




 , Suganya Kannan 1, Kathiresan Shanmugam 2, Dhavamani Sugasini 3,*
, Suganya Kannan 1, Kathiresan Shanmugam 2, Dhavamani Sugasini 3,*1 Central Research Laboratory for Biomedical Research, Vinayaka Mission’s Medical College and Hospital, Vinayaka Mission Research Foundation (A unit of VMRF-DU), 609609 Karaikal, Puducherry, India
2 Department of Biotechnology, School of Integrative Biology, Central University of Tamil Nadu, 610005 Thiruvarur, Tamil Nadu, India
3 Department of Medicine, University of Illinois, Chicago, IL 60612, USA
Abstract
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease that is conventionally characterized by amyloid-β and tau pathology. There is growing evidence, however, that lipid metabolic disturbances are part of the biology of the disease, and not a secondary phenomenon. Lipid signaling controls membrane organization, amyloid precursor protein, tau phosphorylation, mitochondrial energetics, neuroinflammatory signaling, and synaptic stability. The accumulating genetic evidence, including risk variants in the APOE (apolipoprotein E), ABCA1 (ATP-binding cassette subfamily A member 1), ABCA7 (ATP-binding cassette subfamily A member 7), and TREM2 (Triggering receptor expressed on myeloid cells 2) genes, further makes lipid transport and lipid-sensing pathways central to late-onset AD vulnerability. Recent developments in lipidomics based on mass spectrometry have revealed concerted changes in phospholipids, sphingolipids, sterols, and oxidized lipid derivatives in brain tissue and peripheral biofluids. Instead of single abnormalities, directional metabolic imbalance is indicated by pathway changes, including decreased sphingomyelin-to-ceramide ratios and decreased polyunsaturated phospholipids. Co-analysis of lipidomic, genomic, and proteomic data has shown the existence of metabolically different subgroups, which aids genotype stratified risk evaluation and the lipid responder phenotype concept. Protein-centered therapies are complemented by therapeutic strategies that focus on lipid homeostasis, such as the regulation of cholesterol efflux, sphingolipid metabolism, pro-resolving lipid mediators, and metabolic reprogramming. There is also emerging evidence that implicates peroxisomal dysfunction and compromised glymphatic clearance in interfering with lipid balance. Although this field of research has come a long way, the issues of proving causality, standardizing lipidomic techniques, and converting pathway signatures into clinically useful resources persist. Restructuring AD as a lipid network instability disorder offers a systems level model of earlier diagnosis and targeted treatment.
Keywords
- Alzheimer’s disease
- lipid metabolism
- lipidomics
- apolipoprotein E
- neuroinflammation
- mitochondria
- precision medicine
Alzheimer’s disease (AD) is the most prevalent neurodegenerative condition (the major cause of dementia) in the world, with an estimated 60–80% of all cases of dementia [1]. The World Health Organization (2023) notes that there are over 55 million people living with dementia in the world today, and this figure is expected to rise to over 150 million by 2050 because of the aging population and extended lifespan. AD has a high socioeconomic impact, and the cost incurred by the world is much higher at USD 1.3 trillion annually, and there is a dire necessity for effective disease-modifying therapeutic measures.
AD has a neuropathological basis of extracellular amyloid-
Although decades of interdisciplinary research have produced limited and
inconsistent clinical benefits, therapeutic interventions directly affecting
A
The human brain is highly lipid, and lipids constitute about 50–60% of the dry weight [11]. In addition to structural functions of lipids in cellular membranes, lipids control synaptic transmission, receptor trafficking, membrane fluidity, mitochondrial bioenergetics, myelination, and neuroimmune homeostasis [12]. Strict control of cholesterol, phospholipids, sphingolipids and polyunsaturated fatty acids, thus, plays a key role in ensuring neuronal integrity. The modification of these pathways is directly related to the amyloid precursor protein processing, tau phosphorylation, oxidative stress reactions, and inflammatory signaling [8, 9, 13]. Interestingly, lipidomic analyses reveal that changes in ceramides, sphingomyelins, plasmalogens, and oxidized phospholipids at the early stages of the AD progression, in many cases, take place before neurons start to be lost and cognitive impairments are evident.
Genetic evidence also highlights the pre-eminence of lipid biology in AD. The
most powerful genetic risk factor of late-onset AD is the apolipoprotein E (APOE)
This review is a synthesis of existing evidence that lipid metabolism is an upstream and integrative cause of Alzheimer’s. We explore the basic guidelines of brain lipid biology, cell-type-specific lipid metabolic programs and mechanistic connections between lipid dysregulation and essential AD pathologies. Genetic modulators of lipid homeostasis, especially APOE and associated pathways, are addressed and lipidomics and biomarker discovery results are involved in the discussion. Lastly, we consider the new therapeutic approaches to lipid pathways and summarize the problems and future perspectives of precision lipid-based therapy in Alzheimer’s disease.
The classical amyloid cascade hypothesis has played a leading role in AD
research but has not been able to account for the heterogeneity of AD in sporadic
and late-onset cases or the discordance between amyloid load and cognitive
impairment in a minority of individuals [7, 23]. It is emerging that the
metabolic and lipid abnormalities play an upstream role in A
The metabolism of lipids intersects with the AD biology on several levels. The
localization and activity of
Lipid biology is associated with disease susceptibility, which is connected to
genetic factors, especially the APOE
Lipid metabolism has a fundamental effect on the metabolic formation, maturation and maintenance of the CNS. In addition to playing structural roles in cellular membranes, lipids control the membrane fluidity, vesicles in the synaptic cleft, intracellular movement, and intracellular signal transmission. The second-richest organ of the body in terms of lipid content is the brain, in which lipids constitute about 50–60% of the dry mass [8, 11, 12]. Phospholipids, sphingolipids, cholesterol and fatty acids are major lipid classes in the CNS, and each have their specific physiological roles to play. The structural framework of neuronal and glial membranes is made up of phospholipids, including phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, and phosphatidylinositol that also act as the precursors of intracellular signaling molecules [36]. The specific subclass of phospholipids, plasmalogens, is an enriched form of phospholipids, which is found in large amounts in neural tissue and plays a role in membrane stability, vesicular fusion, and antioxidant defense [37]. Sphingolipids such as sphingomyelin and gangliosides are inbuilt parts of membrane microdomains, which structure receptor localization and signal transmission [38]. Cholesterol synthesized de novo in the CNS plays a role in membrane organization, myelination and functioning of neurotransmitter receptors [39], and polyunsaturated fatty acids, including docosahexaenoic acid (DHA) in the maintenance of the structure of synaptic membranes and membrane-associated processes [40]. Table 1 (Ref. [8, 10, 13, 15, 18, 22, 28, 32, 33, 34, 38, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52]) summarizes the major lipid classes, their mechanistic roles, and key study outcomes in Alzheimer’s disease pathophysiology.
| Lipid class/Molecule | Mechanistic role in AD pathophysiology | Experimental/Clinical evidence | Key outcomes | References |
| Cholesterol and oxysterols | Regulate APP processing in lipid rafts; altered cholesterol efflux promotes amyloidogenic cleavage. | Brain and CSF studies show decreased 24S-hydroxycholesterol, disrupted CYP46A1 activity, and reduced cholesterol turnover. | LXR/RXR agonists enhance cholesterol efflux and reduce amyloid burden. | [8, 10, 32] |
| Ceramides and sphingolipids | Promote apoptosis, oxidative stress, and BACE1 stabilization; impair mitochondrial function. | Elevated plasma ceramides correlate with cognitive decline; early accumulation in hippocampus and cortex. | Ceramide synthesis inhibitors reverse neurodegeneration in models. | [13, 22, 38, 51] |
| Plasmalogens | Antioxidant phospholipids critical for membrane stability and synaptic signaling. | Depletion observed in AD plasma and brain; correlated with oxidative stress markers. | Plasmalogen replacement restores antioxidant defenses and improves cognition in mice. | [28, 46, 50] |
| Phosphatidylcholine (PC) and Phosphatidylethanolamine (PE) | Maintain membrane curvature, vesicle fusion, and neurotransmission. | Loss of DHA-containing PC/PE species detected in postmortem AD cortex and plasma. | PUFA supplementation improves membrane dynamics and cognition in early AD. | [10, 34] |
| Apolipoprotein E (APOE) and lipid transport proteins | Mediate cholesterol/phospholipid transport and amyloid clearance. | APOE |
APOE-targeted therapies enhance lipid homeostasis and reduce A |
[15, 18, 45] |
| Sphingosine-1-phosphate (S1P) | Regulates neurogenesis, synaptic plasticity, and inflammation. | Reduced S1P levels in AD brain and CSF; linked to microglial dysfunction. | S1P receptor modulators improve cognition and reduce neuroinflammation. | [42, 43] |
| Fatty acids (DHA, EPA, ARA) | Structural components of neuronal membranes; precursors of pro- and anti-inflammatory mediators. | Reduced brain DHA in AD; supplementation improves synaptic function and reduces tau phosphorylation. | Omega-3 and ketogenic therapies improve energy metabolism and cognition. | [33, 49, 52] |
| Lipid droplets | Reflect glial metabolic stress; regulate lipid storage and immune responses. | LD-accumulating microglia exhibit impaired phagocytosis and chronic inflammation. | Restoring lipid droplet turnover improves glial function. | [10, 41, 44] |
| Gut microbiota-derived lipids | Influence bile acid metabolism, systemic inflammation, and brain lipid signaling. | AD-associated dysbiosis alters circulating bile acids and short-chain fatty acids. | Modulation via probiotics or diet improves cognition. | [47, 48] |
AD, Alzheimer’s disease; APP, amyloid precursor protein; CSF, cerebrospinal
fluid; CYP46A1, cholesterol 24-hydroxylase; LXR, liver X receptor; RXR, retinoid
X receptor; BACE1,
Since most circulating lipids do not enter the CNS due to the blood-brain barrier, autonomous lipid production, transport, and recycling processes assist the CNS to stay at homeostasis [39]. The production of cholesterol and phospholipids and their transport into neurons occurs through lipoprotein particle-containing apolipoproteins produced by astrocytes and transported to the neurons by receptor-mediated endocytosis [53]. ATP (adenosine triphosphate) binding cassette transporters such as ABCA1 (ATP-binding cassette subfamily A member 1) and ABCG1 (ATP-binding cassette subfamily G member 1) mediate cholesterol efflux and loading lipids onto apolipoproteins and have central roles in the maintenance of lipid balance across neural cell types [54]. Other than the transport of lipids, lipid storage is also another facet of CNS lipid homeostasis. Lipid droplets are cytoplasmic organelles that store neutral lipids and serve as dynamic stores that compensate for changes in lipid supply and safeguard cells against lipotoxic stress [55].
The lipid brain metabolism involves the well-coordinated mechanisms of
synthesis, turnover, and consumption. The cholesterol biosynthesis is regulated
by rate limiting enzymes and transcriptional regulators that are responsive to
cellular lipid requirements through the mevalonate pathway [56, 57]. Cholesterol
24-hydroxylase (CYP46A1) converts excess cholesterol to 24S-hydroxycholesterol,
and this allows the body to regulate the amount of cholesterol across the
blood-brain barrier [56]. Fatty acid synthesis is done by fatty acid synthase and
elongation-desaturation systems, and the breakdown of fatty acids is done by
There are several different types of neural cells with lipid metabolic programmes that are dependent on the CNS functions. Astrocytes are the main place of cholesterol production and lipid transport that provides neurons with the necessary lipids to turnover and maintain their membrane and synapses [59]. The neurons are extremely dependent on lipids delivered to support the rapid membrane restructuring and synaptic vesicle cycle [60]. It is observed that microglia change the dynamic organization of lipid metabolism depending on the environmental cues, which are indicators of the change in the cellular energetic and functional state [61]. As oligodendrocytes, which are highly cholesterol and sphingolipid-enriched, rely on a strong lipid biosynthesis to sustain and/or form myelin, their presence in the brain influences myelin regeneration [62]. Interconnectedness of these cell-type-specific lipid metabolic pathways is the basis of CNS structural integrity and functional resilience. Fig. 1 summarizes the lipid metabolic programs of neurons, astrocytes, microglia, and oligodendrocytes, and their roles in maintenance of synapses, neuroinflammation, and maintenance of myelin.
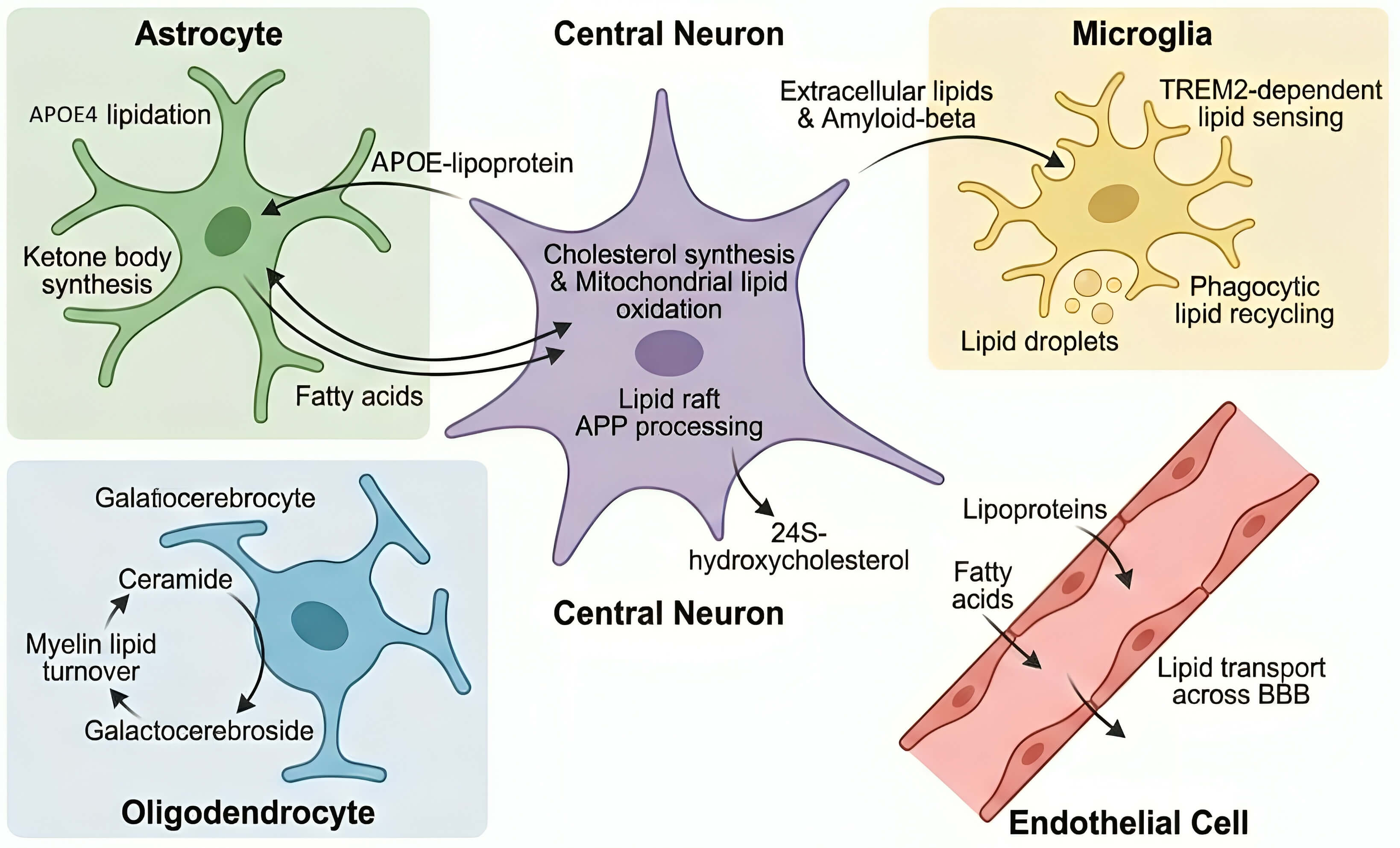 Fig. 1.
Fig. 1.
Cellular crosstalk of lipid metabolism in the Alzheimer’s brain. Astrocytes, neurons, microglia, oligodendrocytes, and endothelial cells coordinate lipid trafficking essential for brain homeostasis. Astrocytes supply cholesterol and fatty acids via APOE lipoproteins, while neurons regulate lipid oxidation and APP processing. Microglia sense extracellular lipids and amyloid through TREM2, forming lipid droplets during activation. Oligodendrocytes maintain myelin lipid turnover, and endothelial cells mediate lipid transport across the blood–brain barrier. Disruption of this lipid network contributes to neurodegeneration and inflammation in Alzheimer’s disease. TREM2, Triggering receptor expressed on myeloid cells 2; BBB, blood–brain barrier.
AD is a progressive neurodegenerative disease that is linked to the deposition
of A
The amyloid cascade hypothesis postulates that aberrant cleavage of APP by
Sphingolipid imbalance also regulates amyloidogenic mechanisms. Ceramide
stabilization makes BACE1 steady and increases its enzymatic performance. High
levels of ceramide species are observed in the preclinical AD and associated with
amyloid load and cognitive impairment [42, 43]. Ceramides, mechanistically, make
membranes stiffer and change endosomal trafficking, which promotes amyloidogenic
APP pathways. Notably, lipid changes can lead to dysfunction of A
Tau is a microtubule related protein that stabilizes the architecture of the
axonal cytoskeleton. During AD, tau hyperphosphorylates, dissociates off
microtubules and forms NFT [63]. Tau pathology can be modified by lipid
perturbations via kinase activation, oxidative stress, and membrane signaling
changes. The activation of the stress-responsive kinases such as glycogen
synthase kinase 3 beta (GSK3
AD is characterized by chronic neuroinflammation, which is mainly caused by the
activation of the microglial and astrocytic functions [65]. Metabolism of lipids
is critical in the establishment of the tone of inflammatory processes. Nuclear
factor kappa B (NF-
Excessive production of reactive oxygen species (ROS) and dysfunctional antioxidant defenses are the causes of oxidative stress. The polyunsaturated fatty acids in the neuronal membranes are also highly susceptible to peroxidation, which produces the reactive aldehydes like 4-HNE and malondialdehyde [67]. These products of lipid peroxidation alter the proteins of synaptic transmission and mitochondrial respiration. The lipid composition of the mitochondria plays a vital role in the bioenergetic stability. Anchor Electron transport chain complexes are anchored by cardiolipin, a mitochondria-specific phospholipid. The oxidative damage of cardiolipin interferes with the respiratory chain assembly, decreases the production of ATP and facilitates the release of cytochrome c [68]. Synaptic terminal energy breakdown is a precursor to neuronal loss and leads to cognitive impairment. The further effect of the ceramide accumulation is the impairment of the mitochondrial membrane integrity and the amplification of the apoptotic signaling pathways. A change in the sterol structure affects the fluidity of the mitochondrial membrane and respiratory efficiency, too [10]. Accordingly, lipid dysregulation is a feed-forward mechanism: oxidative damage to lipids causes lipid damage, lipid damage causes mitochondrial impairment, and mitochondrial impairment produces further ROS.
Loss of synapses is more strongly related to cognitive impairment than plaque or
tangle load [69]. The phospholipids and cholesterol in synaptic membranes are
very abundant in DHA, which control vesicle fusion, receptor movement and
dendritic spine structural formation [12, 40]. Membrane fluidity is reduced by
lowering DHA and plasmalogen loss, which inhibits the assembly of the soluble
N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex,
which inhibits neurotransmitter release, and reduces the plasticity of the
synapse. The N-methyl-D-aspartate (NMDA) and
These giant pathological characteristics of Alzheimer’s disease are described in section 3. In this paper, we consider the mechanisms by which disruptions in cholesterol transport, sphingolipid remodeling, phospholipid composition, and lipid mediated signalling have a direct effect on amyloid processing, tau phosphorylation, mitochondrial fitness, neuroinflammation, and synaptic resilience. These lipid-directed processes do not work in isolation, but they constitute feedback loops, which increase neurodegeneration. Fig. 2 shows the integrated molecular pathways connecting lipid dysregulation and amyloid processing, tau phosphorylation, mitochondrial dysfunction, oxidative stress, and neuroinflammation.
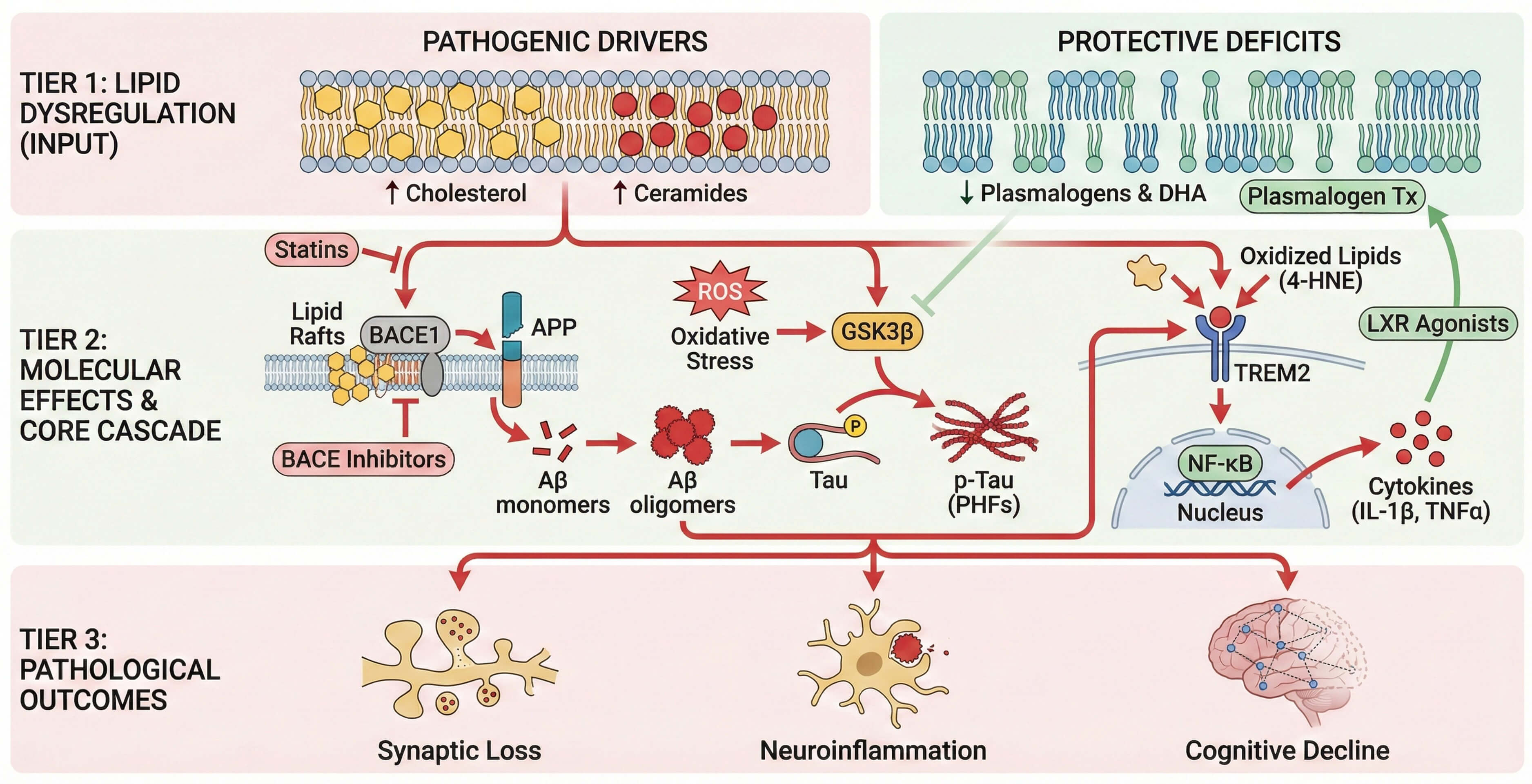 Fig. 2.
Fig. 2.
Mechanistic pathways linking lipid dysregulation to Alzheimer’s
pathology. Lipid imbalances such as increased cholesterol and ceramides or
reduced plasmalogens and DHA initiate amyloidogenic APP processing, tau
phosphorylation, and inflammation. Ceramides stabilize BACE1 and enhance
oxidative stress, while oxidized lipids activate microglial NF-
The distribution of cholesterol in the membranes of neurons is a vital regulator
of APP processing. More cholesterol in the membrane prefers
The metabolism of sphingolipids has a strong impact on the survival of neurons and inflammatory signaling. The sphingomyelin hydrolysis or de novo synthesis produces ceramides that aid amyloidogenic processing and at the same time stimulate the mitochondrial permeability, oxidative stress, and apoptotic signalling [42, 43]. Early lipidomic profiles in preclinical AD show that the species of ceramides such as C16:0 and C18:0 accumulate early [22]. These species disrupt membranes of mitochondria, elevate the production of reactive oxygen species and enhance inflammatory cascades. Conversely, the pro-survival sphingolipid metabolite, sphingosine-1-phosphate (S1P), is lower in AD, the loss of which hinders neurogenesis and synaptic plasticity [72]. Sphingomyelin to bioactive ceramide species transition is thus a directional metabolic imbalance, which strengthens amyloid toxicity and inflammatory susceptibility.
Membrane curvature, vesicle fusion, and receptor trafficking is supported by phospholipids which are phosphatidylcholine (PC), phosphatidylethanolamine (PE), and plasmalogens. Polyunsaturated PC and PE species depletion impairs the fluidity of the membrane and decreases the formation of synaptic vesicles in AD [22]. Reactive aldehydes produced through oxidative modification of phospholipids (4-hydroxynonenal) covalently modify APP and tau and promote aggregation propensity [10, 73]. The depletion of plasmalogens also impairs antioxidant buffering capacity, making one more vulnerable to lipid peroxidation. These structural changes of phospholipids destabilize synaptic membranes and provide a biochemical environment that promotes protein misfolding and neuronal stress.
Lipid rafts are used in receptor clustering and intra-cellular signaling.
Cholesterol and sphingolipid changes in composition impair raft integrity, which
influences the localization of NMDA receptors, Tropomyosin receptor kinase B
(Trk
Neuroinflammation and lipid metabolism are closely coupled with lipid-sensing receptors and bioactive lipid mediators. Toll-like receptor 2 (TLR2), TLR4, and the lipid-sensing receptor Triggering receptor expressed on myeloid cells 2 (TREM2) are activated by oxidized phospholipids and ceramides to induce the activation of microglia and the assembly of inflammasomes [65, 77]. TREM2 mutations affect lipid uptake and phagocytic capacity and cause the build-up of extracellular debris and increased inflammatory signaling. At the same time, the elimination of specialized pro-resolving lipid mediators based on omega-3 fatty acids impairs the resolution stage of inflammation, which prolongs chronic microglial activation [78]. Therefore, lipid imbalance not only initiates inflammatory activation but also inhibits its measurement, establishing a self-sustaining neuroimmune response cycle.
Lipid composition is very sensitive in mitochondria. Mitochondria-specific
phospholipid (cardiolipin) stabilizes respiratory chain complexes. Its oxidation
interferes with the efficiency of electron transport and favors the release of
cytochrome c [68]. An increase in the levels of ceramides in the mitochondrial
membranes increases permeability transition and apoptotic signaling, whereas a
breakdown in
The lipid dysregulation offers a mechanistic intermediation of the amyloid and
tau pathology. Ceramides and oxidized lipids stimulate oxidation-stimulated
lipids GSK3
The peripheral lipid metabolism and gut-derived metabolites have an impact on central lipid homeostasis. Gut microbiota control the bile acid composition and production of short-chain fatty acids that adjust the lipid metabolism in the system and neuroinflammatory tone [47]. Bile acids and trimethylamine-N-oxide (TMAO) cross the blood-brain barrier and encourage oxidative stress and microglial activation [48]. Lipid changes related to dysbiosis thus play roles in systemic-central metabolic interrelation with dietary habits and peripheral lipid dysbalance, being related to neurodegenerative susceptibility.
In addition to the individual lipid species, coordinated changes in the lipid pathways give information on the AD progression. Directional metabolic flux ratios, e.g., sphingomyelin-to-ceramide, are associated with cognitive deterioration [79]. Lipidomic analyses combined with other techniques have shown that cholesterol turnover, sphingolipid remodeling, and phospholipid oxidation are not cascades but networks of interrelated processes [10]. These perturbations at the pathway level affect the amyloid processing, phosphorylation of tau, and activation of inflammation. The mechanistic basis of metabolic intervention, along with stratification of precision, of lipid metabolism can be achieved by seeing lipid metabolism as a dynamic systems network, as opposed to a set of discrete abnormalities.
Genetic studies firmly position lipid metabolism at the core of late-onset Alzheimer’s disease. While mutations in APP, PSEN1 (Presenilin 1), and PSEN2 (Presenilin 2) underlie early-onset familial forms, most sporadic AD cases arise from polygenic risk networks that converge on cholesterol transport, membrane remodeling, endosomal trafficking, and microglial lipid sensing [8, 9, 15]. Rather than acting independently, these loci define interconnected lipid–immune–metabolic circuits that influence amyloid processing, tau vulnerability, and neuronal resilience. Genome-wide association studies consistently implicate APOE, ABCA1, ABCA7, TREM2, CLU (Clusterin), and SORL1 (Sortilin-related receptor 1) genes central to lipid transport or membrane dynamics. The convergence of risk alleles on lipid regulatory pathways supports the conceptualization of AD as a disorder of impaired lipid homeostasis embedded within inflammatory and mitochondrial stress networks.
APOE
Other susceptibility loci support the lipid regulation vicinity in AD. ABCA1 and
ABCG1 regulate cholesterol and phospholipid efflux that is essential in the
normal APOE lipidation. The decreased ABCA1 activity leads to the production of
lipidated APOE particles of poor quality and to disruption of cholesterol
distribution in the membrane [84, 85]. ABCA7 combines lipid translocation and
microglial phagocytosis; the loss-of-function variants disrupt the uptake of
A
Metabolic conditions and exposure to the environment make significant
adjustments to genetic susceptibility. Lipid metabolism is a primary nexus by
which diet, insulin sensitivity, and endocrine contributions to the disease path
are mediated and especially in APOE
Multi-omics studies indicate that AD is caused by the concerted breakdown of lipid-immune-metabolic modules, but not individual gene mutations. The co-expression network of APOE, ABCA1, SORL1, and CLU is associated with inflammatory and mitochondrial genes in large data sets, such as Alzheimer’s Disease Neuroimaging Initiative (ADNI) [10, 22]. The modules linked to ceramide are also related to complement activation and synaptic vulnerability, whereas the decrease in phosphatidylcholine and plasmalogen species correlates with the inhibition of oxidative phosphorylation genes. These synchronized changes can be identified before more complex neurodegeneration, and this shows that lipid network destabilization is a systems-level process. Lipid metabolism is also further tied to the process of gene regulation by epigenetic means. The histone acetylation under the influence of acetyl-CoA affects the transcription of lipid and mitochondrial genes [93]. The products of lipid peroxidation can alter transcriptional regulators, which strengthens expression designations by oxidative stress [94]. Lipid-sensitive nuclear receptors are activated to remodel chromatin accessibility and orchestrate the cholesterol efflux and immune regulation. There is another modulatory layer of sex hormones. The estrogen controls the expression of APOE and ABCA1 and modulates the activity of the mitochondrial function [95]. Reduced estrogen levels can thus increase lipid regulation in those genetically predisposed, which explains sex-specific differences in AD risk. Taken together, these results place AD as a disease of genetically regulated lipid network instability, which is influenced by metabolic and epigenetic contexts.
Lipidomics using mass spectrometry can now be used to quantitatively profile hundreds of lipid species in brain tissue, cerebrospinal fluid (CSF) and plasma. Rather than being alone, lipid abnormalities are coordinated in the plasma of Alzheimer’s disease to occur in conjunction with phospholipids, sphingolipids, sterols, and oxidized products. Notably, a lot of these changes occur on preclinical or prodromal stages, meaning that lipidomic disturbance is a manifestation of early metabolic disequilibrium but not end-stage neuronal death. Modern lipidomics, as opposed to simple measurements, focuses on the direction of pathways and network-level understanding. Combination with genetic and proteomic data is gradually pioneering lipid profiles as the mechanistic outputs of membrane remodeling, mitochondrial strain, inflammatory stimulation, and lipid transport proficiency.
Consistent with a large-scale decrease in PC and PE species enriched with DHA, such as PC 38:6 and PE 40:6, postmortem lipidomic analyses of AD cortex and hippocampus show such decreases [20, 28]. Plasmalogens, especially plasminyl-PE species, are significantly decreased, that weaken the stability of the membrane curvature and antioxidant buffering activity. Ceramide species, including Cer d18:1/16:0 and Cer d18:1/18:0, on the contrary, are increased [22, 42, 50], indicating increased sphingomyelin hydrolysis and transition to pro-apoptotic lipid signaling. The sphingomyelin-to-ceramide ratio is consequently shifted in favor of pathway activation that is congruent with inflammatory and mitochondrial stress. The total cholesterol can be quite stable, but the decreased level of 24S-hydroxycholesterol and the enhanced level of oxidized sterols such as 7-ketocholesterol show evidence of the disrupted cholesterol turnover and oxidative modification [96]. Spatial lipidomics proves the idea of enrichment of ganglioside GM1 in amyloid plaques and peri-plaque areas, accumulation of ceramide to show the anatomically localized lipid remodeling related to the pathological lesions [97]. Together, brain tissue lipidomics can show a predictable course of lipid degradation, plasmalogen disappearance, ceramide increase, and sterol oxidation that may be observed before severe neuronal damage.
Peripheral lipid profiling detects consistent patterns that are related to cognitive decline and phenoconversion. Lipidomic profiles reported by Huynh K et al. [98] (2020), encompassing multiple lipid classes such as phospholipids and sphingolipids, are associated with both prevalent Alzheimer’s disease and future disease risk. Ceramide species are one of the strongest predictors of the plasma. The high Cer d18:1/16:0 and Cer d18:1/18:0 levels are associated with hippocampal atrophy and progressive acceleration [99]. Notably, the pathway-level measures are better than those based on individual species. A decrease in the ratios of sphingomyelin-to-ceramide, especially SM d18:1/24:0, reflects a higher activity of the sphingomyelinase and metabolic directionality [51]. AD plasma raises the levels of oxidized phospholipids including POVPC (1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine) and PGPC (1-palmitoyl-2-glutaroyl-sn-glycero-3-phosphocholine) [100], which is associated with systemic oxidative stress and the activation of inflammation. Reduced levels of 24S-hydroxycholesterol and disturbed levels of desmosterol in CSF indicate impaired neuronal cholesterol metabolism [101]. Even though the effect of peripheral lipids on predictive performance can be enhanced by systemic metabolism, their combination with genotype and longitudinal imaging data is more effective.
The genotype of APOE plays a significant role in altering the
systemic and central lipidomic architecture that can modify the baseline lipid
transport efficiency and the susceptibility to metabolic stress.
Lipid species do not tend to work individually. Systems-level analyses of ADNI
and associated multi-cohort data reveal that lipid changes come together in
coordinated metabolic modules, which combine inflammatory, mitochondrial, and
complement pathways [30, 103]. Such modules are more biologically resolved than
individual biomarkers and represent pathway-wide dysregulation. A reproducible
module is based on ceramide enrichment and complement cascade proteins (C1q, C3),
and inflammatory mediators. Higher levels of ceramide species are associated with
higher complement activation levels and microglial inflammatory signals, which
are predictors of faster cognitive impairment and increased cortical atrophy.
Mechanically, the presence of ceramide facilitates rigidification of membranes,
the activation of the inflammasome, and mitochondrial strain, which strengthens
complement-mediated synaptic pruning. A second modular association is between
loss of polyunsaturated phosphatidylcholine and plasmalogen species in connection
with decreased oxidative phosphorylation proteins and abnormal acylcarnitine.
This trend indicates that there is impaired membrane remodeling and reduced
mitochondrial
Regardless of technological progress, the clinical application of lipidomics is limited by the lack of methodological consistency. The pre-analytical variables, such as diet, fasting condition, and sample manipulation, cause a lot of variability [104]. Differences between analytical procedures, ionization platforms, and internal standardization cause problems in cross-study comparability. Biological heterogeneity also makes interpretation difficult. Lipid baseline is dependent on age, sex, metabolic comorbidities, and APOE genotype. Clinical implementation is restrained by the lack of standardized reference ranges. Efforts are being undertaken to standardize nomenclature, reporting, and data processing systems like the Lipidomics Standards Initiative [104]. A reproducible panel of pathways at the panel level, and established cutoffs will be required to move lipidomics past exploratory profiling and establish lipidomics as the precision diagnostic and therapeutic monitoring instrument in AD.
The identification of lipid dysregulation as a mechanistic cause of AD has broadened therapeutic conceptualization of amyloid and tau as sole targets to include therapies that alter cholesterol flux, membrane composition, inflammatory lipid mediators and cerebral energy metabolism. Some of these strategies are outlined below and include pharmacologic manipulation of lipid metabolism, metabolic reprogramming through dietary interventions and new forms of precision based on lipidomic and genetic stratification. The relationship between pharmacologic modulation, dietary methods, lipidomic stratification, and organelle-targeting at the precision framework is summarized in Fig. 3.
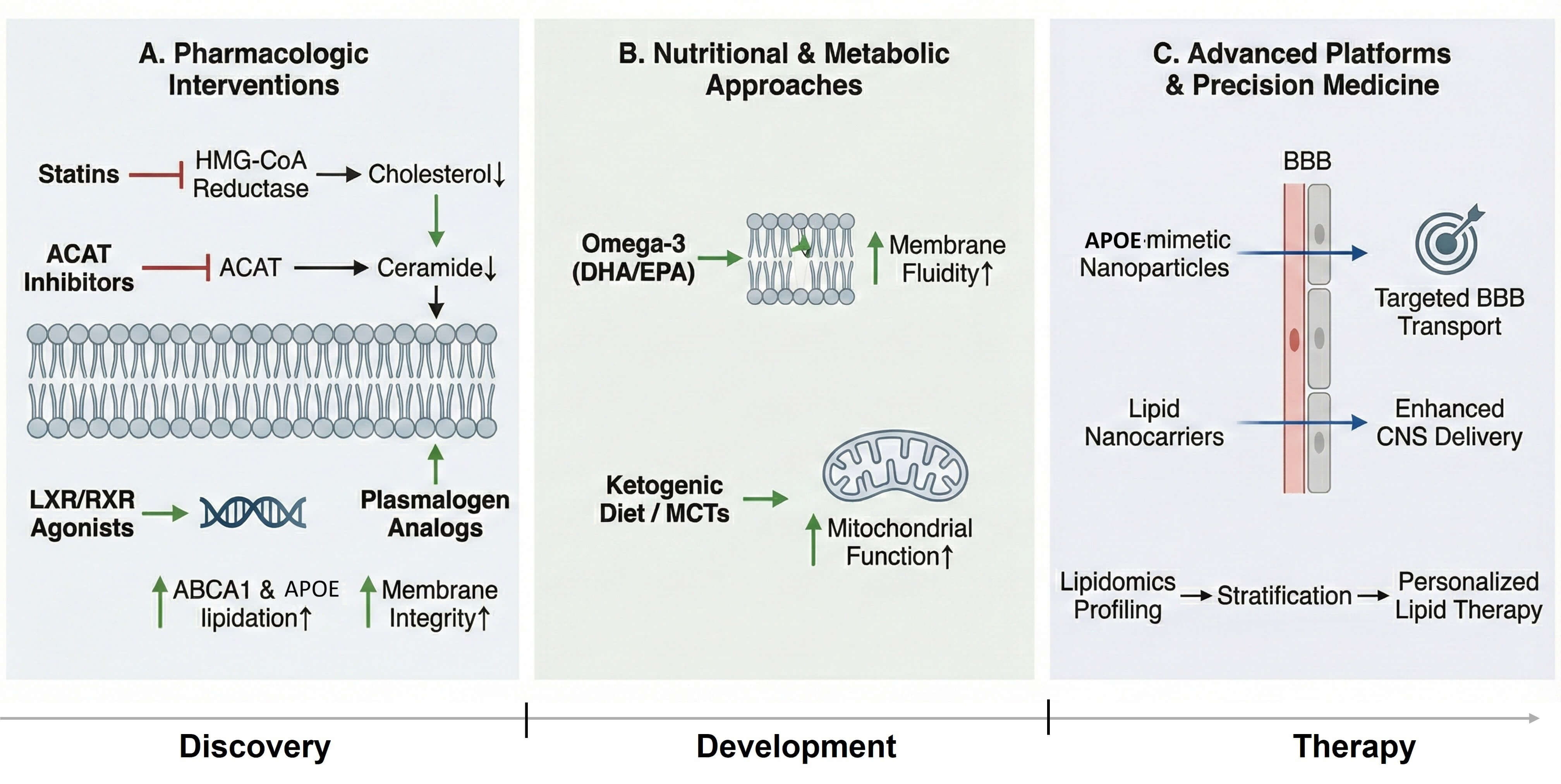 Fig. 3.
Fig. 3.
Translational and therapeutic landscape of lipid-targeted
interventions. Lipid-based therapeutic strategies span pharmacologic,
nutritional, and nanotechnological approaches. Statins, ACAT inhibitors, and
LXR/RXR agonists modulate cholesterol and ceramide metabolism. Nutritional
interventions such as omega-3 supplementation and ketogenic diets improve
membrane and mitochondrial function. Advanced lipid nanocarriers and APOE-mimetic
nanoparticles enhance targeted brain delivery, while lipidomic profiling supports
personalized treatment strategies in Alzheimer’s disease. Black arrows represent
biochemical or mechanistic pathways. Green arrows indicate beneficial or
enhancing effects, whereas red blunt-ended lines denote inhibition. Upward
(
Statins, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors,
have also been suggested as possible neuroprotective agents due to their effects
on the lowering of peripheral cholesterol and their possible impact on cerebral
cholesterol turnover. Epidemiological research indicates that statins can prevent
the onset of dementia over the long term, and greater evidence indicates this
effect is stronger in APOE
Cholesterol efflux, APOE lipidation and inflammatory tone are controlled by LXRs
and RXRs. Expressions of ABCA1 and ABCG1 are augmented by pharmacologic
activation, which boosts the export of cholesterol and possibly facilitates
A
The goal of ketogenic dietary interventions is to reverse the cerebral
hypometabolism in AD by enhancing the levels of circulating
The Mediterranean and MIND diets, which are based on monounsaturated fats,
omega-3 polyunsaturated fatty acids, antioxidants, and polyphenol-rich diets, are
linked with decreased AD risk and slower cognitive deterioration [48].
Mechanistically, oleic acid of olive oil and omega-3 fatty acids are
membrane-stabilising in nature and can re-establish lipid mediator balance
towards anti-inflammatory eicosanoids. The components of Polyphenols and
antioxidants minimize lipid peroxidation and oxidative alteration of membrane
phospholipids. The MIND diet also focuses on factors that are associated with
vascular and metabolic well-being (leafy greens, berries, whole grains) and
minimizes the intake of saturated fats, which can indirectly lower the levels of
insulin resistance and lipid-induced inflammatory priming. Nevertheless,
causality is challenging to determine because of confounding lifestyle factors,
whereas evidence in the form of randomized trials is still scarce. Individual
effects of genotypes can be different; APOE
Neuroprotectin D1 (NPD1) is a DHA-derived specialized pro-resolving mediator
that is produced through the action of 15-lipoxygenase and serves as a bioactive
signal to counteract the action of A
Membrane phospholipid-released arachidonic acid (AA) is a key precursor of
bioactive eicosanoids that regulate inflammatory and excitotoxic signalling.
Higher activity of PLA2 (Phospholipase A2) and free AA was also reported in
AD-affected areas [116]. AA oxidation through cyclooxygenase (COX) and
lipoxygenase (LOX) produces prostaglandins and leukotrienes that encourage
microglial action, cytokine production, and oxidative stress, which amplifies inflammatory
responses that depend on NF-
Lipid responder phenotypes, where the exceptional pathway-wide lipid dysfunction
can be used to predict treatment responses, are supported by integrating the
lipidomics of plasma with genetic stratification [10, 22]. High levels of
ceramides and low levels of sphingomyelin-ceramide ratios accompanied by
complement activation and rapid progression are known to be associated with a
ceramide-dominant inflammatory phenotype, often based on APOE
Peroxisomes control very-long-chain fatty acid
The glymphatic system helps in the exchange of CSF-interstitial fluid and the
clearance of soluble metabolites. Although it is best examined in the context of
A
Despite the fast development of lipidomics and neurobiology, some important gaps are still present in the definition of the role of lipid dysregulation in AD onset and progression. Recent evidence incriminates lipid pathways in amyloid processing, tau susceptibility, neuroinflammation, as well as organelle stress, yet a host of conceptual and methodological constraints still impede mechanistic inference and clinical translation. One major question that is yet to be answered is causality. Several lipidomic discoveries are based upon cross-sectional, case-control comparisons, and it is hard to determine whether lipid changes are initiating events, early parallel processes, or secondary effects of proteinopathy. Stage-resolved biospecimen collection combined with longitudinal cohorts is vital to establishing a temporal order. Recent methods, such as single-cell and spatial lipidomics, can be used to identify the localization of lipid remodeling to cell types and pathological niches at different stages of the disease [22]. Complementary stable isotope tracer experiments may help clarify the direction-dependent pathway flux and at which point the cholesterol turnover, sphingolipid remodeling and phospholipid depletion are directionally perturbed over the course of progression [10].
The second difficulty is biological heterogeneity. There are diverse lipid signatures among individuals depending on the genotype, metabolic status, diet, vascular risk, and endocrine factors. The modification influence of the APOE genotype is well-identified, whereas other loci (e.g., ABCA7, TREM2, CLU) probably determine lipid-mediated susceptibility due to variations in lipid transportation and immune-metabolic adjustment [124]. Sex-specific lipid regulation (estrogen-linked lipid transport and mitochondrial activity) can also be a source of different risk patterns and that ought to be included in stratified analyses [30, 125]. Subgroup-specific lipid architectures are going to have to be solved with large, ethnically diverse cohorts with harmonized phenotyping in the future.
A major impediment to the validation of biomarkers is methodological standardization. Variations in sample treatment, extraction chemistry, setting of a platform, and reference standards reduce cross-study comparability and complicate cutoff definitions actionable in the clinic. Efforts like the Lipidomics Standards Initiative (LSI) and wider metabolomics quality-control work to coordinate nomenclature, reporting and analytical pipelines [104]. Advances in this field will be determining in bringing lipidomics to the discovery to clinical phases.
Multi-omics integration is becoming a requirement, particularly under systems biology. Lipid pathways are tightly interacting with proteomic, transcriptomic, mitochondrial and immune networks and single-modality biomarkers may not reflect this coupled biology. Integrative modelling systems that relate lipid patterns with genetic framework and inflammatory systems will probably offer more powerful and biologically comprehensible stratification [10]. Models generated by machine learning can help determine predictive signatures and dominant pathway phenotypes; however, must be validated in external cohorts and confounded by medications, diet, and metabolic disease [125].
Therapeutic translation is at an early phase. Several lipid-modulating approaches demonstrate preclinical efficacy, but human trials are typically constrained by safety, lack of interaction with brain targets, and intervention timing as well as disease heterogeneity [126]. A further step in this direction will probably be brain selective modulators, pathway-informed combination regimens and biomarker directed trial designs that will enrich against people with the desired lipid aberration. Lipidomics combined with genotype and metabolic profiling could be used to create precision strategies to enhance the detection of therapeutic signals and to help define responder subgroups [34, 48, 49]. Another boundary is that of peripheral-central coupling, especially the gut-brain axis. The effects of gut microbiota-derived metabolites on systemic lipid metabolism and neuroimmune tone are possible as well as central lipid remodeling [47, 48]. Precision of metabolite-lipid pathway interactions can provide available biomarkers and intervention targets, which could supplement brain-directed therapies.
Lastly, sealing the clinical implementation gap will need synchronous work to create normative lipidomic reference ranges, validate pathway-level panels in diverse populations, and use lipidomic endpoints in longitudinal studies and clinical trials. When rigorously standardized, stage-resolved validated, and pathway-stratified studies of lipid metabolism, lipid metabolism can become more than an associative marker to provide a mechanistically based platform of early disease detection and target therapy in AD.
AD is showing growing acceptance as a pathology of lipid homeostasis disturbance that overlaps with amyloid pathogenesis, tau pathology, neuroinflammation and/or mitochondrial malfunction. The converged genetic risk factors that include APOE, ABCA1, ABCA7 and TREM2 are implicated in transporting lipids and remodeling of the membrane, which emphasizes the centrality of lipid regulation in disease susceptibility. Lipidomic analyses also indicate a perturbation of pathways such as ceramide enrichment, plasmalogen loss, and sterol peroxidation that predict overt neurodegeneration and are coordinated by metabolic changes. An intervention mechanism with mechanical foundations is provided by therapeutic interventions that regulate lipid metabolism in response to nuclear receptor modulation to ketogenic interventions and pro-resolving lipid mediators. A lipid framework at the systems level thus offers a harmonizing framework that bridges genetic vulnerability, metabolic strain and synaptic hardening, and is a promising future direction of diagnosis and disease-modulating treatments in AD.
JB contributed to the conceptualization, methodology, investigation, data curation, visualization, and drafting of the original manuscript. SK—literature review, investigation, data curation, methodology, drafting of supporting sections, and critical review and editing of the manuscript. KS contributed to supervision, validation of scientific content, methodology, and critical revision and editing of the manuscript. DS—supervision, methodology, validation, and critical review and editing of the manuscript. All authors read and approved the final version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Authors acknowledge Vinayaka Mission’s Research Foundation-Deemed to be University (VMRF-DU) and Vinayaka Mission’s Medical college and Hospital, Karaikal, Puducherry, India for the financial support (Seed Money Ref no: VMRF/Research/SeedMoney/AY2025-26/VMMC/1).
This research received no external funding.
The authors declare no conflict of interest. This research was funded by the Vinayaka Mission’s Research Foundation, the institution where several authors are affiliated. The authors declare that the funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
During the preparation of this work, the authors used Consensus AI tool to assist in the identification and summarization of relevant peer-reviewed literature. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.