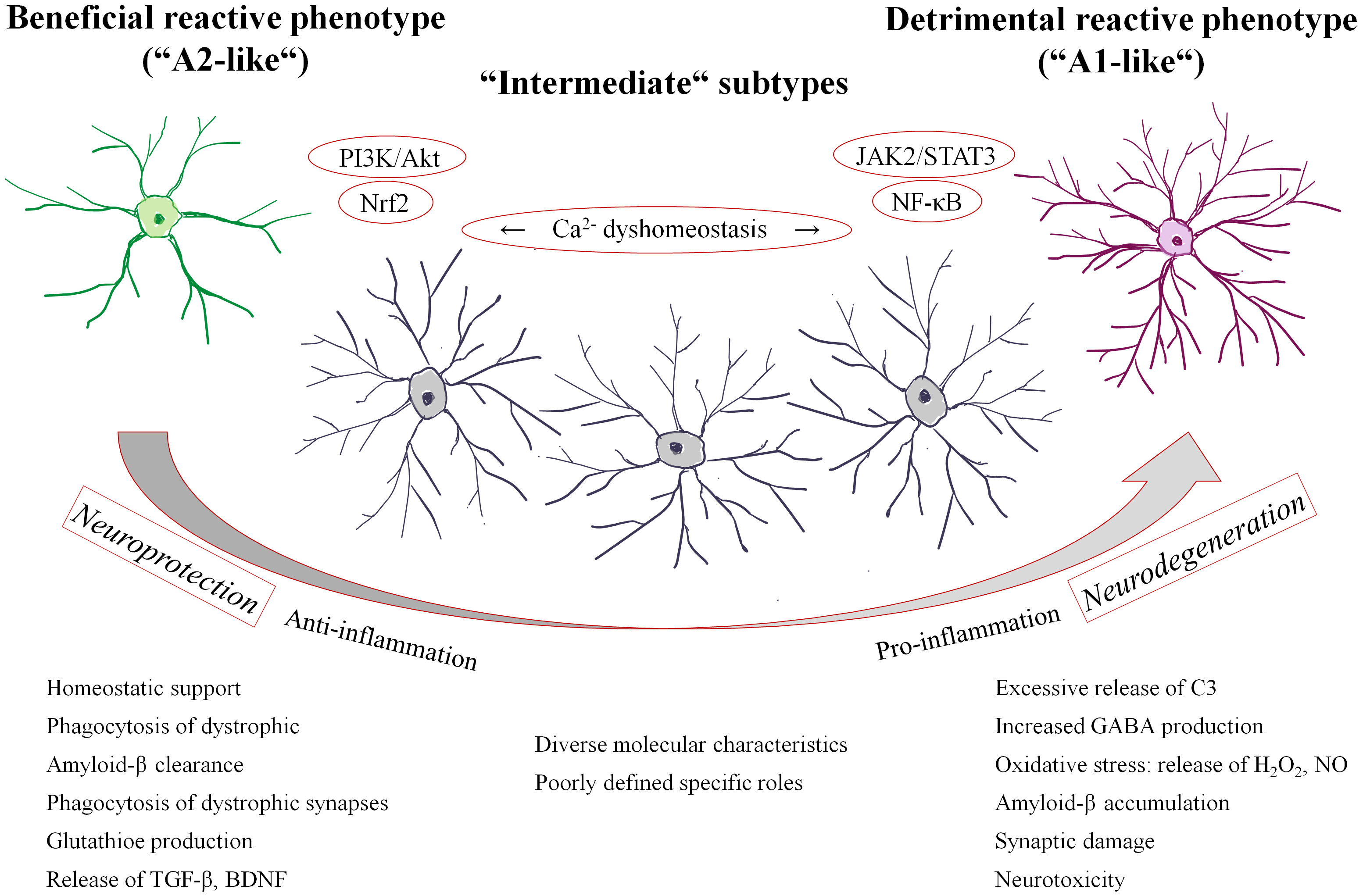 Fig. 2.
Fig. 2.
A simplified schematic illustrating the dual roles of
reactive astrocytes in AD and the regulation of their phenotypic states. Astrocyte polarization is governed by a balance of intracellular signaling
pathways, in which JAK/STAT3 and NF-
Calcium dyshomeostasis is a common feature of many neurodegenerative diseases,
including AD [93]. Dysregulated calcium signaling often arises early in disease
progression and contributes to pathological alterations in neuronal synaptic
activity [94, 95]. In particular, oligomeric A
Although the mechanisms underlying astrocytic dysfunction in AD are not fully
understood, disruption of Ca2+ signaling is considered a key contributing
factor. A
Disrupted astrocytic calcium signaling also affects neurovascular regulation and
neuronal network activity. Under normal conditions, astrocytic Ca2+
transients trigger the release of vasoactive molecules that regulate local
cerebral blood flow [106]. Impairment of this mechanism contributes to
neurovascular dysfunction in AD. Indeed, astrovascular decoupling observed in AD
mouse models has been associated with altered astrocytic calcium signaling and
reduced astrocyte functional connectivity [107]. Reactive astrocytes in amyloid
models also exhibit abnormal Ca2+ dynamics that disrupt communication
between astrocytic endfeet and cerebral arterioles, potentially contributing to
the cerebral hypometabolism characteristic of AD [108]. Importantly,
normalization of astrocytic calcium signaling has been shown to reduce neuronal
network hyperactivity and improve cognitive performance in
Calcium dysregulation further contributes to neuronal excitotoxicity by
disrupting glutamate homeostasis. Under physiological conditions, astrocytes
remove synaptic glutamate through excitatory amino acid transporters and convert
it to glutamine via glutamine synthase. The Wnt/
Another critical signaling mechanism implicated in AD is the Janus kinase/signal
transducer and activator of transcription (JAK/STAT) pathway, particularly the
JAK2/STAT3 axis, which plays a central role in regulating neuroinflammation and
astrocyte reactivity. Activation of JAK2/STAT3 signaling promotes astrocyte and
microglial activation and stimulates the release of pro-inflammatory cytokines,
thereby contributing to the inflammatory environment characteristic of AD [113].
Increased STAT3 activity has been associated with A
The JAK/STAT pathway is a key mediator of astrocyte reactivity in several
neurodegenerative disorders, including AD, Huntington’s disease, and Parkinson’s
disease [115]. Activation of STAT3 regulates multiple aspects of astrocyte
behavior, including morphological remodeling, migration, proliferation, and the
expression of reactive markers such as GFAP, as well as the secretion of
inflammatory cytokines. Through these mechanisms, STAT3 acts as a master
regulator that drives the formation of distinct reactive astrocyte phenotypes.
Experimental studies further demonstrate that activation of the JAK2/STAT3 axis
induces reactive astrocyte formation in AD models, whereas SOCS3, an endogenous
inhibitor of JAK/STAT signaling, functions as a negative regulator that restrains
astrocyte reactivity [86]. Manipulation of this pathway has revealed important
functional consequences in experimental models. In the APP/PS1
Although STAT3 regulates many reactive astrocyte states following inflammatory insults [117], recent single-cell and single-nucleus RNA sequencing studies have identified astrocyte subpopulations that appear to be independent of STAT3 signaling [118, 119]. Alternative regulators of astrocyte reactivity include the chromatin remodeler SMARCA4 [120] and microRNAs such as miR-146a, miR-145, and miR-125b [121]. While these microRNAs may not directly initiate the transcription of reactive genes, they likely stabilize reactive transcriptional programs and contribute to the sustained transition from physiological to reactive astrocyte states [122]. These observations suggest that neuroprotective and neurotoxic astrocyte phenotypes may coexist within the same cellular populations, reflecting the complex regulatory networks that govern astrocyte responses during neurodegeneration.
The PI3K/Akt pathway, which is upstream of NF-
The NF-
Several studies have demonstrated that Nrf2 plays a key role in regulating redox
homeostasis and exerts anti-inflammatory effects in various neurodegenerative
disorders [135]. The importance of astrocytes in this context is underscored by
findings showing that astrocyte-specific activation of Nrf2 is sufficient to
attenuate disease progression in multiple experimental models, including AD
[136]. Consistently, Nrf2 deficiency promotes the activation of reactive
astrocytes in brain tissue from 5xFAD mice, whereas Nrf2 upregulation suppresses
the induction of reactive astrocyte gene expression by inhibiting the recruitment
of the NF-
To better understand the functional changes in astrocytes that may contribute to
the initiation and progression of inflammatory responses and neurodegenerative
diseases, several important questions still need to be addressed. Different
neurodegenerative conditions appear to affect distinct anatomical regions of the
brain. In AD, pathology initially targets the hippocampus and entorhinal cortex,
before later spreading to the neocortex [139]. This suggests that certain brain
regions may be inherently more vulnerable to internal pathological changes,
external toxic insults, or natural processes such as those occurring during
aging [140]. Aging—one of the major risk factors for neurodegenerative
diseases—has been shown to influence astrocytic immune responses and other key
functions [141]. However, further research is needed to determine whether
astrocyte heterogeneity and region-specific dysregulation actively contribute to
the neurodegenerative process. It is therefore essential to investigate whether
astrocytic changes vary depending on brain region or proximity to pathological
features. For instance, it would be of interest to determine whether the
reduction in glutamate transporter currents and other neurosuppressive functions
observed in the AD brain occurs predominantly in astrocytes located near
A
It is also important to examine the role of astrocytes in disrupting the signaling pathways between neurons, astrocytes, and blood vessels that underlie neurovascular coupling. Furthermore, identifying which astrocytic alterations have the greatest impact on cognitive performance is essential for pinpointing the astrocyte phenotypes that should be targeted by future therapies. However, these questions may be overly simplistic, as multiple functional disturbances can contribute to circuit dysfunction, synapse loss, or neuronal death. In addition, the stage of the disease may critically determine whether a given therapeutic intervention will be effective.
Astrocyte functional heterogeneity under different pathological conditions is
closely linked to dysregulated intracellular signaling pathways, including
Ca2+ dynamics, JAK/STAT, PI3K/Akt, NF-
Translational gaps and limitations between rodent models and human studies
should also be considered. Rodent astrocytes differ from their human counterparts
in morphology, gene expression, and functional complexity; human astrocytes are
larger and exhibit greater structural and transcriptional diversity [142, 143].
Another limitation is the incomplete representation of AD pathology in
experimental models. Most transgenic mouse models rely on amyloid precursor
protein overexpression and primarily reproduce A
Astrocyte heterogeneity itself represents a critical translational barrier. Single-cell and single-nucleus transcriptomic studies have identified multiple astrocyte subpopulations with distinct molecular signatures that vary across brain regions and disease stages [145]. As discussed above, in AD, disease-associated astrocytes exhibit context-dependent phenotypes that may exert both protective and detrimental effects. This functional duality complicates therapeutic strategies, as indiscriminate modulation of astrocytes could disrupt essential homeostatic functions, including neurotransmitter recycling and ion balance. Temporal dynamics further complicate translation. Astrocytes may initially contribute to amyloid clearance and synaptic support but later acquire pro-inflammatory and neurotoxic properties as disease progresses [12].
The lack of robust and specific biomarkers of astrocyte function in living patients represents another major gap. While GFAP levels in cerebrospinal fluid and plasma show promise as indicators of astrocyte reactivity, they lack specificity for distinct functional states and do not capture the full complexity of astrocyte responses [146]. This limitation hampers patient stratification and the assessment of target engagement in clinical trials. Astrocytes also operate within a tightly interconnected cellular network involving neurons, microglia, and vascular cells. For example, microglia-derived cytokines can induce specific reactive astrocyte phenotypes, underscoring the importance of intercellular signaling in disease progression [19]. Consequently, targeting astrocytes in isolation may be insufficient, and combinatorial or systems-level therapeutic approaches may be required. An additional complication arises from the difficulty of identifying the optimal therapeutic window in human patients, due to the long preclinical phase of AD and the limited tools available for monitoring astrocyte activity in vivo.
Astrocytes are critical regulators of CNS homeostasis, far beyond their traditional role as support cells. They maintain neuronal function through metabolic regulation, ion signaling, synaptic modulation, clearance of toxic molecules, and contributions to the blood–brain barrier. In response to physiological stress or pathological insults, astrocytes undergo functional and morphological changes that enable them to modulate immune signaling, preserve tissue integrity, and limit neuronal damage.
A hallmark of AD is astrogliosis, characterized by structural remodeling and
altered gene expression. While initially protective, excessive or prolonged
astrocyte reactivity can become detrimental, amplifying neuroinflammation,
promoting neuronal apoptosis, and facilitating A
The heterogeneity and intercellular crosstalk of astrocytes position them as promising therapeutic targets in AD. Effective strategies are likely to be combinatorial, enhancing neuronal resilience while modulating astrocyte reactivity. Interventions could prevent the emergence of harmful reactive phenotypes, promote clearance of toxic proteins, restore glutamate homeostasis, support glymphatic function, and optimize astrocytic metabolic capacity. Emerging technologies—spatial transcriptomics, single-cell sequencing, and chromatin accessibility profiling—are beginning to reveal the transcriptional and epigenetic programs underlying astrocyte heterogeneity and plasticity. When integrated with physiologically relevant models, including organoids, organotypic slices, and in vivo imaging, these insights offer the potential to translate mechanistic understanding into targeted interventions. Ultimately, mapping astrocyte state transitions and intercellular interactions will be essential for developing precision therapies that harness their full therapeutic potential in AD.
Importantly, it cannot be ruled out that neurotoxic and neuroprotective functional changes, driven by distinct transcription factors, may occur simultaneously within the same astrocyte. From a therapeutic standpoint, strategies aimed at enhancing adaptive and protective astrocytic responses while concurrently inhibiting pathways that promote neurodegeneration or functional neglect could shift the balance of astrocyte sub-states. Such an approach may result in reactive astrocytes exerting a net disease-modifying and potentially neuroprotective effect. This implies the necessity of developing strategies that selectively modulate specific astrocyte functions in a stage-dependent manner, as certain astrocytic activities may be beneficial during early disease stages but become detrimental as pathology progresses. For instance, during the early inflammatory phase of AD—when neuronal stress pathways are first activated—the temporary removal of compromised neurons from active circuits might help preserve overall network stability and provide time for neuronal recovery. In this context, transient suppression of astrocytic synaptic maintenance functions could be advantageous. Conversely, in later stages of AD, when synapse density is already severely compromised due to excessive microglial pruning, the preservation and support of remaining synapses by astrocytes becomes critically important. These complex and dynamic shifts suggest that broadly targeting astrocyte reactivity may not yield therapeutic benefit. Instead, focusing on defined astrocyte sub-states or isolating specific functional pathways may offer more precise and effective therapeutic opportunities.
IS conceived the project, conducted the analysis of previous studies, and wrote the preliminary draft. ZB performed the literature search and revised the manuscript. JN contributed to the study the conception, the design of the content framework, and the preparation of the final manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was supported by the Czech Grant Foundation (grant no. 23-07184S).
The authors declare no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.