






 , Chao Fang 1,2,†, Chengcheng Feng 1, Zuomeng Wu 1,2, Chenhao Zhao 1,2, Cailiang Shen 1,2,3,*
, Chao Fang 1,2,†, Chengcheng Feng 1, Zuomeng Wu 1,2, Chenhao Zhao 1,2, Cailiang Shen 1,2,3,*
1 Department of Orthopedics, The First Affiliated Hospital of Anhui Medical University, 230032 Hefei, Anhui, China
2 Laboratory of Spinal and Spinal Cord Injury Regeneration and Repair, The First Affiliated Hospital of Anhui Medical University, 230032 Hefei, Anhui, China
3 Anhui Province Research Center for the Clinical Application of Digital Medical Technology, The First Affiliated Hospital of Anhui Medical University, 230022 Hefei, Anhui, China
†These authors contributed equally.
Abstract
Fibrous scar formation significantly inhibits axonal regeneration and functional recovery following spinal cord injury (SCI). Atractylodin (ATD), an active constituent of traditional Chinese medicine, exhibits broad pharmacological properties, including anti-inflammatory and anti-fibrotic effects. Nevertheless, the potential therapeutic role of ATD in SCI and its underlying molecular mechanisms remain to be fully elucidated.
An SCI model was established in C57 mice. Motor function was assessed using the Basso Mouse Scale scoring system, inclined plane test, swimming test, and footprint analysis. Immunohistochemical staining was performed to evaluate fibrotic scar formation and neuronal survival. Western blotting and quantitative real-time PCR (qPCR) were also employed to investigate the molecular mechanisms underlying ATD-mediated regulation of fibroblasts following SCI.
ATD administration significantly enhanced motor function in SCI mice, reduced the area of fibrotic scars, and suppressed the expression of fibrotic markers. Mechanistically, ATD inhibited Mothers Against Decapentaplegic Homolog 2/3 (SMAD2/3) phosphorylation and nuclear translocation, thereby suppressing fibroblast activation and extracellular matrix deposition, while promoting neuronal survival and axonal regeneration.
ATD mitigates fibrotic scar formation by targeting the Transforming Growth Factor Beta (TGF-β)/SMAD pathway, thereby facilitating axonal regeneration and functional recovery. This offers a promising therapeutic strategy for SCI.
Graphical Abstract
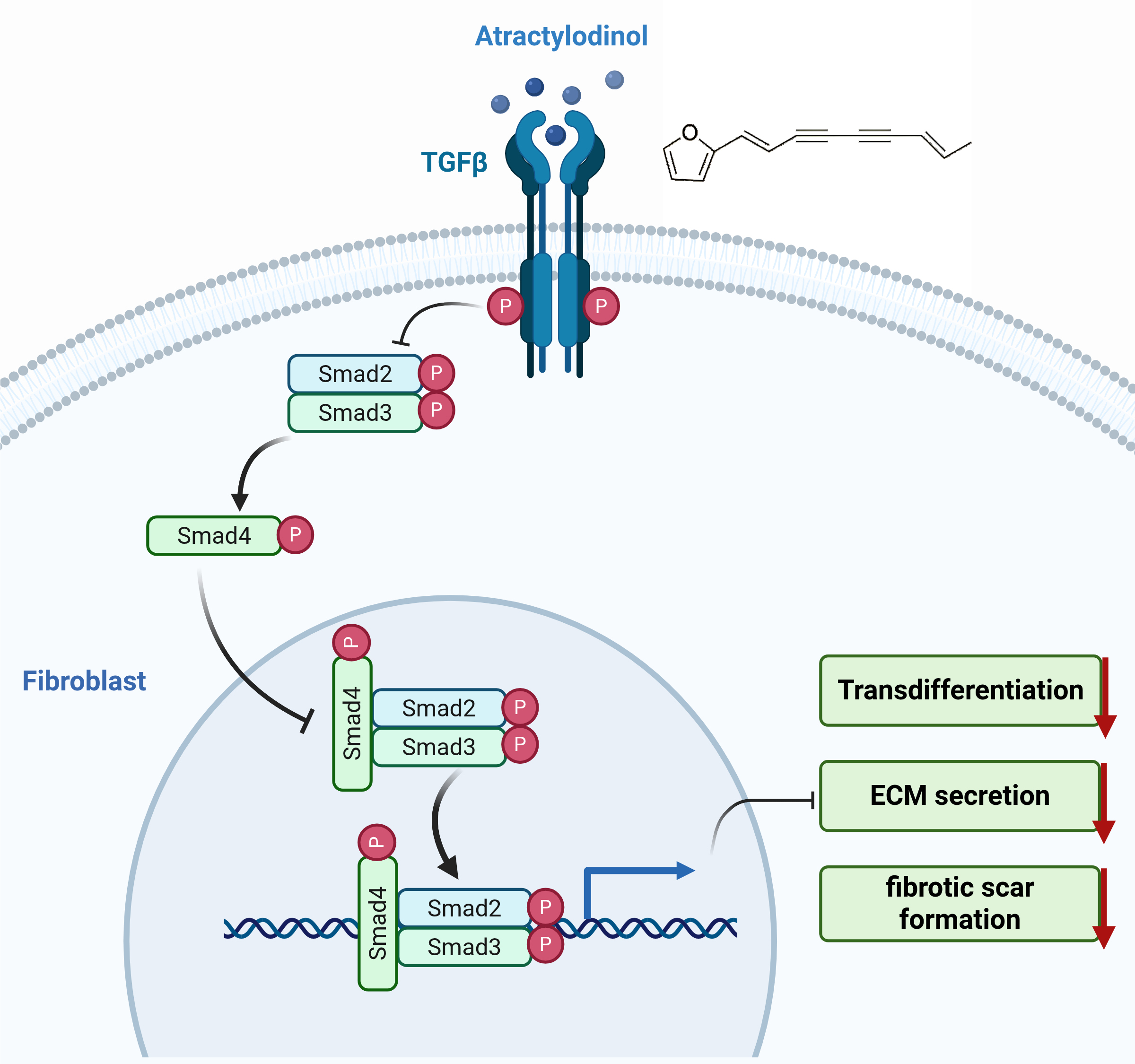
Keywords
- spinal cord injuries
- atractylodin
- fibrosis
- transforming growth factor beta
- smad proteins
Spinal cord injury (SCI) is a profoundly disabling condition characterized by acute onset and catastrophic outcomes. Its clinical refractoriness creates a significant burden for patients, their families, and society at large [1]. Currently, no efficacious treatment is available for SCI [2]. In the acute stage, extensive neuronal death at the lesion epicenter triggers microglial/macrophage recruitment [3, 4]. These cells engulf cellular debris, secrete numerous cytokines and inflammatory mediators, and recruit astrocytes and fibroblasts to participate in the subsequent repair process [5, 6, 7]. This cascade establishes an inflammatory microenvironment at the injury site. As astrocytes and fibroblasts undergo reactive activation, they secrete substantial amounts of chondroitin sulfate proteoglycans (CSPGs) and extracellular matrix (ECM) proteins, resulting in the formation of dense fibrous and glial scars at the periphery of the lesion [8, 9, 10]. These scars significantly impede axonal regeneration and functional recovery. While fibrotic scar formation may have a protective role, it is increasingly recognized that excessive deposition after SCI is ultimately detrimental. Therefore, its moderate inhibition is considered a promising strategy to support neural repair and functional restoration [11, 12]. Therefore, targeting fibrotic scar formation after SCI may represent an effective strategy to reduce neuron death and promote axon regeneration and functional recovery.
The Transforming Growth Factor Beta/Mothers Against Decapentaplegic Homolog
(TGF-
Atractylodin (ATD) is a polyacetylene compound extracted from the dried rhizome
of Atractylodes lancea or Atractylodes chinensis, which are members of the
Asteraceae family in traditional Chinese medicine [23]. ATD has important and
extensive pharmacological effects. It has been found to play an anti-inflammatory
role by inhibiting pro-inflammatory factors and the NF-
Therefore, we sought to determine if ATD-mediated inhibition of fibrous scar
formation enhances neurological recovery in a mouse model of SCI and, further, to
uncover the mechanistic basis of this effect through in
vitro experiments. We hypothesized that ATD reduces the formation of fibrous
scar tissue after SCI by inhibiting TGF-
Female C57BL/6 mice (8–10 weeks old, 20–22 g weight) were supplied by the Animal Center of Anhui Medical University, China. The animals were acclimatized and housed under standard specific pathogen-free conditions (22 °C, 30–50% humidity, 12-h light/dark cycle; 4 mice/cage) with free access to food and water. After final assessments, all mice were humanely euthanized with carbon dioxide. Animal surgeries were performed under the same approved protocol as a prior study investigating a different therapeutic compound and mechanism.
Mice were randomly assigned to the sham operation group, vehicle group, or ATD treatment group. SCI was induced using a severe compression model. Briefly, after anesthesia induction with intraperitoneal 1% pentobarbital sodium (MERCK, Rahway, NJ, USA, CAS:57-33-0), mice underwent a T9–T10 laminectomy to expose the spinal cord. The exposed cord was then subjected to a 5-second compression using a calibrated Dumont #5 forceps (Fine Science Tools, Heidelberg, Germany, Cat1125220). The muscle and skin layers were then sutured sequentially. Postoperatively, all mice received antibiotic treatment to prevent infection and manual bladder expression twice daily until spontaneous urination was restored. Mice in the sham group underwent a laminectomy without spinal cord compression. The ATD treatment group received daily intraperitoneal injections of 20 or 40 mg/kg ATD (MCE, New Jersey, USA, HY-N0238) starting immediately after injury and continuing until day 7 post-injury. The vehicle group received an equivalent volume of the solvent to control for potential solvent-related effects. All surgical procedures were performed by a single, experienced, and independent surgeon to ensure consistency and minimize procedural variability.
Hindlimb motor function was assessed using the Basso Mouse Scale (BMS) score, the slant board test and footmark analysis test, as described previously. Mice were placed in an open field, and their hind limb movement was observed before operation and at 1, 3, 7, 14, 21, and 28 days post-injury (dpi). The footprint analysis test was conducted at 28 dpi to further evaluate the recovery of motor function. The front and back paws were soaked in green and red dyes, respectively, and the animals were placed on white paper to walk. All assessments were conducted in a blinded fashion by two independent, experienced investigators to ensure objectivity.
After the accomplishment of the necessary assessments, all mice were euthanized using carbon dioxide. The flow rate of CO₂ should be controlled at 50% of the volume of the euthanasia chamber per minute. The animals should be exposed to CO₂ for at least 5 minutes. Mice were transcardially perfused with 0.9% saline and then 4% paraformaldehyde (PFA) (Servicebio, Wuhan, Hubei, China, G1101). The spinal cord segment encompassing the injury epicenter was dissected and post-fixed overnight in 4% PFA at 4 °C. After sucrose gradient cryoprotection, the specimens were embedded in paraffin wax. Subsequently, longitudinal sections were cut at 5 µm for the following histological analyses. For hematoxylin and eosin (H&E) staining, sections first underwent hematoxylin staining for 5 minutes, were sequentially rinsed with double-distilled water, and were then subjected to eosin counterstaining for 1 minute. Digital images were acquired under a light microscope. Sections designated for Nissl staining were stained using a Nissl stain solution (Beyotime, Shanghai, China, C0117) at 37 °C for a duration of 10 minutes, briefly washed with double-distilled water, differentiated with 95% ethanol for 5 seconds, and then dehydrated and cleared as appropriate. Representative images were captured using a light microscope VS200 Panoramic Tissue Quantitative Analysis System (OLYMPUS, Tokyo, Japan, VS200). Positive Nissl staining was identified by the presence of distinct, dark purple granules (Nissl bodies) within the neuronal cytoplasm. Viable motor neurons with large, clear nuclei and rich Nissl substance were counted.
For immunofluorescence, mice were transcardially perfused with 4%
paraformaldehyde, and the spinal cord tissues were harvested and processed for
paraffin embedding. Prior to staining, the sections underwent deparaffinization
in xylene and sequential rehydration through a graded alcohol series to aqueous
conditions. Antigen retrieval was performed using citrate buffer (pH 6.0),
followed by blocking with 5% bovine serum albumin (BSA) (Servicebio,Wuhan,
Hubei, China, GC305010) in phosphate-buffered saline (PBS) for 1 h at room
temperature to reduce nonspecific binding. To label astrocytes and neurons,
sections were incubated overnight at 4 °C with rabbit anti-Glial Fibrillary Acidic Protein (GFAP) antibody
(1:1000; Abcam, Cambridge Biomedical Campus, Cambridge, UK, ab68428) and rabbit
anti-TUJ1 antibody (1:1000; Abcam, ab18207), respectively. Sections then received fluorescent secondary antibodies
for detection: green-fluorescent Alexa Fluor 488
goat anti-rabbit Ig (Gat 1:50 from Elabscience, Wuhan, Hubei, China, E-AB-1055) and red-fluorescent Cy3 goat anti-mouse IgG (Gat 1:50 from Elabscience, E-AB-1011). Nuclei were counterstained with
4′,6-diamidino-2-phenylindole (DAPI) (Beyotime, Shanghai, China, P0131) Fluorescent
signals were visualized and images captured using a Leica DM-6B fluorescence
microscope (Leica, Wetzlar, Hesse, Germany, DM-6B). For quantification of positive
cells or stained areas, digital images were analyzed using ImageJ software (1.54r, NIH,
Bethesda, MD, USA). Quantification data are presented as mean
Mice were transcardially perfused for tissue fixation, and paraffin-embedded tissue sections were subsequently prepared. Tissue sections were deparaffinized and rehydrated through a xylene-alcohol series, followed by antigen retrieval in citrate buffer. Subsequently, nonspecific binding was blocked by incubation with 5% BSA for 1 hour at room temperature. Apoptosis was then assessed by terminal deoxynucleotidyl transferase-mediated 2′-Deoxyuridine-5′-Triphosphate (dUTP) nick-end labeling (TUNEL) staining, which was carried out accordingly to the manufacturer’s instructions (Beyotime, C1089).
Primary fibroblasts derived from mouse spinal cord were acquired from Procell.
Cells were expanded in the provided complete growth medium (Procell, Wuhan,
Hubei, China, CM-M162) according to the supplier’s instructions. All cultures
were kept in a humidified incubator at 37 °C with 5% CO2. Primary
fibroblasts were characterized by vimentin immunofluorescence staining,
confirming a purity of
We seeded the fibroblasts into 96-well plates (5
For Western blot analysis, proteins were extracted from cells or tissue samples
using RIPA lysis buffer (Beyotime, P0013B). Following lysis, the protein concentration was
quantified with a BCA assay kit (Beyotime, P0010) prior to further
processing. After separation by SDS-PAGE (20 µg protein per lane) and
transfer to PVDF membranes, the membranes were incubated in a blocking buffer
(5% non-fat milk in Tris-Buffered Saline with Tween®
20 (TBST) (Servicebio, Wuhan, Hubei, China, G0001)) for 2 h at room temperature.
This was followed by an overnight incubation at 4 °C with the respective
primary antibodies. Membranes then received incubation with the appropriate
HRP-conjugated secondary antibodies for 2 hours at room temperature. Detailed information for all antibodies involved in WB:
FN (1:1000, Immunoway, SuZhou, JiangSu, China, YM8309), Collagen (1:1000, Immunoway, YT6135),
At 14 days post-injury, mice were perfused, and a 5-mm segment of spinal cord tissue centered precisely on the injury epicenter was dissected under a surgical microscope. This segment, encompassing the primary lesion and peri-lesion area, was immediately snap-frozen for subsequent total RNA extraction. RNA was extracted from tissues, cells, and exosomes with TRIzol reagent (Gibco, Grand Island, NY, USA, 15596026CN). For cDNA synthesis, the Superscript III RT Reaction Mix (Invitrogen, Carlsbad, CA, USA, 18080085) was used. Quantitative PCR was performed with SYBR Green PCR Master Mix on a 7900 Fast Real-Time PCR System (7900, Applied Biosystems, Foster City, CA, USA). Relative gene expression was determined by normalizing to GAPDH and applying the 2-ΔΔCT calculation. The primer sequences were listed below:
Fn: 5′–CCATTCCACCTTACAACAC–3′, 5′–CAAGCCAGACACAACAAT–3′;
Col1
GAPDH: 5′–TGTCTCCTGCGACTTCAACA–3′, 5′–GGTGGTCCAGGGTTTCTTACT–3′.
All statistical analyses were performed with SPSS (version 16.0; IBM, Chicago,
IL, USA) and GraphPad Prism (version 8.0, GraphPad Software, Boston, MA, USA).
Quantitative data are expressed as means
To evaluate the therapeutic efficacy of ATD after SCI, mice underwent surgical induction of SCI followed by intraperitoneal administration of ATD post-operation (Fig. 1A). A series of behavioral assessments were subsequently performed to evaluate functional recovery, with the experimental timeline summarized in Fig. 1B. Recovery of motor function represents a critical endpoint for assessing therapeutic outcomes. Therefore, locomotor performance was systematically evaluated using the Basso Mouse Scale (BMS) at multiple time points post-injury. Mice in the ATD-treated groups exhibited significantly improved motor function starting on day 14 after SCI, with the most pronounced recovery observed in the high-dose group (Fig. 1C,D). Consistent with these findings, swimming test results demonstrated enhanced hindlimb motor coordination and propulsion in the ATD treatment groups compared to the vehicle group, particularly in the high-dose cohort (Fig. 1E,F). Footprint analysis confirmed that ATD treatment led to improved gait symmetry and stride patterns, indicating enhanced functional recovery of both hindlimbs (Fig. 1G–I). Collectively, these data demonstrate that ATD promotes robust recovery of motor function following SCI in mice.
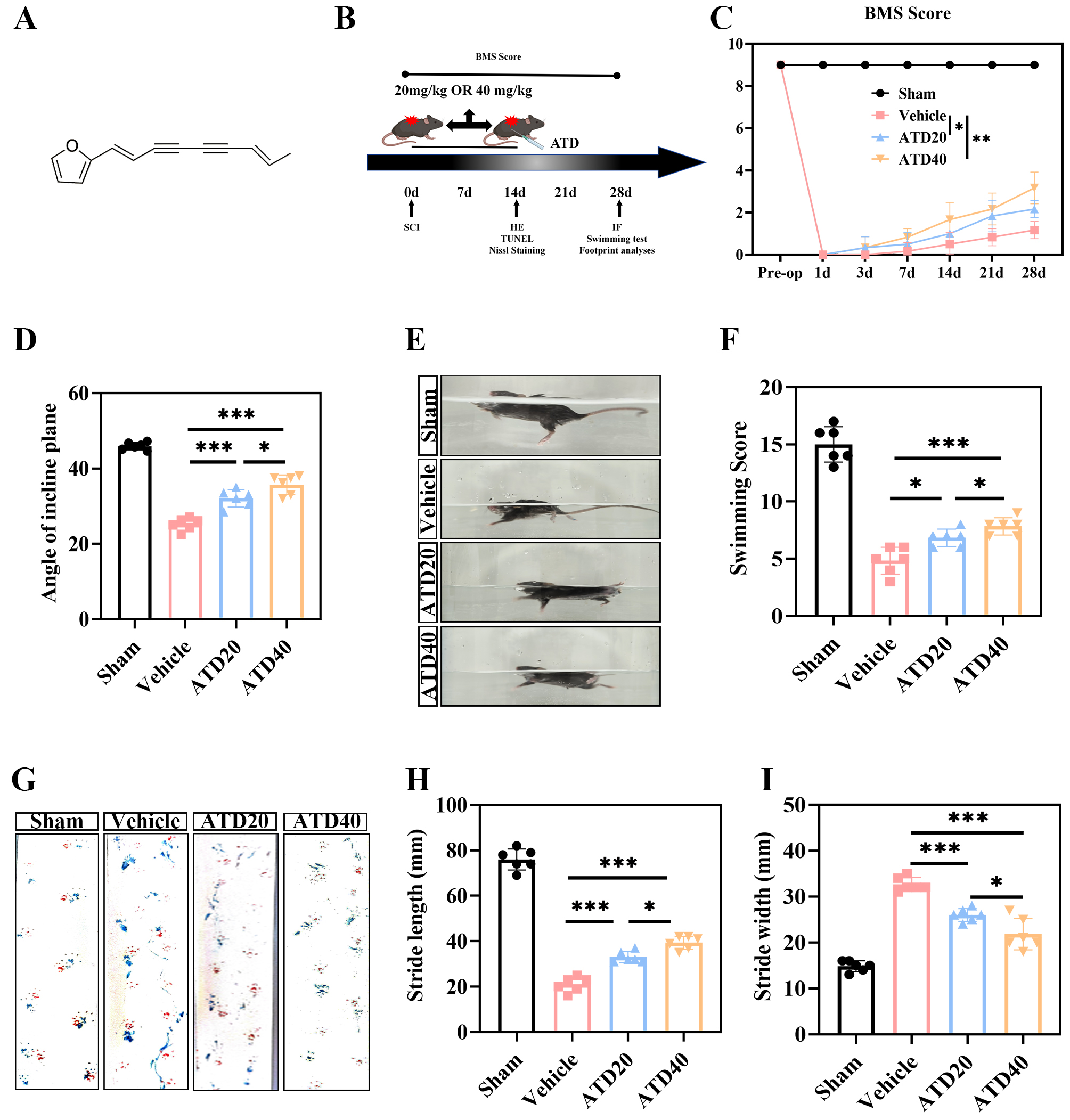 Fig. 1.
Fig. 1.
Atractylodin significantly improve the motor function of mice
after SCI. (A) Chemical structure of ATD. (B) Schematic representation of the
timeline for behavioral experiments conducted at various time points following
SCI. (C) Results from the BMS test showing the recovery of hindlimb motor
function after SCI (n = 12 animals per group). (D) Results from the Santing board
test showing recovery of hindlimb motor function after SCI (n = 6 in each group).
(E,F) Results from the swimming test showing recovery of hindlimb motor function
after SCI (n = 6 in each group). (G–I) Results from footprint analysis showing
the recovery of hindlimb motor function after SCI (n = 6 in each group). Data
shown are the mean
Following SCI, astrocytes and fibroblasts collaborate to form a dense
glial-fibrotic scar surrounding the lesion core, which acts as a major barrier to
axonal regeneration. This study further investigated the impact of ATD on
glial-fibrotic scar formation after SCI (Fig. 2A). Quantitative real-time PCR
(qPCR) analysis revealed that ATD treatment significantly downregulated the mRNA
expression levels of key fibrosis-related markers, Fn and
Col1
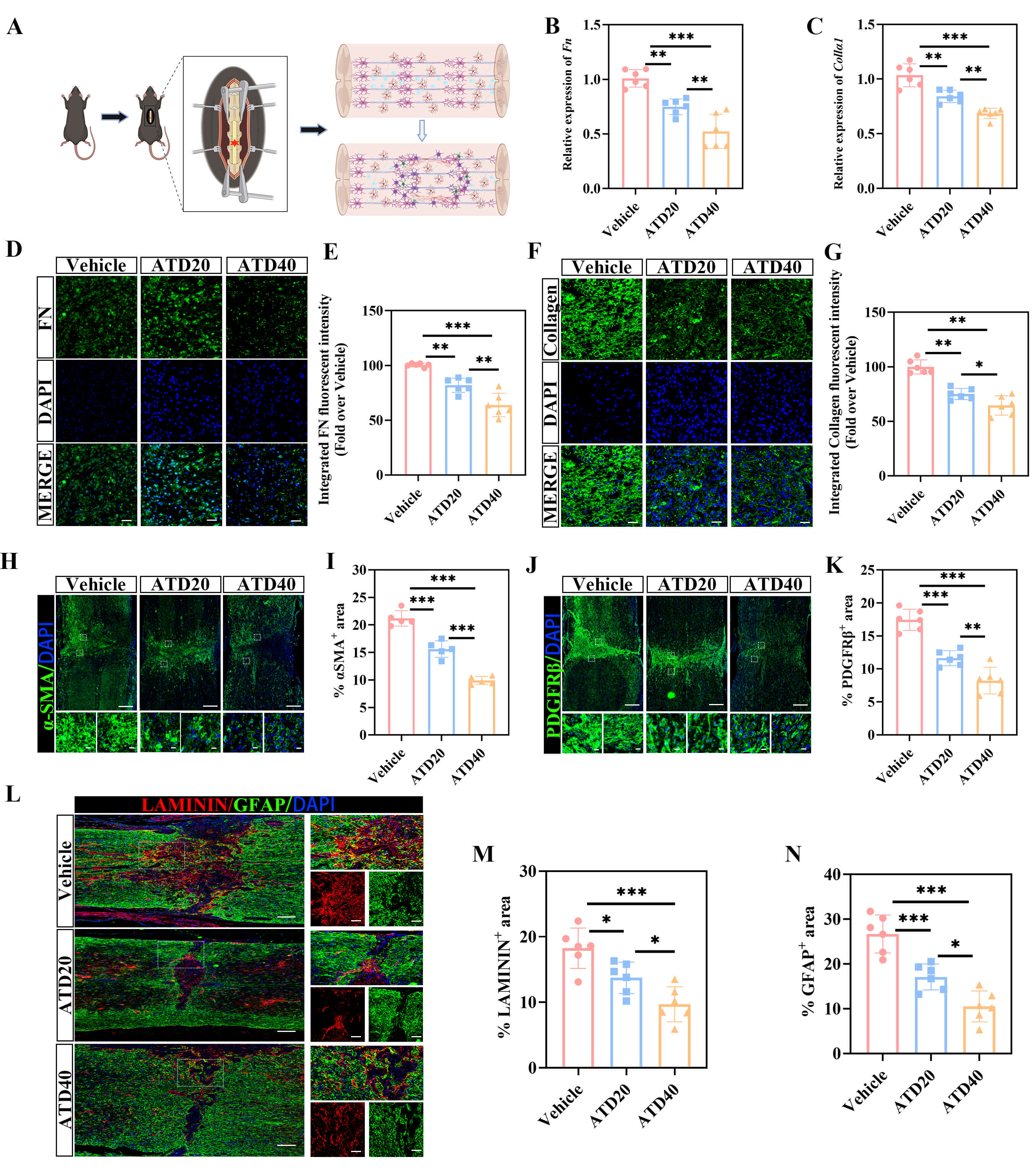 Fig. 2.
Fig. 2.
Atractylodin inhibits fibrous scar formation following SCI. (A) Schematic illustration of fibrous glial scar formation in the injured spinal cord of mice following SCI modeling. (B) Quantitative gene expression analysis of Fn at the lesion center in the Vehicle, ATD20, and ATD40 groups at 14 dpi (n = 6 animals per group). (C) Quantitative gene expression analysis of Col1
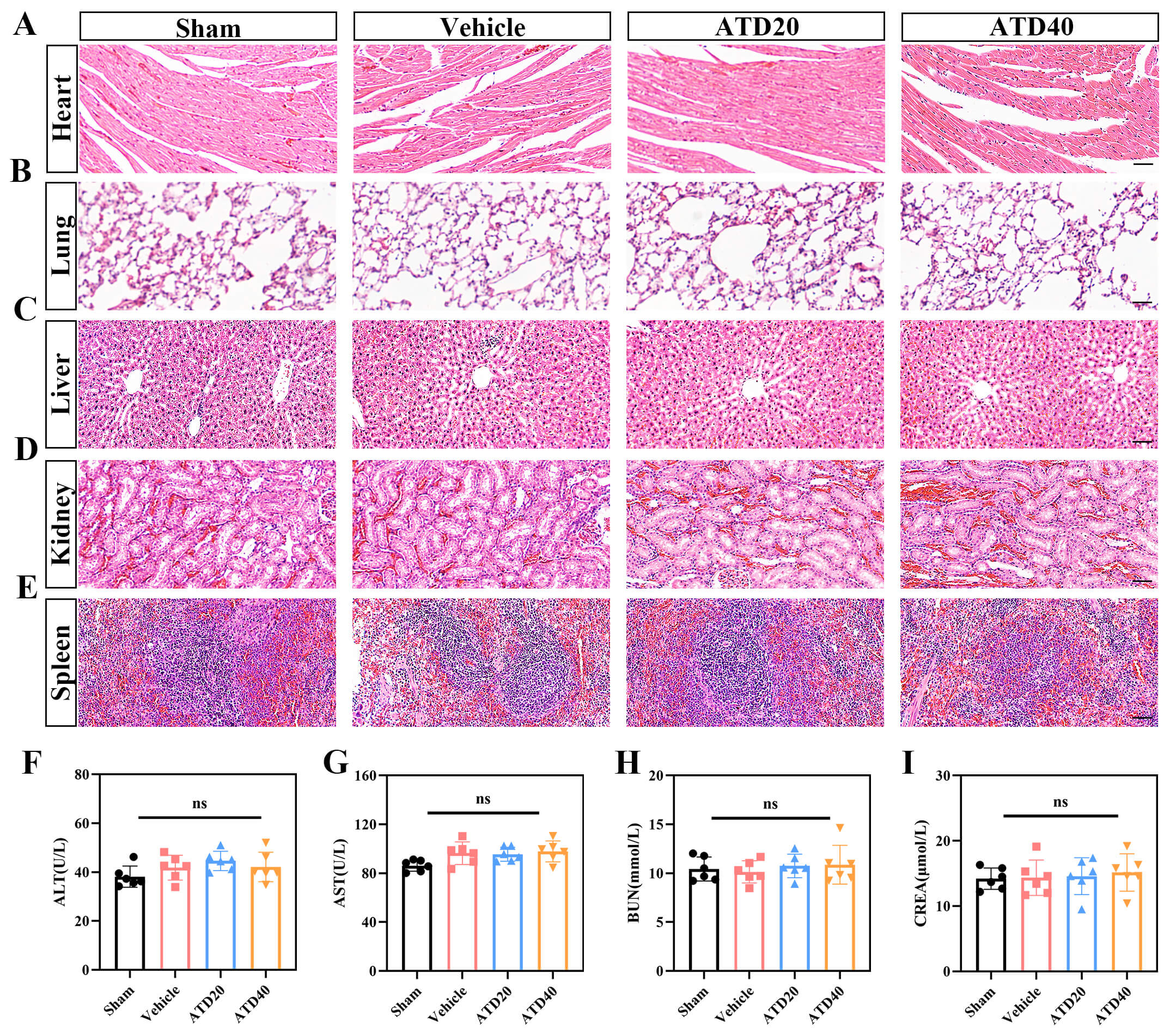 Fig. 3.
Fig. 3.
Atractylodin exhibits favorable biocompatibility in
vivo. (A–E) Histopathological analysis (H&E staining) of major organs was
performed on day 28 after SCI. No significant pathological alterations were
observed in the heart (A), lungs (B), liver (C), kidneys (D), or spleen (E)
across the sham, Vehicle, ATD20, and ATD40 groups. All tissue sections displayed
normal architecture and cellular morphology, with no signs of inflammation,
necrosis, or tissue damage (n = 6 animals per group; scale bar = 100 µm).
(F–I) Evaluation of systemic biosafety via serum biochemistry. At 7 days
post-injury, key markers for hepatic and renal function were measured. Serum
levels of (F) alanine aminotransferase (ALT), (G) aspartate aminotransferase
(AST), (H) blood urea nitrogen (BUN), and (I) creatinine (CRE) showed no
significant differences among the sham, Vehicle, ATD20, and ATD40 groups (n = 6
animals per group). ns p
We investigated the effects of ATD on the dynamic regulation of neurons and axons following SCI. H&E staining and Nissl staining were performed on spinal cord tissue sections to assess structural integrity and neuronal morphological changes after injury. The results revealed extensive neuron loss and cavity formation at the lesion epicenter in SCI mice. The Vehicle group displayed characteristic features of severe neurodegeneration, such as neuronal pyknosis, Nissl body disintegration, and nuclear dissolution. These pathological features were substantially alleviated by ATD treatment (Fig. 4A–D). Next, TUNEL staining combined with NeuN immunofluorescence was conducted to evaluate neuronal apoptosis and survival. In the sham group, neurons were regularly aligned and showed minimal apoptotic activity. In stark contrast, the SCI group suffered substantial neuronal loss, disorganized cytoarchitecture, and extensive apoptosis. However, ATD treatment markedly increased the number of surviving NeuN-positive neurons in the peri-lesion areas (Z1–Z3) compared to the Vehicle group (Fig. 4E–H). Collectively, these results indicate that ATD potently inhibits neuronal apoptosis and promotes neuronal survival following SCI. To investigate whether ATD further contributes to axonal regeneration by attenuating fibrotic scarring, we performed immunofluorescence staining for TUJ1 and NF at 28 dpi. Quantitative analysis revealed that four weeks of ATD treatment significantly expanded the area of TUJ1-positive labeling relative to the Vehicle group (Fig. 4I,J), implying the promotion of axonal sprouting. Furthermore, the ATD group exhibited a marked increase in the number of NF-positive axons penetrating the lesion core (Fig. 4K,L), indicating successful axonal regeneration across the scar boundary.
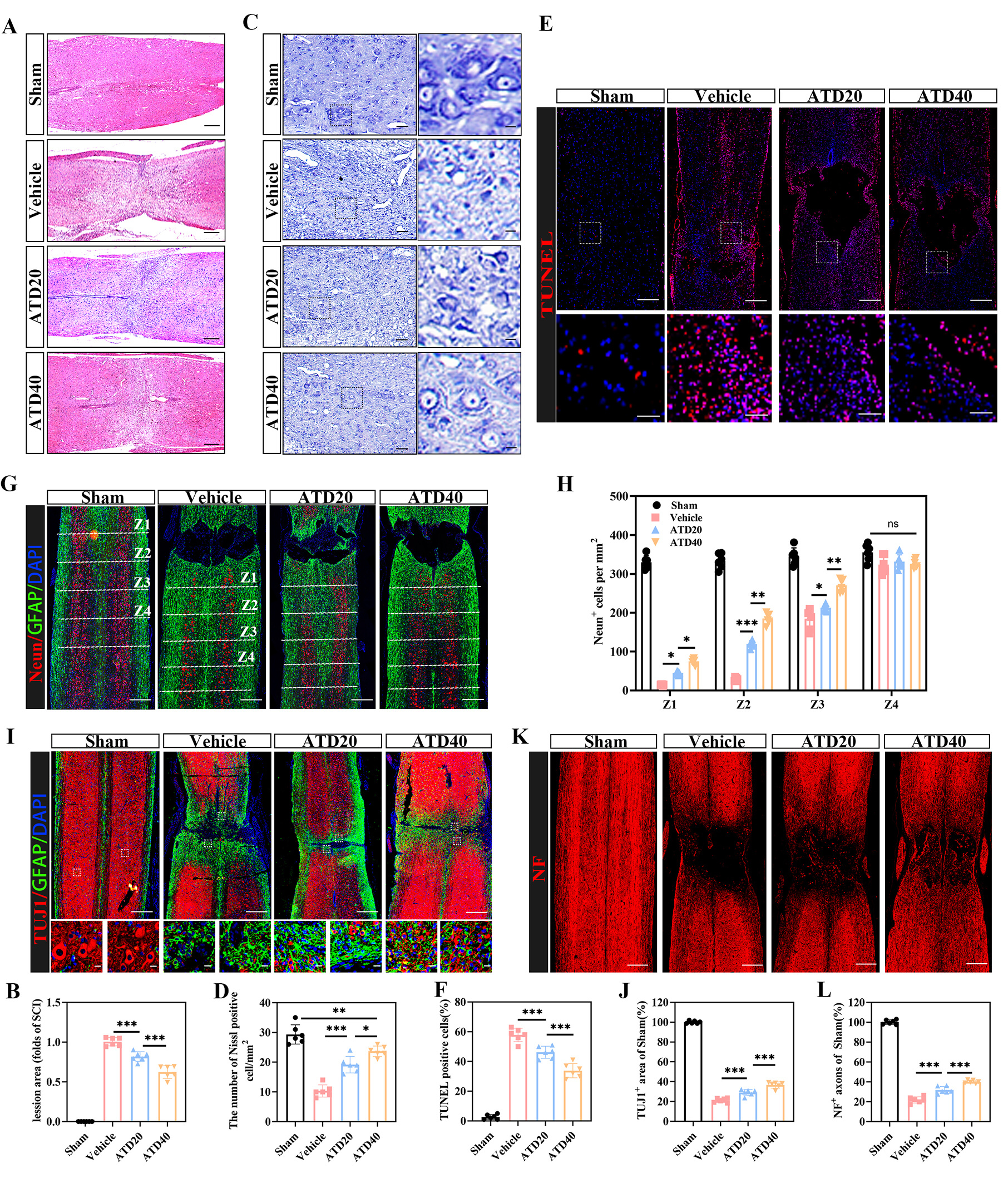 Fig. 4.
Fig. 4.
Atractylodin inhibits neuronal apoptosis and promotes axonal
regeneration following SCI. (A) Representative H&E-stained images of the lesion area at 28 days post-SCI in Sham, Vehicle, ATD20, and ATD40 groups (scale bar = 500 µm). (B) Corresponding quantitative analysis of lesion size from panel (A). (n = 6 animals per group). (C) Nissl staining was used to evaluate the morphological integrity and distribution of Nissl-positive neurons in the injured spinal cord tissues across all experimental groups at 28 dpi (scale bar = 100 µm, enlarged view scale bar = 20 µm). (D) Quantitative analysis of the number of Nissl-positive neurons from panel (C) (n = 6 animals per group). (E) Representative images of TUNEL assay detecting apoptotic cells in the lesion area (scale bar = 500 µm, enlarged view scale bar = 100 µm). (F) Quantification of TUNEL-positive cells from panel (E) (n = 6 animals per group). (G) Immunofluorescence labeling of NeuN+ neurons was performed to assess neuronal survival (scale bar = 500 µm). (H) Quantification of NeuN+ neuron counts from panel (G) (n = 6 animals per group). (I) Representative double immunofluorescence images of TUJ1+ axons (green) and GFAP+ astrocytes (red) (scale bar = 500 µm, enlarged view scale bar = 20 µm). (J) Quantification of the relative positive areas of TUJ1 and GFAP from panel (I). (K) Immunofluorescence images showing NF+ neurofilaments (regenerating axons) in the injured spinal cord (scale bar = 500 µm). (L) Quantified fluorescence intensity of NF+ signals from panel (K). (n = 6 animals per group). Data shown are the mean
To investigate the anti-fibrotic effects of ATD on spinal cord fibroblasts, we
first confirmed its biocompatibility. The CCK-8 assay revealed no significant
cytotoxicity at a concentration of 100 µM (Fig. 5A). We then modeled
fibrotic activation using TGF-
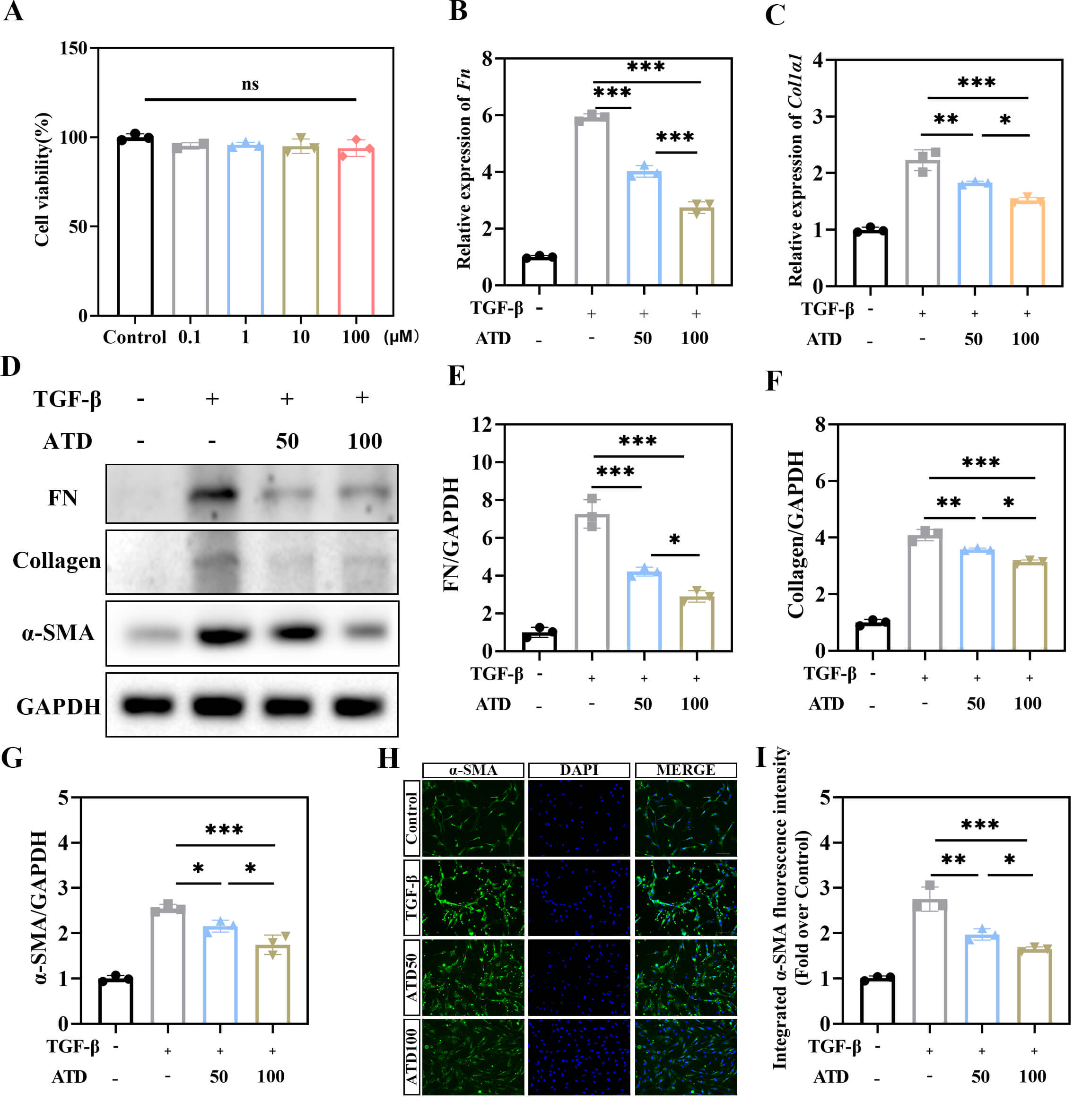 Fig. 5.
Fig. 5.
ATD inhibits TGF-
Following SCI, TGF-
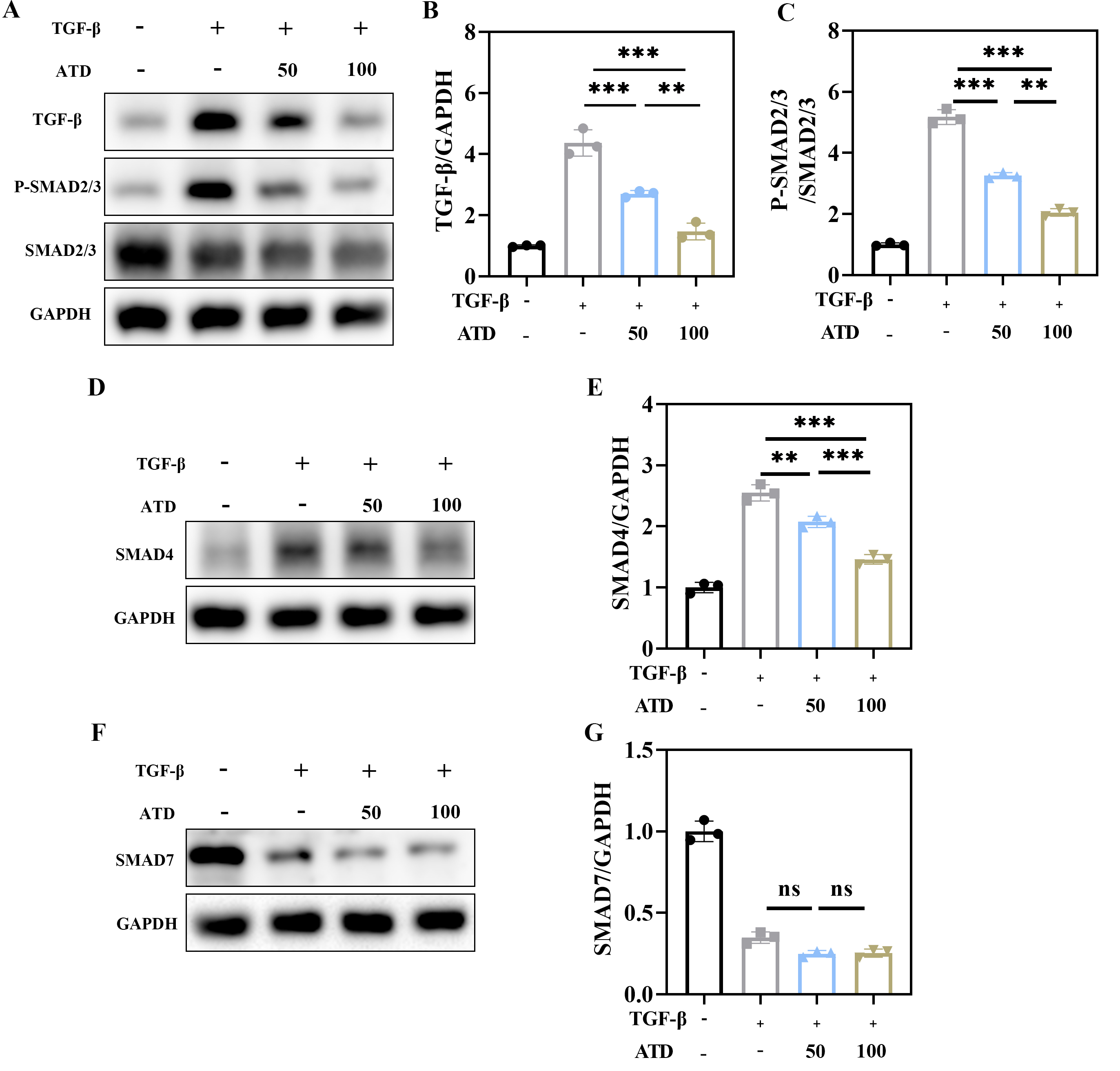 Fig. 6.
Fig. 6.
ATD inhibits the TGF-
This study employed a well-established mouse model of SCI to systematically
investigate the inhibitory effect of ATD on fibrotic scar formation, as well as
the underlying mechanisms. In vivo, intraperitoneal administration of
ATD significantly reduced the fibrotic scar area at the lesion site, suppressed
the expression of glial scar and fibrosis-related markers, and promoted the
recovery of hindlimb motor function in mice. In vitro, ATD effectively
inhibited TGF-
Fibrotic scar formation constitutes a dynamic and multicellular pathological
process driven primarily by the aberrant activation of pro-fibrotic signaling
within the injured microenvironment following SCI. The acute injury phase is
characterized by widespread death of neuronal and glial cells, thereby
propagating the release of damage-associated molecular patterns (DAMPs). These
molecules recruit and activate microglia/macrophages, prompting them to secrete
pro-inflammatory mediators such as TGF-
A vicious cycle of bidirectional regulation occurs between fibrous scar formation and neuronal death. On the one hand, fibroblasts and activated astrocytes in the scar continuously secrete pro-inflammatory factors and neurotoxic molecules to directly induce neuronal apoptosis [37, 38]. On the other hand, excessive accumulation of ECM within the fibrotic scar disrupts the blood-spinal cord barrier in the lesion site, impedes the delivery of neurotrophic factors, and exacerbates neuronal ischemia and hypoxia via mechanical compression [39]. ATD treatment after SCI significantly reduced neuronal pyknosis and apoptosis and increased the number of Neun positive neurons, which may be due to an improved local microenvironment following inhibition of fibrous scar formation. In addition, CSPGs in fibrous scars inhibit axonal regeneration and induce growth cone collapse by binding to neuronal surface receptors, while ATD indirectly attenuates the inhibitory effects on neuronal survival and axon repair by reducing the scar area and deposition of CSPGs [40]. In the current study, the number of serotonergic axons passing through the scar area was significantly increased in the ATD treatment group, further confirming that inhibition of the fibrous scar can promote neuronal survival and reconstruction of functional connectivity by improving the neural microenvironment.
TGF-
ATD, an active constituent of the traditional Chinese medicinal herb
Atractylodes, has been previously reported to exert multiple pharmacological
activities, including anti-inflammatory, metabolic regulatory, and anti-fibrotic
effects [24, 25, 26, 27]. The therapeutic potential of bioactive compounds derived from
Atractylodes species in central nervous system disorders, including SCI, is
gaining recognition. Notably, atractylenolide III, a sesquiterpene lactone from
the same genus, has demonstrated efficacy in experimental SCI, primarily by
modulating neuroinflammatory responses involving microglia/macrophages [44].
While this prior work provides valuable context, our study
introduces atractylodin—a chemically distinct polyacetylene
from Atractylodes—and defines its unique therapeutic niche. Unlike the
established anti-inflammatory profile of atractylenolides, our investigation is
the first to systematically establish atractylodin’s potency in specifically
mitigating fibrotic scar formation, a pivotal and mechanically obstructive
pathology in the subacute to chronic phases of SCI. The chemical distinction
between these compound classes suggests divergent molecular interactions and
target profiles. Our focus on the TGF-
In the current study, ATD showed a significant inhibitory effect on fibrosis
after SCI. The mechanism of action was different from that of traditional
anti-inflammatory drugs, and was achieved by specifically interfering with the
TGF-
This study has several limitations that warrant careful consideration. First, although our in vitro data demonstrate that ATD suppresses SMAD2/3 phosphorylation, its direct molecular target remains unknown. It is unclear whether ATD acts directly on TGF-
In summary, our data indicate that ATD acts as an effective inhibitor of the
TGF-
The data used to support the findings of this study are available from the corresponding author upon request.
CLS supervised the research. ZWL, CF and CLS designed and performed the experiments, ZWL interpreted the data and wrote the manuscript. CCF, ZMW, and CHZ analyzed the data. All authors critically reviewed and revised the academic content of the article. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experimental protocols were reviewed and approved by the Anhui Medical University Ethics Committee (LLSC20242471) in accordance with the ARRIVE guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
We would like to thank the Center for Scientific Research of Anhui Medical University for valuable help in our experiment.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN47565.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.