


 , Yang Pan 1, Min Shu 1, Li Zou 1,*
, Yang Pan 1, Min Shu 1, Li Zou 1,*
1 Department of Neurology, Zhongnan Hospital of Wuhan University, 430071 Wuhan, Hubei, China
Abstract
Lysosomal-associated protein transmembrane (LAPTM) family members—including LAPTM4A, LAPTM4B, and LAPTM5—are key regulators of lysosomal integrity, autophagy-lysosome flux, lipid metabolism, and immune responses. Dysregulation of LAPTM proteins contributes to neurological disorders such as Alzheimer’s disease, Parkinson’s disease, ischemic stroke, and gliomas, affecting neuronal survival, glial homeostasis, neuroinflammation, and tumor progression. In this review, we summarize recent insights into the structural features and molecular mechanisms of LAPTM proteins in the nervous system and highlight their therapeutic potential in promoting protein aggregate clearance, mitigating oxidative stress, regulating microglial polarization, and enhancing tumor immunotherapy. Future research integrating gene therapy, small-molecule modulators, multi-omics profiling, and advanced delivery platforms may enable translation of LAPTM-targeted interventions into clinical practice, offering new avenues for diagnosis, prognosis, and treatment of neurological diseases.
Keywords
- lysosomal membrane proteins
- autophagy
- lysosomes
- neurodegenerative diseases
- glioma
- brain ischemia
- genetic therapy
- molecular targeted therapy
Neurological disorders span a broad spectrum, including neurodegenerative diseases, cerebrovascular disorders, brain tumors, and neuroinflammatory conditions. These diseases, which have complex and multifactorial etiologies, represent a major threat to global health and a significant cause of impaired quality of life. The accelerating aging of the global population is driving a continuous increase in the incidence of Alzheimer’s disease (AD) and Parkinson’s disease (PD), posing a major public health challenge worldwide [1]. Meanwhile, primary brain tumors such as gliomas are characterized by high invasiveness and poor prognosis, placing a heavy burden on patients [2]. Despite remarkable progress in molecular pathology and neuroscience, effective therapeutic strategies directly targeting the root causes of these diseases remain limited. Therefore, elucidating the molecular mechanisms underlying neurological disorders and identifying novel biomarkers and therapeutic targets have become critical research priorities.
Lysosomes serve as the primary degradative and recycling compartments within cells and are essential for maintaining homeostasis in the nervous system. Due to their post-mitotic state and high metabolic demands, neurons heavily rely on an efficient autophagy–lysosome system to clear abnormal protein aggregates and damaged organelles, thereby ensuring long-term survival and synaptic integrity [3]. Thus, lysosomal dysfunction — leading to impaired protein balance, mitochondrial damage, and bioenergetic deficits — is considered a hallmark of neurodegeneration and a key contributor to broader neuropathology [4, 5]. Therefore, elucidating the molecular regulators of lysosomal homeostasis holds promise for advancing our understanding of neurological disease mechanisms and identifying novel therapeutic targets.
The lysosomal-associated protein transmembrane (LAPTM) family represents an important group of multi-pass transmembrane proteins localized to lysosomal or late endosomal membranes. Among them, LAPTM4B and LAPTM5 are the most extensively studied [6]. LAPTM4B contains canonical lysosomal targeting signals and localizes predominantly to late endosomes/lysosomes [7]. Functionally, it regulates lysosomal stability, promotes autophagosome maturation, and enhances cellular adaptation to metabolic stress [8]. Gene amplification of LAPTM4B has been frequently observed in various human cancers, where it promotes autophagy flux and contributes to tumor cell survival under metabolic and chemotherapeutic stress [9]. Moreover, LAPTM4B can interact with the epidermal growth factor receptor (EGFR) pathway, enhancing downstream signaling such as phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB/AKT), thereby supporting tumor cell survival and proliferation [10].
In contrast, LAPTM5 is predominantly expressed in hematopoietic and immune cells
[11]. It plays a key role in regulating immune and inflammatory responses. A study
has shown that LAPTM5 positively regulates nuclear factor
Emerging evidence implicates that LAPTM family members in neurological disorders
through several mechanisms. First, the impaired autophagy–lysosome pathway is
recognized as a hallmark of AD and PD [13]. As regulators of lysosomal membranes,
LAPTM proteins may influence autophagy flux, lysosomal degradation efficiency,
and the clearance of pathogenic proteins, ultimately affecting neuronal survival
and death. Second, in central nervous system tumors, overexpression of LAPTM4B has been associated with enhanced tumor proliferation, angiogenesis, and poor patient outcomes, highlighting its potential as a therapeutic target [14]. Third,
given the central role of neuroinflammation in neurological diseases,
LAPTM5—through its regulatory functions in immune and microglial cells—may
offer novel molecular insights for modulating neuroinflammatory processes [15].
Furthermore, LAPTM proteins have been associated with the regulation of critical
signaling pathways, including mammalian target of rapamycin (mTOR), PI3K/AKT, and
NF-
Despite growing recognition of the importance of LAPTM proteins in cancer biology and immunology, research on their roles in the nervous system is still in its infancy. The cell-type–specific functions of LAPTM family members in neurons, glial cells, and immune cells remain poorly defined. Moreover, the causal links between LAPTM-mediated regulation of autophagy–lysosome pathways and neurological pathology require further experimental and clinical validation. Systematic investigation of LAPTM molecular functions in neurological contexts will not only enhance mechanistic understanding but may also yield novel diagnostic biomarkers and therapeutic strategies.
In this review, we summarize the molecular functions of LAPTM family proteins, focusing on their roles in neurodegenerative diseases, cerebrovascular disorders, central nervous system tumors, and neuroinflammatory conditions. We also discuss potential underlying mechanisms and highlight the translational implications of targeting LAPTM proteins in clinical practice. By synthesizing current research progress, this review aims to provide theoretical support and inspiration for future basic and clinical studies.
A comprehensive literature search was conducted using PubMed (https://pubmed.ncbi.nlm.nih.gov/), Web of Science (https://www.webofscience.com), and Scopus (https://www.scopus.com) databases from inception through May 2025. Search terms included “LAPTM” OR “lysosomal-associated protein transmembrane” combined with “nervous system” OR “neurodegenerative” OR “neurological” OR “brain” OR “neuron” OR “glia” OR “stroke” OR “glioma” OR “Alzheimer” OR “Parkinson”. Additional searches were performed for each family member (LAPTM4A, LAPTM4B, LAPTM5) combined with relevant disease terms. Reference lists of identified articles were manually screened for additional relevant publications. Studies were included if they provided mechanistic insights, clinical associations, or therapeutic implications of LAPTM proteins in nervous system contexts. Review articles, editorials, and non-English publications were excluded from primary analysis but were consulted for background information.
From a clinical perspective, the significance of LAPTM research extends beyond basic mechanistic understanding. First, LAPTM family members, particularly LAPTM4A and LAPTM4B, have emerged as valuable diagnostic and prognostic biomarkers in gliomas and other central nervous system (CNS) malignancies, with their expression levels strongly correlating with tumor grade, chemotherapy resistance, and overall patient survival. Second, LAPTM-targeted therapies represent a promising avenue for conditions with limited treatment options, including glioblastoma multiforme and treatment-refractory neurodegenerative diseases. Third, the development of non-invasive imaging modalities targeting LAPTM proteins enables real-time assessment of disease status and therapeutic efficacy, facilitating personalized treatment strategies. These translational opportunities underscore the urgent need for a comprehensive investigation of LAPTM biology in the nervous system.
The LAPTM family is an evolutionarily conserved group of small membrane proteins. These proteins share common structural features and transport signals, enabling them to interact with ubiquitin ligases, adaptor proteins, and endosomal sorting mechanisms. Although most studies on LAPTM proteins have focused on their role in cancer [17, 18], recent evidence indicates that these proteins are also key regulators of lysosomal signal transduction, autophagy, lipid processing, and receptor transport, all of which are crucial for the homeostasis of nerve cells. Dysfunction of lysosomal-related pathways is a recognized feature of neurodegenerative diseases, suggesting that LAPTM proteins are also significant outside the field of oncology [19]. This section will review the molecular structure, transport mechanisms, and signaling roles of LAPTM family members, with a focus on their correlations in neurons, glial cells, and microglia. Integrating LAPTM-mediated lysosomal regulation with neurotrophic signals, amino acid sensing, and immune homeostasis reveals their emerging importance in physiological brain function and neurodegenerative diseases.
LAPTM4A, also known as lysosomal-associated protein transmembrane 4 alpha, is a four-transmembrane-spanning protein encoded by the LAPTM4A gene, primarily residing in late endosomes and lysosomes [20]. Its structure includes four hydrophobic transmembrane domains, with the N-terminus and C-terminus exposed to the cytosol, facilitating interactions with cytoplasmic proteins [21]. The protein contains several putative lysosomal targeting signals, such as dileucine motifs and tyrosine-based sorting signals, which direct its trafficking from the Golgi apparatus to endolysosomal compartments. Additionally, LAPTM4A possesses proline-tyrosine (PY) motifs in its C-terminal region, enabling recruitment of ubiquitin ligases like neural precursor cell expressed developmentally downregulated 4-1 (NEDD4-1), which promotes ubiquitination and subsequent lysosomal degradation pathways [22]. This structural feature is conserved across species, underscoring its evolutionary importance in membrane dynamics.
In the context of the nervous system, LAPTM4A’s structural features are particularly relevant to neuronal homeostasis. Its four-transmembrane domain architecture enables stable integration into lysosomal and late endosomal membranes, which is essential for the high-turnover protein degradation demands of post-mitotic neurons. The cytosolic PY motifs facilitate recruitment of ubiquitin ligases such as NEDD4-1, promoting the ubiquitin-dependent degradation of misfolded proteins and damaged organelles that would otherwise accumulate in neurons. Furthermore, LAPTM4A’s interaction with human organic cation transporter 2 (hOCT2) has implications for neuronal detoxification, as this transporter mediates the clearance of cationic neurotoxins and metabolic byproducts from brain tissue. In glial cells, particularly microglia, LAPTM4A modulates polarization states, with its presence promoting macrophage 2 (M2) anti-inflammatory phenotypes that support neuronal survival and tissue repair.
Functionally, LAPTM4A acts as a regulator of endosomal sorting and lysosomal function. It interacts with various transporters and enzymes, modulating their activity and localization. For instance, LAPTM4A associates with hOCT2, influencing its endocytic recycling and thereby affecting cellular uptake of cationic compounds [23]. This interaction enhances hOCT2 trafficking between the plasma membrane and intracellular compartments, impacting drug transport and cellular detoxification. In lipid metabolism, LAPTM4A modulates globotriaosylceramide (Gb3) synthase activity post-transcriptionally, reducing Gb3 levels in lysosomes and preventing accumulation-related pathologies [24]. Loss of LAPTM4A leads to decreased Gb3 synthase function, highlighting its role in glycosphingolipid homeostasis. In immune and cancer contexts, LAPTM4A influences macrophage polarization. It promotes M2 polarization of tumor-associated macrophages (TAMs), enhancing tumor proliferation and invasion in gliomas by fostering an immunosuppressive microenvironment [25]. This is achieved through signaling pathways that support anti-inflammatory responses. Furthermore, LAPTM4A is involved in toxin resistance mechanisms; genome-wide clustered regularly interspaced short palindromic repeats (CRISPR) screens have identified it as a factor in Shiga toxin and ricin susceptibility, where it facilitates toxin entry or processing in endosomes [26]. Structurally, its transmembrane domains enable ceramide interactions, potentially stabilizing lysosomal membranes and preventing leakage.
LAPTM4A’s expression correlates with prognosis in gliomas, including glioblastoma and low-grade glioma, where higher levels are associated with advanced grades, histological aggressiveness, and poorer outcomes [27]. Mechanistically, it interacts with microRNAs and circular RNAs, such as in the hsa_circ_0042260/miR-4782-3p/LAPTM4A axis, which promotes hepatocellular carcinoma progression by enhancing cell proliferation and invasion [28]. In ubiquitination pathways, LAPTM4A undergoes self-ubiquitination via ring finger protein 152 (RNF152), contributing to a conserved endosomal sorting complexes required for transport (ESCRT)-dependent internalization process that regulates membrane protein turnover [22]. Overall, LAPTM4A’s structure supports its multifaceted functions in trafficking, metabolism, and disease modulation, making it a key player in endolysosomal biology.
LAPTM4B, or lysosomal-associated protein transmembrane 4 beta, is a tetratransmembrane protein overexpressed in various cancers, including hepatocellular carcinoma, where it serves as a prognostic marker [29]. Structurally, LAPTM4B comprises four transmembrane helices, with N- and C-terminal domains in the cytosol, and includes PY motifs that interact with ubiquitin ligases like NEDD4 [30]. It exists in two isoforms, LAPTM4B-35 and LAPTM4B-24, differing in their N-terminal regions, with LAPTM4B-35 being the predominant form in tumors [31]. The protein localizes to late endosomes and lysosomes, where its transmembrane domains facilitate lipid binding, particularly ceramides and phosphatidylinositol 4,5-bisphosphate [32].
Functionally, LAPTM4B regulates lysosomal ceramide export, preventing ceramide accumulation and promoting cell death resistance in tumors [33]. It interacts with ceramide to facilitate its removal from late endosomes, independently of sphingomyelin synthase activity, thus maintaining lysosomal integrity [33]. In autophagy, LAPTM4B promotes lysosomal acidification and maturation, essential for autophagosome-lysosome fusion, and its amplification enhances tumor growth by supporting autophagic flux [34]. LAPTM4B also recruits the leucine transporter L-type amino acid transporter 1 (LAT1)-4F2hc to lysosomes, enabling leucine uptake and mechanistic target of rapamycin complex 1 (mTORC1) activation, which drives cell proliferation under nutrient stress [35].
In cancer progression, LAPTM4B enhances multidrug resistance by promoting cell
migration via integrin
In hepatocellular carcinoma, LAPTM4B is regulated by histone deacetylase 2 (HDAC2) and ETS translocation variant 1 (ETV1), enhancing autophagy and stemness [40]. It counteracts ferroptosis by stabilizing solute carrier family 7 member 11 (SLC7A11), reducing lipid peroxidation [41]. Serum LAPTM4B levels serve as diagnostic markers for breast cancer [42]. In glioblastoma, high LAPTM4B-35 expression correlates with poor prognosis and angiogenesis [14]. Amplification with tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ) contributes to chemotherapy resistance. In nasopharyngeal and bladder cancers, LAPTM4B drives migration and invasion [43].
Its role in lung adenocarcinoma prognosis underscores its oncogenic potential [44]. In B-cell acute lymphoblastic leukemia, LAPTM4B promotes progression and immune evasion. Polymorphisms associate with breast cancer risk, though not always significantly [45]. In colorectal cancer, overexpression predicts poor outcomes [46]. Noninvasive imaging targets LAPTM4B for hepatocellular carcinoma detection [47]. Overall, LAPTM4B’s structure enables its broad regulatory functions in lysosomal metabolism, signaling, and cancer biology.
LAPTM5, lysosomal-associated protein transmembrane 5, is a five-transmembrane-domain protein with three PY motifs and one ubiquitin-interacting motif (UIM), enabling interactions with substrates and ubiquitin ligases. Its structure includes cytosolic loops and termini, facilitating supramolecular assemblies and really interesting new gene (RING) domain interactions for cellular signaling [11]. The PY and UIM motifs are critical for recruiting enzyme 3 (E3) ligases like NEDD4, promoting protein degradation [48].
Functionally, LAPTM5 regulates lysosomal function by interacting with
mucolipin-1 (MCOLN1), modulating ion channels and preventing mucolipidosis type
IV-like phenotypes [49]. It promotes proinflammatory signaling in macrophages,
enhancing cytokine secretion via NF-
In cancers, LAPTM5 drives malignant phenotypes; in breast cancer, forkhead box protein 3 (FOXP3)-regulated LAPTM5 enhances proliferation, migration, and invasion [52]. In clear cell renal cell carcinoma, it promotes progression via elevated expression. LAPTM5 confers resistance to lenvatinib in hepatocellular carcinoma through CRISPR-identified mechanisms [53]. Zinc finger with KRAB and SCAN domains 5 (ZKSCAN5) activates LAPTM5 by recruiting SET domain-containing protein 7 (SETD7) for histone H3 lysine 4 trimethylation (H3K4me3) modification, supporting pancreatic ductal adenocarcinoma growth [54, 55]. In multiple myeloma, LAPTM5 promotes venetoclax resistance by enhancing myeloid cell leukemia 1 (MCL-1) stability [56]. It confers cisplatin resistance in non-small cell lung cancer by suppressing ferroptosis via cathepsin D release [57].
In kidney injury, LAPTM5 induces tubular senescence via WW domain-containing E3 ubiquitin protein ligase 2 (WWP2)/Notch receptor 1 (Notch1) signaling, exacerbating fibrosis [58]. Its role in autophagy activation links it to disease processes, including immunity and inflammation [11]. In chronic rhinosinusitis, LAPTM5 is part of M2 macrophage signatures [59]. Overall, LAPTM5’s unique structure underpins its functions in degradation, signaling, and pathology, with broad implications for therapeutic intervention. The structural features, expression patterns, and primary biological functions of the three LAPTM family members are comparatively summarized in Table 1.
| Feature | LAPTM4A | LAPTM4B | LAPTM5 |
| Structure | 4 TM domains; PY motifs | 4 TM domains; PY motifs; 2 isoforms | 5 TM domains; 3 PY motifs; UIM |
| Localization | Late endosomes/lysosomes | Late endosomes/lysosomes | Lysosomes; late endosomes |
| CNS Expression | Neurons; microglia; glioma cells | Neurons; glioma cells; overexpressed in tumors | Predominantly microglia; immune cells |
| Key Functions | Endosomal sorting; lipid metabolism; microglial polarization | Ceramide export; mTORC1 activation; autophagy promotion | Proinflammatory signaling; lysosomal degradation; TCR regulation |
| Disease Links | Glioma; PD (mitophagy); neuroinflammation | Glioblastoma; AD (DLB); hearing loss | I/R injury; AD; ICH; glioblastoma |
| Therapeutic Role | Immunotherapy target (inhibition enhances anti-PD1) | Biomarker; therapeutic target; anti-ferroptosis | Neuroprotection target (overexpression beneficial) |
Note: LAPTM, lysosomal-associated protein transmembrane; TM, transmembrane; PY, proline-tyrosine; UIM, ubiquitin-interacting motif; CNS, central nervous system; mTORC1, mechanistic target of rapamycin complex 1; TCR, T cell receptor; PD, Parkinson’s disease; AD, Alzheimer’s disease; DLB, dementia with Lewy bodies; I/R, ischemia-reperfusion; ICH, intracerebral hemorrhage; PD1, programmed cell death protein 1.
Mounting evidence implicates lysosomal dysfunction as a convergent pathological mechanism in a wide spectrum of neurological disorders, including neurodegenerative diseases, ischemic brain injury, brain tumors, and neuroinflammatory conditions [5]. The LAPTM family is pivotal in regulating lysosomal functions, including acidification, membrane trafficking, autophagy, and the degradation of cellular components. As central regulators of lysosomal function, the LAPTM family is therefore increasingly implicated in the maintenance of nervous system cell homeostasis [49]. In neural tissues, where post-mitotic neurons and dynamic glial cells must sustain long-term functionality amidst high metabolic demands, lysosomal integrity prevents the accumulation of damaged proteins, lipids, and organelles that could precipitate neurodegeneration [60]. Dysfunctions in LAPTM proteins have been linked to various neurological conditions, such as ischemia-reperfusion injury, intracerebral hemorrhage, gliomas, and age-related hearing loss, underscoring their role in neural homeostasis. LAPTM family members interact with ubiquitin ligases and ion channels, modulating endolysosomal trafficking and ion homeostasis, which are critical for neuronal survival and glial support functions [61]. In neurons, these proteins facilitate the clearance of misfolded proteins via autophagy-lysosome pathways, mitigating proteotoxic stress [62]. In glial cells, they regulate inflammatory responses and metabolic crosstalk with neurons, ensuring tissue homeostasis [63]. Emerging evidence from mouse models and human studies highlights how LAPTM dysregulation disrupts these processes, leading to apoptosis, inflammation, and impaired neural circuits. This section delves into the specific contributions of LAPTM proteins to homeostasis in neurons and glial cells, drawing on recent research to elucidate their mechanisms and implications for neurological health.
Neurons, as highly specialized, post-mitotic cells, rely heavily on efficient lysosomal systems to maintain homeostasis by degrading damaged organelles and proteins through autophagy [62]. LAPTM proteins enhance this process by regulating lysosomal membrane stability and trafficking. For instance, LAPTM5 deficiency in mouse models exacerbates cerebral ischemia/reperfusion injury, leading to increased neuronal apoptosis and inflammation via heightened c-Jun N-terminal kinase (JNK)/p38 signaling and impaired lysosomal function [64]. Overexpression of LAPTM5 mitigates these effects, suggesting its protective role in neuronal stress responses [65]. In intracerebral hemorrhage models, LAPTM5 is upregulated in neurons surrounding hematomas, where it modulates lysosomal degradation to prevent secondary brain injury and support neuronal survival [66]. This protein interacts with apoptosis signal-regulating kinase 1 (ASK1) to suppress pro-apoptotic pathways, maintaining neuronal homeostasis under hemorrhagic stress.
LAPTM4A also contributes to neuronal homeostasis by interacting with transporters like hOCT2, which regulates cationic compound uptake in the brain, potentially aiding in detoxification and ion balance [23]. In Parkinson’s disease models, LAPTM4A is involved in mitochondrial-lysosomal crosstalk, where miR-5701 modulates its expression to regulate neuronal death by preventing excessive mitophagy and lysosomal overload [28]. Furthermore, LAPTM4A protects neurons from prion protein fragment-induced apoptosis, possibly through pathways mediated by imatinib (STI571) that stabilize lysosomal membranes and enhance autophagic clearance [67]. Its role in glycosphingolipid metabolism helps prevent lipid accumulation that could disrupt neuronal membrane integrity and signaling [24].
LAPTM4B supports neuronal homeostasis by facilitating ceramide export from lysosomes, preventing ceramide-induced apoptosis and maintaining autophagic flux [33]. In neuronal ceroid lipofuscinosis, LAPTM4B mutants contribute to abnormal triaging of misfolded proteins, leading to lipofuscin accumulation and disrupted proteostasis [68]. Copy number variations in LAPTM4B are associated with dementia with Lewy bodies, where they impair lysosomal degradation of alpha-synuclein, exacerbating neuronal toxicity [69]. In age-related hearing loss, LAPTM4B influences auditory neuron homeostasis by regulating gene expression profiles linked to lysosomal function [70].
Beyond individual members, the LAPTM family collectively ensures neuronal ion homeostasis by interacting with channels like mucolipin-1, preventing lysosomal storage disorders with neurological manifestations. In neuroblastomas, LAPTM5 promotes spontaneous regression through lysosomal destabilization and autophagic cell death, highlighting its role in neuronal progenitor homeostasis. LAPTM5 also regulates T-cell receptor degradation in neurons, indirectly supporting synaptic plasticity and homeostasis [71]. In spinocerebellar ataxia models, related transmembrane proteins underscore the importance of lysosomal integrity for Purkinje neuron survival [72]. Overall, LAPTM proteins safeguard neuronal homeostasis by optimizing lysosomal degradation, mitigating stress-induced apoptosis, and supporting metabolic balance, with deficiencies leading to vulnerability in ischemic, hemorrhagic, and degenerative states.
The intricate mechanisms involve LAPTM5’s PY motifs facilitating ubiquitin-dependent sorting, essential for neuronal protein turnover [12]. In low-grade gliomas, LAPTM expression correlates with prognosis, implying a role in neuronal-glial interactions during oncogenesis [8]. LAPTM4A’s upregulation in gliomas promotes tumor progression but may reflect compensatory mechanisms for neuronal homeostasis [25]. In chronic pain models, LAPTM5 in dorsal horn neurons modulates gene networks for inflammatory homeostasis. These findings emphasize LAPTM’s multifaceted contributions to neuronal stability.
Glial cells, including astrocytes, microglia, and oligodendrocytes, provide structural, metabolic, and immune support to neurons, with lysosomal functions critical for phagocytosis, cytokine regulation, and myelin maintenance. LAPTM proteins in glia regulate these processes to sustain nervous system homeostasis. LAPTM4A loss inhibits M2 polarization of tumor-associated macrophages in glioblastoma, promoting immune activation and enhancing anti-programmed cell death protein 1 (PD1) therapy [25]. This suggests LAPTM4A modulates microglial phenotypes, balancing pro- and anti-inflammatory states for glial homeostasis. Chemokine-like factor (CKLF) induces microglial activation via defective mitophagy, involving LAPTM4A in lysosomal dysfunction and neuroinflammation. In gliomas, high LAPTM4A expression correlates with poor prognosis, indicating disrupted glial homeostasis in malignancy [73].
LAPTM4B in glial cells regulates exosome release induced by ceramide, influencing intercellular communication and homeostasis in the tumor microenvironment [74]. In glioblastoma, LAPTM4B-35 overexpression is a prognostic factor, linked to glial proliferation and angiogenesis [14]. Its role in chemotherapy resistance in breast carcinomas extends to glial tumors, where copy number gains impair response to treatments. In hypomyelinating disorders, related transmembrane (TMEM) proteins like TMEM106B highlight LAPTM4B’s potential in oligodendrocyte myelin homeostasis [75].
LAPTM5 is prominently expressed in microglia, where it enhances proinflammatory
signaling via NF-
A critical unresolved question concerns the seemingly paradoxical dual roles of
LAPTM5 in the nervous system. On one hand, LAPTM5 deficiency exacerbates cerebral
ischemia-reperfusion injury and increases neuronal apoptosis, suggesting a
neuroprotective function. On the other hand, LAPTM5 promotes proinflammatory
signaling through NF-
In astrocytes, LAPTM proteins facilitate lactate shuttling and cholesterol homeostasis, essential for neuronal support. Aquaporin-4 interactions in gliomas suggest LAPTM involvement in water-ion balance [79]. In neurodegenerative models, LAPTM5 promotes glial phagocytosis of neuronal debris, maintaining tissue integrity. Overall, LAPTM proteins in glial cells ensure lysosomal efficiency, inflammation control, and metabolic support, vital for nervous system homeostasis.
To elaborate further, in microglia, LAPTM5’s role in chronic rhinosinusitis parallels its function in brain inflammation, where it marks M2 signatures for resolution [11]. In renal cell carcinoma models, LAPTM5’s proliferative effects may be analogous to glial activation in brain tumors. LAPTM4B’s association with inositol status implies links to neural signaling homeostasis [53]. In frontotemporal lobar degeneration, TMEM-related proteins underscore lysosomal roles in glial pathology. Semaphorin family interactions with immune regulation highlight cross-talk between nervous and glial systems. Neurotrophins in energy homeostasis involve glial support, potentially modulated by LAPTM. Lysophospholipid receptors in glia regulate motility and differentiation. Innate lymphoid cells’ interaction with the nervous system implicates glial immune homeostasis. N-methyl-D-aspartate (NMDA)-independent long-term potentiation (LTP) in glial-neuronal circuits may involve lysosomal stability. Lysosome-associated membrane protein (LAMP) in neurite outgrowth suggests analogous roles for LAPTM in glial processes. In mucolipidosis type IV, LAPTM-mucolipin interactions affect glial ion channels. These diverse mechanisms illustrate LAPTM’s integral role in glial homeostasis [80]. These multifaceted molecular functions of the LAPTM protein family in neural cell homeostasis and their dysregulation in neurological disorders are illustrated in Fig. 1.
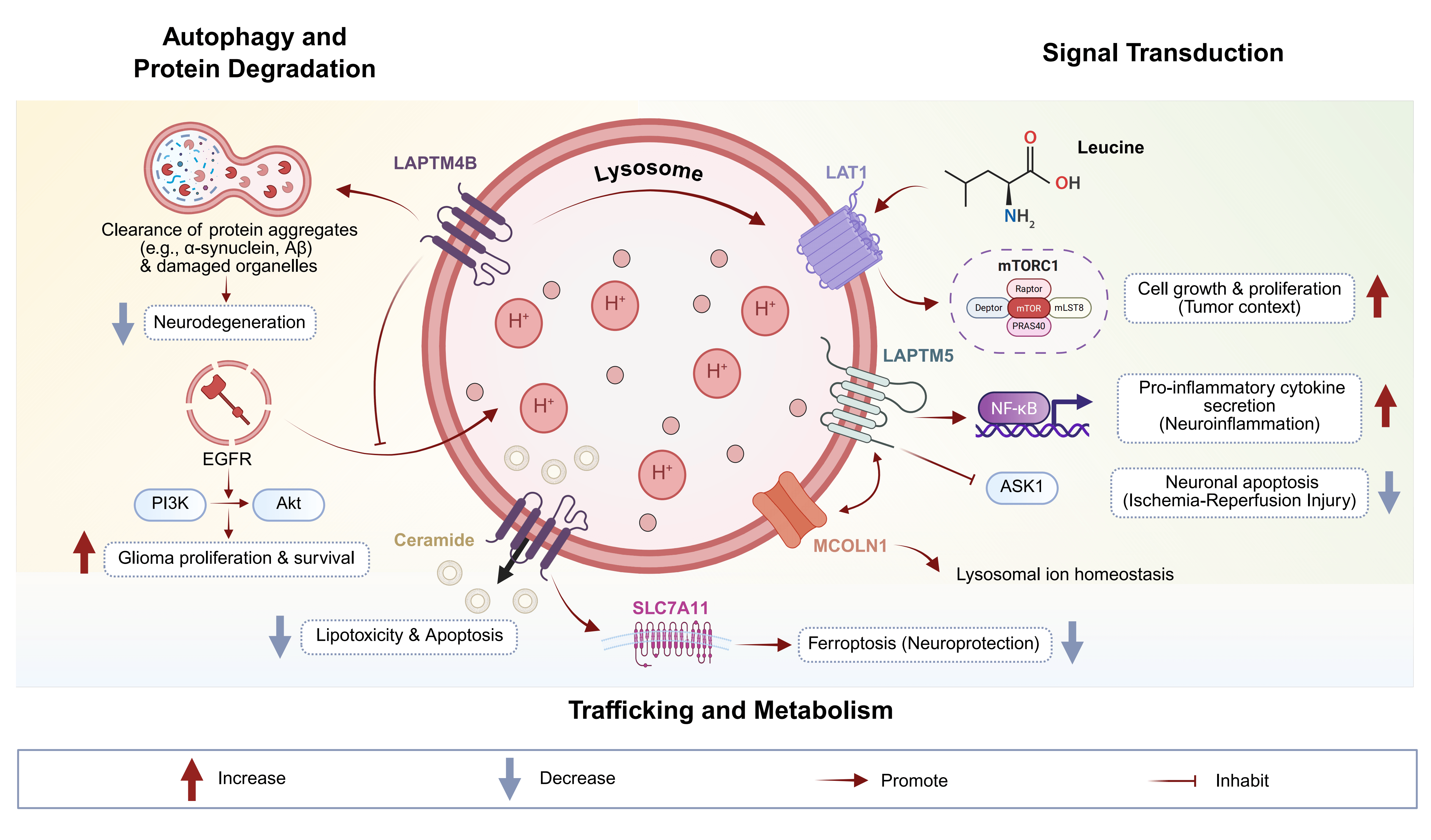 Fig. 1.
Fig. 1.
Molecular functions of the LAPTM protein family in neural cell
homeostasis and neurological disorders. LAPTM family proteins are key regulators
of lysosomal function that sustain neural health through multiple interconnected
mechanisms. They maintain cellular homeostasis by facilitating the autophagic
clearance of pathogenic protein aggregates (e.g., alpha-synuclein, A
To fully appreciate the functional significance of LAPTM proteins in neurological disorders, it is essential to position them within the broader landscape of lysosomal regulators. Lysosomes are governed by a complex network of membrane proteins, transcription factors, and ion channels that collectively maintain organelle homeostasis. Understanding how LAPTM family members compare and interact with other key lysosomal regulators—including LAMP1/LAMP2, transcription factor EB (TFEB), and MCOLN1—provides critical context for their therapeutic targeting.
Lysosome-associated membrane proteins 1 and 2 (LAMP1 and LAMP2) are the most abundant lysosomal membrane proteins, constituting approximately 50% of all lysosomal membrane proteins [81]. While both LAMP and LAPTM families localize to lysosomal membranes, their functions are distinct yet complementary. LAMP proteins primarily serve structural roles, protecting the lysosomal membrane from degradation by luminal hydrolases through their heavily glycosylated luminal domains. In contrast, LAPTM proteins function predominantly as regulators of membrane trafficking, substrate transport, and signaling pathway modulation. For instance, whereas LAMP2A serves as the receptor for chaperone-mediated autophagy (CMA), LAPTM4B facilitates ceramide export and mTORC1 activation [33, 35]. In the context of neurological diseases, LAMP2 deficiency causes Danon disease with prominent neurological manifestations, while LAPTM dysregulation contributes to more diverse pathologies, including gliomas and ischemic injury. Importantly, LAPTM5 interacts functionally with LAMP1 to stabilize lysosomal membranes and sustain autophagic flux, as demonstrated in cisplatin resistance models [57]. This functional interplay suggests that LAPTM and LAMP proteins may represent complementary therapeutic targets for lysosomal modulation in neurological disorders.
TFEB is the master regulator of lysosomal biogenesis and autophagy gene expression, coordinating the transcription of over 500 genes in the CLEAR (Coordinated Lysosomal Expression and Regulation) network [13]. While TFEB operates at the transcriptional level to increase lysosomal capacity, LAPTM proteins function post-translationally to modulate lysosomal membrane dynamics and cargo processing. These two regulatory layers are interconnected: LAPTM4B-mediated mTORC1 activation influences TFEB phosphorylation and nuclear translocation, thereby affecting lysosomal gene expression [35]. Conversely, TFEB activation may upregulate LAPTM expression as part of the coordinated lysosomal response. In neurodegenerative diseases, TFEB activation has shown therapeutic promise for enhancing aggregate clearance [13]. Combining TFEB activators with LAPTM modulators could potentially produce synergistic effects by simultaneously increasing lysosomal capacity (via TFEB) and optimizing lysosomal function (via LAPTM). Furthermore, as discussed in Section 4.1.1, Sirtuin 1 (SIRT1) deacetylates TFEB to promote its nuclear translocation, suggesting a SIRT1-TFEB-LAPTM regulatory axis that warrants therapeutic exploration [82, 83].
Mucolipin-1, encoded by the MCOLN1 gene, is a lysosomal cation channel essential for calcium signaling, membrane fusion, and lysosomal trafficking [49]. Mutations in MCOLN1 cause mucolipidosis type IV, a lysosomal storage disorder with severe neurological manifestations. Critically, LAPTM family members physically interact with mucolipin-1 to regulate lysosomal ion homeostasis [49]. This interaction is particularly relevant for neural cells, where precise calcium signaling is essential for synaptic function and neuronal survival. LAPTM5’s interaction with MCOLN1 modulates lysosomal calcium release, affecting downstream processes including autophagosome-lysosome fusion and TFEB activation. Disruption of LAPTM-MCOLN1 interactions may contribute to the lysosomal dysfunction observed in neurodegenerative diseases. Therefore, therapeutic strategies targeting LAPTM proteins must consider their effects on MCOLN1-dependent ion homeostasis to avoid unintended consequences on neuronal calcium signaling.
The positioning of LAPTM proteins within the lysosomal regulatory network has important implications for therapeutic development. Unlike TFEB, which broadly upregulates lysosomal function, or LAMP proteins, which primarily serve structural roles, LAPTM family members offer opportunities for more selective modulation of specific lysosomal processes—such as ceramide metabolism (LAPTM4B), inflammatory signaling (LAPTM5), or immune cell polarization (LAPTM4A). This functional specificity may enable more precise therapeutic interventions with fewer off-target effects compared to broader lysosomal modulators. However, the interconnected nature of lysosomal regulation also means that LAPTM-targeted therapies may produce cascading effects on TFEB activity, MCOLN1 function, and overall lysosomal homeostasis, necessitating careful evaluation in preclinical models.
In nervous system pathologies, LAPTM proteins influence neuroinflammation,
apoptosis, and tumor progression through interactions with ubiquitin ligases, ion
channels, and signaling pathways like NF-
LAPTM proteins’ structural features, such as multiple transmembrane domains and PY motifs, enable their localization to lysosomes and interactions with key molecules like ceramides and transporters, influencing disease progression [76]. In neurodegenerative contexts, LAPTM5 acts as a putative risk gene, co-expressing with established AD genes in microglial networks [84]. LAPTM4A modulates mitochondrial-lysosomal crosstalk in Parkinson’s models, regulating neuronal death [85]. These interactions underscore the family’s broad relevance in CNS pathologies, where lysosomal dysfunction is a common hallmark. Clinical study reveals correlations between LAPTM expression and disease prognosis, suggesting potential biomarkers [86]. For example, in low-grade gliomas, LAPTM genes are diagnostic markers. Overall, LAPTM’s involvement spans inflammatory, ischemic, and oncogenic processes in the nervous system. The specific roles and molecular mechanisms of LAPTM proteins across various neurological conditions are detailed in Table 2 (Ref. [14, 23, 25, 27, 64, 65, 66, 68, 69, 70, 72, 73, 74, 77, 81, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93]).
| Disease category | Specific disease | LAPTM member | Key mechanisms | Functional outcome | Key references |
| Neurodegenerative Diseases | Alzheimer’s Disease | LAPTM5 | Co-expression with TYROBP, CSF1R in microglial networks; modulation of amyloid beta response | Putative AD risk gene; inflammatory response regulation | [84, 90, 91] |
| Parkinson’s Disease | LAPTM4A | miR-5701-mediated regulation; mitochondrial-lysosomal crosstalk; VCP modulation | Neuronal death regulation; mitophagy defects | [85, 93] | |
| Dementia with Lewy Bodies | LAPTM4B | Copy number variations; impaired alpha-synuclein degradation | Exacerbated neuronal toxicity; disrupted proteostasis | [69] | |
| Neuronal Ceroid Lipofuscinosis | LAPTM4B | Abnormal triaging of misfolded proteins | Lipofuscin accumulation; disrupted proteostasis | [68] | |
| Cerebrovascular Diseases | Ischemic Stroke (I/R Injury) | LAPTM5 | ASK1 interaction; JNK/p38 signaling suppression; lysosomal membrane stabilization | Neuroprotection; reduced apoptosis and inflammation | [64, 65] |
| LAPTM4A | Microglial M2 polarization; mitophagy regulation; ion balance modulation | Reduced neuroinflammation; enhanced survival | [23, 25, 73] | ||
| LAPTM4B | Ceramide export; exosome release regulation | Prevention of lipid-induced apoptosis | [74, 87] | ||
| Intracerebral Hemorrhage | LAPTM5 | ASK1 suppression; lysosomal degradation modulation | Prevention of secondary brain injury; neuronal survival | [65, 66] | |
| CNS Tumors | Glioblastoma | LAPTM4A | M2 TAM polarization via NF-κB; immunosuppressive microenvironment | Tumor progression; poor prognosis; immunotherapy target | [25, 81, 88, 89] |
| LAPTM4B-35 | VEGF upregulation; angiogenesis promotion; autophagy enhancement | Tumor proliferation; chemoresistance; poor prognosis | [14, 87] | ||
| LAPTM5 | CD40 crosstalk; microglial gene expression | Tumor invasion; temozolomide resistance | [77] | ||
| Low-Grade Glioma | LAPTM4A/4B | Prognostic correlation with tumor grade | Diagnostic/prognostic biomarkers | [27, 86] | |
| Other Conditions | Neuropathic Pain | LAPTM5 | Interaction with CD68, C1QC in dorsal horn PPI networks | Neuroinflammation-mediated chronic pain | [92] |
| Age-Related Hearing Loss | LAPTM4B, LAPTM5 | Lysosomal function regulation in auditory neurons and glial networks | Cochlear homeostasis; auditory neuron survival | [70] | |
| Spinocerebellar Ataxia | Related TM proteins | Lysosomal integrity for Purkinje neurons | Early glial activation; neurodegeneration | [72] |
Note: TYROBP, tyrosine kinase-binding protein; CSF1R, colony-stimulating factor-1 receptor; VCP, valosin-containing protein; JNK, c-Jun N-terminal kinase; M2, macrophage 2; TAM, tumor-associated macrophage; VEGF, vascular endothelial growth factor; CD40, cluster of differentiation 40; C1QC, complement C1q C-chain; PPI, protein-protein interaction.
Ischemia-reperfusion (I/R) injury in the brain, often resulting from stroke, involves oxidative stress, inflammation, and apoptosis, where lysosomal integrity plays a protective role. LAPTM5 deficiency exacerbates cerebral I/R injury by promoting neuronal apoptosis and inflammation through enhanced JNK/p38 signaling and impaired lysosomal function [64]. In mouse models, LAPTM5 knockout leads to larger infarct sizes, worsened neurological deficits, and increased proinflammatory cytokine levels post-I/R. Overexpression of LAPTM5 mitigates these effects by suppressing ASK1-mediated pathways, highlighting its anti-apoptotic function in neurons during reperfusion stress. This protein interacts with lysosomal components to maintain acidification and autophagic flux, preventing the release of cathepsins that exacerbate tissue damage.
LAPTM4A contributes to I/R resilience by regulating microglial activation and mitophagy. In chemokine-like factor 1 (CKLF1)-induced models, LAPTM4A deficiency inhibits defective mitophagy, reducing microglial proinflammatory responses and neuroinflammation associated with I/R [73]. This suggests LAPTM4A promotes M2 polarization in microglia, fostering a protective environment during reperfusion [25]. Study shows that LAPTM4A interacts with transporters to modulate ion balance, mitigating calcium overload in ischemic neurons [23]. Its expression is upregulated in I/R-affected brain regions, correlating with survival outcomes.
LAPTM4B’s role in I/R involves ceramide metabolism, where it facilitates ceramide export from lysosomes, preventing lipid-induced apoptosis in neurons. In neuroblastoma models under hypoxic stress mimicking I/R, LAPTM4B regulates exosome release, potentially clearing toxic aggregates from ischemic areas [87]. Deficiency in LAPTM4B leads to accumulated ceramides, exacerbating mitochondrial dysfunction and oxidative stress during reperfusion. Therapeutic targeting of LAPTM4B could enhance lysosomal stability, reducing I/R injury severity.
Collectively, LAPTM proteins protect against I/R by optimizing lysosomal degradation and modulating inflammation, with potential for gene therapy interventions. Clinical correlations in stroke patients show lower LAPTM5 levels associated with poor prognosis, emphasizing their biomarker potential. Future studies may explore LAPTM agonists to mitigate I/R damage.
To elaborate, in cerebral I/R, LAPTM5’s interaction with CD1e in immune cells influences antigen presentation, affecting post-ischemic inflammation. This partnership stabilizes lysosomal membranes, preventing leakage during reperfusion. In related models, LAPTM5’s upregulation in response to neuronal apoptosis activates microglial clearance mechanisms, aiding recovery. LAPTM4A’s role in Parkinson’s-like stress conditions parallels I/R, where it regulates valosin-containing protein (VCP) and ATPase H+ transporting vacuolar subunit D1 (ATP6V0D1) for mitochondrial-lysosomal balance. These mechanisms highlight LAPTM’s multifaceted protection against I/R-induced neurodegeneration.
Nervous system tumors, including gliomas and glioblastomas, are characterized by aggressive growth and resistance to therapy, where LAPTM proteins promote oncogenesis through enhanced autophagy and immune evasion [94]. LAPTM4A is a robust biomarker in gliomas, with high expression correlating with advanced grades, histological aggressiveness, and poor prognosis. Loss of LAPTM4A inhibits M2 polarization of TAMs in glioblastoma, promoting immune activation and enhancing anti-PD1 therapy efficacy [88]. This protein fosters an immunosuppressive microenvironment by supporting TAM infiltration and tumor proliferation. In low-grade gliomas and glioblastomas, LAPTM4A expression is linked to patient age, tumor grade, and survival, making it a prognostic indicator [89].
LAPTM4B is a novel prognostic factor for glioblastoma, with the LAPTM4B-35 isoform strongly associated with tumor proliferation, angiogenesis, and poor outcomes. Overexpression of LAPTM4B-35 correlates with increased vascular endothelial growth factor levels, promoting neovascularization in tumors [14]. In glioblastoma cells, LAPTM4B enhances sensitivity to olaparib by disrupting lysosomal function when combined with selinexor (KPT330), suggesting therapeutic synergies. Its role in ceramide-induced exosome release facilitates tumor-microenvironment communication, aiding metastasis in nervous system cancers.
LAPTM proteins collectively enhance tumor resistance and invasion in nervous system malignancies, with targeting strategies showing promise for improving treatment outcomes. For example, knockdown of LAPTM4A in glioblastoma models reduces invasion and boosts immunotherapy response. Clinical data indicate LAPTM expression as a predictor of recurrence in gliomas [14].
Expanding on mechanisms, LAPTM4A’s promotion of M2 TAMs in glioblastoma involves
NF-
Beyond the specified sub-sections, LAPTM’s roles extend to other nervous system diseases. In Alzheimer’s disease, LAPTM5 is correlated with severity in microglia, acting via gene networks with tyrosine kinase-binding protein (TYROBP) and colony-stimulating factor-1 receptor (CSF1R). Genetic variability in LAPTM5 influences amyloid beta response, affecting AD risk [84]. LAPTM5 is highlighted as a putative AD risk gene in longevity studies, co-expressing with integrin subunit alpha M (ITGAM) and leukocyte immunoglobulin like receptor B4 (LILRB4) [90]. In integrated analyses, diet, apolipoprotein E (APOE) genotype, and sex affect immune networks involving LAPTM5 in AD models [91]. Application of weighted co-expression network analysis identifies LAPTM5 as a key gene in AD through inflammatory responses.
In neuropathic pain, LAPTM5 is a crucial node in gene expression profiles of the rat dorsal horn, interacting with CD68 and complement C1q C-chain (C1QC) in protein-protein interaction (PPI) networks [92]. This suggests LAPTM5 contributes to chronic constriction injury-induced pain via neuroinflammation. In Parkinson’s disease, LAPTM4A modulates neuronal death through miR-5701, affecting VCP and LAPTM4A transcripts. LAPTM4A’s involvement in mitophagy defects links it to PD pathology [93].
With the continuous deepening understanding of the mechanism of action of the LAPTM family in the nervous system, the therapeutic potential of this class of lysosomal transmembrane proteins in various neurological diseases is increasingly prominent. They regulate various core processes such as the autophagy-lysosome pathway, lipid metabolism, ionic homeostasis, and inflammatory response, involving neurodegenerative diseases, cerebral ischemia-reperfusion injury, glioma and other central nervous system tumors, as well as neuroinflammation and other pathological processes. Research on LAPTM not only helps to reveal the disease mechanism but may also provide new targets for drug development, gene therapy, immune intervention and precision medicine.
LAPTM4B and LAPTM5 play crucial roles in regulating lysosomal acidification, membrane stability, and autophagosome-lysosome fusion [13]. In neurodegenerative diseases, pathological protein aggregation and mitochondrial damage are common. Targeting and enhancing the function of LAPTM can promote the clearance of abnormal proteins and damaged organelles, thereby delaying the progression of the disease. Studies have shown that knocking down LAPTM5 exacerbates cerebral ischemia-reperfusion injury [64], while its overexpression can significantly improve neuronal survival rate and inhibit inflammation [65]. This finding suggests that enhancing LAPTM activity through gene delivery or small molecule agonists may become a novel intervention strategy for stroke and ischemic brain injury.
LAPTM4B promotes the transport of ceramides within nerve cells and maintains lipid homeostasis, preventing apoptosis caused by lipid overload [74]. Additionally, the latest research suggests that LAPTM4B can inhibit ferroptosis by stabilizing SLC7A11 [41]. This mechanism may have potential protective effects in the context of ischemia-reperfusion and neurodegenerative diseases, where oxidative stress is prevalent. Targeted regulation of the LAPTM4B-SLC7A11 axis is expected to become a new approach for future therapies against oxidative stress and ferroptosis.
Notably, the anti-aging gene SIRT1 represents a promising upstream regulator that may modulate LAPTM-mediated pathways. SIRT1 has been extensively documented to regulate autophagy, lysosome function, and exosome secretion through its deacetylase activity on transcription factors and autophagy-related proteins [96, 97]. In the context of neurological diseases, SIRT1 activation prevents amyloid beta aggregation, reduces oxidative stress, inhibits neuronal apoptosis, and suppresses glioma progression [82, 98]. Furthermore, SIRT1 plays a pivotal role in immune regulation and autoimmunity, which may intersect with LAPTM5-mediated inflammatory responses [83, 99]. Given the shared functional targets of SIRT1 and LAPTM proteins in lysosomal integrity and autophagy flux, future therapeutic strategies may benefit from combinatorial approaches that target both pathways. SIRT1 activators, such as resveratrol and its derivatives, could potentially enhance LAPTM-mediated neuroprotection by reinforcing the autophagy-lysosome axis and mitigating oxidative damage [100]. The interplay between SIRT1 and LAPTM warrants further investigation to establish optimal therapeutic regimens.
The success of LAPTM-targeted therapies may critically depend on the coordinated modulation of SIRT1 activity. SIRT1 serves as a primary control point for lysosomal integrity through its deacetylation of TFEB and other transcription factors that govern lysosomal biogenesis and autophagy gene expression. In neurodegenerative diseases where enhanced autophagy flux is desired, combining LAPTM-enhancing strategies with SIRT1 activators (such as resveratrol or synthetic activators like SRT1720) may produce synergistic neuroprotective effects. These combinations could simultaneously enhance LAPTM4B-mediated lysosomal stability, promote autophagosome-lysosome fusion, and reduce oxidative stress through SIRT1’s antioxidant signaling [83]. However, in glioma and other CNS tumors where LAPTM4B overexpression promotes tumor survival, SIRT1 inhibition may be a more appropriate strategy. A study has shown that SIRT1 knockdown inhibits glioma cell proliferation and potentiates temozolomide toxicity through facilitation of reactive oxygen species generation [82]. Therefore, the therapeutic context—neuroprotection versus tumor suppression—must guide the decision between SIRT1 activation and inhibition in LAPTM-targeted treatment regimens.
LAPTM5 can activate NF-
The high expression of LAPTM4A/4B is closely related to the grading, prognosis and chemotherapy resistance of glioma [14]. The detection of its mRNA or protein levels can serve as a new biomarker for early diagnosis and efficacy assessment. Non-invasive imaging techniques have enabled the visualization of LAPTM4B expression in liver cancer. This approach, using tools such as radiolabeled peptide positron emission tomography (PET) probes, is expected to be extended to central nervous system tumors in the future. Meanwhile, LAPTM5 is highly co-expressed with the risk genes of microglial cell networks in AD [47], suggesting its value in the early diagnosis of neurodegenerative diseases.
Based on the transmembrane structure and ligand-binding characteristics of LAPTM, developing small molecules or monoclonal antibodies to regulate its activity is a feasible strategy. For instance, targeting LAPTM4B can inhibit its promotion of prolonged EGFR signaling and enhanced autophagy, thereby reducing tumor resistance [32]. In the future, through high-throughput screening to find LAPTM agonists or inhibitors, or using PROteolysis targeting chimera (PROTAC) technology for specific degradation, all have potential application value.
CRISPR/Cas9 has been used to screen the function of LAPTM and reveal its role in drug resistance [54]. Gene editing is expected to achieve precise regulation of LAPTM in animal models to verify its role in neuroprotection or tumor suppression. In addition, vectors targeting the nervous system, such as adeno-associated virus, can be used to deliver the LAPTM gene or regulatory elements to specific nerve cells, providing new ideas for the treatment of ischemic or neurodegenerative diseases [71].
LAPTM regulates the tumor microenvironment and the polarization of immune cells, suggesting its potential value in combined immunotherapy with immune checkpoint inhibitors or chimeric antigen receptor T-cell (CAR-T) therapy [88]. In a glioblastoma mouse model, interfering with LAPTM expression significantly increased the response rate to anti-PD1 treatment. In the future, it can be explored to combine LAPTM inhibitors with oncolytic viruses, immune adjuvants, etc., to improve the immune tolerance of refractory CNS tumors.
Despite the promising therapeutic potential of LAPTM-targeted interventions, several significant challenges must be addressed for successful clinical translation. First, blood-brain barrier (BBB) penetration remains a fundamental obstacle for most neurological therapeutics. Small-molecule LAPTM modulators must be designed with appropriate lipophilicity and molecular weight to cross the BBB, or alternative delivery strategies such as nanoparticle encapsulation, focused ultrasound-mediated BBB opening, or intranasal administration must be employed.
Second, achieving lysosomal targeting presents unique pharmacological challenges. Therapeutic agents must traverse the plasma membrane and endosomal compartments to reach their intracellular targets without premature degradation. Lysosomotropic agents that accumulate in acidic compartments may provide one solution, but their non-specific accumulation in all lysosomes raises concerns about off-target effects on healthy cells.
Third, the ubiquitous expression of LAPTM proteins across multiple cell types raises concerns about systemic toxicity. Cell-type–specific delivery systems or prodrug approaches that are activated only in target cells may be necessary to achieve therapeutic selectivity. Additionally, the distinct and sometimes opposing functions of LAPTM family members (e.g., LAPTM4A promoting tumor immunity while LAPTM4B promotes tumor survival) necessitate isoform-specific modulators that can selectively target individual family members without affecting others.
Fourth, biomarker validation for LAPTM-targeted therapies requires substantial additional work. While preclinical studies suggest that LAPTM expression levels correlate with disease severity and treatment response, prospective clinical validation studies with standardized assays are needed to establish their utility in patient stratification and treatment monitoring.
Finally, current preclinical models have limitations in recapitulating human LAPTM biology. Species differences in expression patterns and functional consequences may limit translational applicability. The development of humanized mouse models and patient-derived organoid systems will be essential for more accurate preclinical evaluation of LAPTM-targeted interventions.
Current research mainly focuses on tumor cells or immune cells. The specific functions of LAPTM in neurons, astrocytes, oligodendrocytes, etc. still need to be further explored. Through new technologies such as single-cell transcriptomics and spatial multi-omics, a more comprehensive cell map of LAPTM can be constructed.
LAPTM plays a significant role in neurodegenerative diseases, tumors, ischemic injuries, and neuroinflammation. In the future, multiple disease models should be integrated to reveal the common and specific mechanisms from multiple dimensions such as energy metabolism, immunity, and types of cell death (such as ferroptosis and pyroptosis), thereby finding more broad-spectrum treatment strategies.
It is necessary to systematically evaluate the association between LAPTM gene polymorphisms, expression levels and disease risk or therapeutic efficacy in a large sample population, and establish predictive models using machine learning to provide evidence for precision medicine [84]. At the same time, multi-center clinical trials should be designed to assess the safety and efficacy of LAPTM-related interventions.
The delivery barriers for drugs in the nervous system are strict. In the future,
we can explore nanoparticle, exosome carriers, and trans-blood-brain barrier
delivery systems to effectively deliver LAPTM regulators or nucleic acid drugs to
the central nervous system [61]. Additionally, a multi-target therapeutic
strategy that jointly regulates LAPTM and pathways such as mTOR and
NF-
Blood or cerebrospinal fluid biomarkers based on LAPTM can be used for the dynamic monitoring of disease progression and therapeutic efficacy. Through multi-time point, longitudinal follow-up studies, the changes in LAPTM levels with disease progression or treatment response can be evaluated, thereby guiding individualized medication [101].
Despite the significant advances summarized in this review, several limitations warrant acknowledgment. First, the majority of LAPTM research has been conducted in cancer biology contexts, with extrapolation to neurological settings often based on limited direct evidence. The cell-type–specific functions of LAPTM family members in neurons, astrocytes, and oligodendrocytes remain incompletely characterized. Second, no clinical trials have specifically evaluated LAPTM-targeted interventions in neurological diseases, limiting our understanding of therapeutic feasibility and safety in human patients. The translational gap between preclinical promise and clinical reality remains substantial. Third, methodological heterogeneity across studies—including variations in model systems, experimental conditions, and outcome measures—complicates direct comparison of findings. Standardization of experimental approaches and outcome metrics would facilitate more robust conclusions. Fourth, distinguishing primary LAPTM-mediated effects from secondary consequences of general lysosomal dysfunction remains technically challenging. Genetic rescue experiments and acute modulation approaches are needed to establish causality. Fifth, species differences in LAPTM expression patterns and functional consequences may limit the translational applicability of findings from rodent models. Human-relevant model systems are urgently needed. Finally, the current absence of highly selective LAPTM modulators hampers precise functional interrogation and therapeutic development. Addressing these limitations through rigorous, neurology-focused studies will be essential for advancing LAPTM-based therapeutic strategies.
In conclusion, LAPTM family proteins represent a convergent node linking lysosomal dysfunction to neurological pathology across neurodegenerative diseases, ischemic injury, and brain tumors. The most promising near-term opportunities include: (1) validation of LAPTM4B and LAPTM5 as diagnostic/prognostic biomarkers in glioma and Alzheimer’s disease through prospective clinical studies; (2) development of LAPTM-SIRT1 combinatorial therapies that synergistically enhance autophagy flux in neurodegeneration while targeting tumor survival mechanisms in CNS malignancies; and (3) creation of cell-type–specific LAPTM modulators using advanced delivery platforms to overcome BBB limitations. As the field advances toward clinical translation, LAPTM-targeted strategies hold transformative potential for addressing the unmet therapeutic needs in neurological disorders, offering hope for patients with currently intractable conditions.
ZW conceived the study, conducted the initial literature search, and drafted the manuscript. YP, MS, and LZ contributed to the conceptualization and structural design of the review, and participated in the critical interpretation of the synthesized literature. All authors contributed to editorial changes in the manuscript. All other authors rigorously reviewed the paper and approved the final version. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by grants from the National Natural Science Foundation of China (No. 82201590), the Natural Science Foundation of Hubei Province (No. 2022CFB721), the Translational Medicine, the Interdisciplinary Research Joint Fund of Zhongnan Hospital of Wuhan University (Grant No. ZNJC202536) and the Science and Technology Innovation Cultivation Fund of Zhongnan Hospital of Wuhan University (No. CXPY202535) to LZ.
The authors declare no conflict of interest.
During the preparation of this work, the authors used ChatGPT-5.0 solely for the purpose of checking spelling and grammar in the manuscript. After using this tool, the authors thoroughly reviewed and edited the content as needed. The authors take full responsibility for the entire content of this publication, including any portions that were processed with the assistance of the AI tool.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.