








 , Xiaomei Liu 1,*
, Xiaomei Liu 1,*1 Jiangsu Key Laboratory of Immunity and Metabolism, Department of Pathogen Biology and Immunology and Laboratory of Infection and Immunity, School of Basic Medical Sciences, Xuzhou Medical University, 221004 Xuzhou, Jiangsu, China
2 Clinical Laboratory, Shiyan Hospital of Integrated Traditional and Western Medicine, 442001 Shiyan, Hubei, China
3 First Clinical Medical College, Xuzhou Medical University, 221004 Xuzhou, Jiangsu, China
4 Department of Human Anatomy, Xuzhou Medical University, 221004 Xuzhou, Jiangsu, China
†These authors contributed equally.
Abstract
Neuroinflammation serves as a pivotal driver of pathology in multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE) – the widely used animal model of MS. A key contributor to this pathological process is neurotoxic A1-like reactive astrocytes, which play an essential role in disease progression. Although the antidiabetic drugs Metformin (Met) and Pioglitazone (Pio) exhibit anti-inflammatory properties, the effects of Met and Pio on A1-like reactive astrocytes in MS, as well as the underlying mechanisms, remain poorly defined. In this study, we investigated whether Met and Pio can attenuate neuroinflammation by modulating A1-like astrocyte activation to uncover the underlying signaling pathways involved in the process.
Primary astrocytes were isolated from mice and then treated with interleukin‑17 (IL-17) to induce an A1-like reactive state. The effects of Met and Pio on A1-like astrocyte activation and inflammatory responses were evaluated. The role of the protein kinase B /mammalian target of rapamycin /signal transducer and activator of transcription 3 (AKT/mTOR/STAT3) signaling pathway was examined using Western blotting and immunofluorescence assay. Meanwhile, the experiments in vivo were performed in EAE mice, where Met and Pio administration was used to assess the therapeutic effects on neuroinflammation, demyelination, and disease progression.
Both Met and Pio significantly suppressed the production of inflammatory cytokines and attenuated A1-like astrocyte activation in IL-17-stimulated primary astrocytes. These effects were mediated by the inhibition of the AKT/mTOR/STAT3 pathway. In EAE mice, drug treatment markedly reduced neuroinflammation and demyelination, thereby leading to a significant alleviation of clinical symptoms and pathological damage.
Our findings suggest that Met and Pio downregulate the activated astrocyte-mediated inflammatory reaction to alleviate EAE pathogenesis through suppression of the AKT/mTOR/STAT3 pathway. Collectively, these results demonstrate a novel mechanism underlying the potential therapeutic effects of these drugs in MS and other neuroinflammatory disorders.
Keywords
- multiple sclerosis
- astrocytes
- neuroinflammation
- metformin
- pioglitazone
Multiple sclerosis (MS) is an autoimmune mediated chronic inflammatory disorder of the central nervous system (CNS), characterized by neuroinflammation, glial cell activation and demyelination.
It is estimated that MS affects approximately 2.8 million people worldwide [1]. Traditionally, MS has been classified into three distinct clinical phenotypes, namely primary progressive MS (PPMS), relapsing-remitting MS (RRMS), and secondary progressive MS (SPMS) [2]. Pathological lesions within MS are distinguished by myelin loss, infiltrating inflammatory cells, reactive gliosis, the blood-brain barrier (BBB) breakdown and axonal damage [3]. The underlying pathological mechanisms of MS involve the activation of auto-reactive lymphocytes. However, glial cells are increasingly recognized as critical mediators in the immunopathology of MS, particularly during the chronic progressive phase [4, 5].
Astrocytes are key players throughout the pathogenesis of MS, exhibiting significant heterogeneity and responding. In response to CNS injuries, astrocytes undergo reactive astrogliosis, which is a dual-functional process that not only drives inflammation but also facilitates the disease remission [6]. However, in MS/experimental autoimmune encephalomyelitis (EAE), reactive astrocytes contribute to disease progression by secreting inflammatory cytokines and mediators, which impair the BBB integrity, promote inflammatory cell infiltration, and accelerate demyelination [4, 7, 8]. Recent studies have elucidated distinct transcriptomic alterations in astrocytes in response to diverse pathological conditions, including MS, particularly with the identification of neurotoxic A1 phenotypes of astrocyte [8, 9]. At present, clinical treatments for MS predominantly modulate the peripheral immune system, and have shown limited efficacy against progressive MS [4]. Consequently, the development of therapeutic strategies focuses on directly targeting astrocyte-mediated pathological cascades. However, progress in both astrocyte-targeted drug development and mechanistic characterization remains limited.
Evidence indicates that cluster of differentiation 4+ T helper 17
(CD4+ Th17) cells are characterized by the production of
interleukin‑17 (IL-17), which is critical in the pathogenesis of MS/EAE. The
disease onset and severity of EAE are delayed and reduced in IL-17-deficient
animals [10, 11], and anti-IL-17 antibody inhibits the expression of chemokine in
the brain and the subsequent development of EAE [12]. Our results show that IL-17
promotes the activation of astrocytes to release proinflammatory cytokines
in vitro and in EAE mice [7, 13]. Further studies indicate that IL-17
activates protein kinase B /mammalian target of rapamycin (AKT/mTOR), and Janus
kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3)
signaling pathways [14, 15]. In addition, Li et al. [16] find that IL-17 mediates
neuroinflammation and astrocyte polarization through nuclear factor-
Metformin (Met) is widely prescribed as a first-line therapy for type 2 diabetes, where its primary mechanism involves AMP-activated protein kinase (AMPK) activation and the reduction of insulin resistance [9, 17]. Furthermore, Met has an anti-inflammatory effect by suppressing AKT in macrophages and promotes wound healing via the AKT/mTOR pathway [18, 19]. Additionally, Met has been found to exert novel immunomodulatory and neuroprotective functions, including inhibiting Th17 cell differentiation and protecting oligodendrocytes to restore CNS integrity [20, 21]. Moreover, Met is recognized as a promising candidate for treating diverse diseases, including cancer and autoimmune diseases, potentially via the mTOR/STAT3 pathway [22, 23]. Notably, Met improves cognitive function by attenuating microglial activation, astrocyte hypertrophy, and the production of pro-inflammatory cytokines in the hippocampus [24]. However, the specific effect and mechanism of Met on astrocytes in MS/EAE remain to be elucidated.
Recent evidence indicates that the antidiabetic drug Pioglitazone (Pio), a
potent agonist of peroxisome proliferator-activated receptor
In this study, we aimed to investigate the effects and mechanisms of Met and Pio in the regulation of neurotoxic A1-like astrocyte activation and the progression of EAE. We demonstrated that Met and Pio inhibit the AKT/mTOR/STAT3 signaling pathway and neurotoxic astrocyte activation both in vitro and in vivo, which in turn reduced pro-inflammatory cytokine production, ultimately ameliorating the pathology in EAE mice.
Female 6 to 8-wk-old C57BL/6 mice were obtained from the Shanghai Experimental Animal Center, Chinese Academy of Sciences. All mice were housed in specific pathogen-free conditions. All animal protocols were approved by the Laboratory Animal Ethics Committee of Xuzhou Medical University. Euthanasia was conducted in compliance with the “AVMA Guidelines for the Euthanasia of Animals”. Mice were euthanized by intraperitoneally injected 150mg/kg pentobarbital sodium (P3761, Sigma-Aldrich, St. Louis, MO, USA).
Antibodies and reagents employed in the present study were as follows:
anti-p-AKT (Ser473, 4060, Cell Signaling Technology, Danvers, MA, USA), anti-AKT
(4685, Cell Signaling Technology), anti-p-mTOR (Ser2448, 2971, Cell Signaling
Technology), anti-mTOR (4517, Cell Signaling Technology), anti-p-STAT3 (Tyr705,
BS4181, Bioworld Technology, Bloomington, MN, USA), anti-STAT3 (12640, Cell
Signaling Technology), anti-glial fibrillary acidic protein (GFAP) (Rabbit,
ab7260, Abcam, Cambridge, UK), anti-GFAP (Mouse, ab4648, Abcam), anti-complement
component 3 (C3) (ab11862, Abcam). Alexa
Fluor® 488 donkey anti-mouse IgG (A21202) and Alexa
Fluor® 594 donkey anti-Rabbit IgG (A21207) antibodies were from
Life Technologies (Carlsbad, CA, USA). Myelin oligodendrocyte glycoprotein (MOG)
amino acids 35–55 (MOG35-55 peptides, MEVGWYRSPFSRVVHLYRNGK) were
purchased from China Peptides Co., Ltd. (051716, Shanghai, China). Met and Pio were
purchased from Med Chem Express (HY-B0627, HY-13956, MCE, Monmouth Junction, NJ, USA). Recombinant
mouse IL-17 was from R&D Systems (7956-ML, Minneapolis, MN, USA). Cytokine and chemokine
detection kits: Interleukin‑6 (IL-6) (88-7064-88, Invitrogen, Vienna, Austria), Tumor necrosis factor-alpha (TNF-
Procedures employed for EAE induction were performed as detailed in prior work [7, 27]. Female C57BL/6 mice aged 6–8 weeks were randomly assigned to groups. For EAE induction, 250 µg of MOG35-55 was dissolved in 0.1 mL of sterile PBS, and 500 µg of inactivated Mycobacterium tuberculosis (H37Ra strain, 231141, Difco, Franklin Lakes, NJ, USA) was emulsified in 0.1 mL of complete Freund’s adjuvant (F5881, Sigma Aldrich). The two solutions were repeatedly aspirated and expelled through a three-way connector in an ice-water bath until a stable water-in-oil emulsion was formed. A total of 0.2 mL of this antigen emulsion was administered subcutaneously to each mouse via 4–6 injection sites along the back. Additionally, 200 ng of pertussis toxin (BP-225, lot: 4376920, Invitrogen) was dissolved in 0.2 mL of sterile PBS and administered intraperitoneally on days 0 and 2 post-immunization. Subsequently, neurological function scores were assessed daily on a 0–5 scale as described previously in a double-blinded way: 0, No clinical signs; 1, Loss of tail tone or limp tail; 2, Hind limb paresis (unilateral or bilateral); 3, Complete paralysis of both hind limbs; 4, Complete hind limb paralysis with forelimb paresis or paralysis; 5, Moribund state or death.
Primary astrocytes from mice were isolated and cultured following the detailed procedures reported in our previous study [6, 20]. Briefly, the cerebral cortex freed of meninges was dissected, minced, and then digested with 0.125% trypsin (T4049, Sigma-Aldrich). Following two rinses with Dulbecco’s modified Eagle’s medium/F-12 (DMEM/F-12, 11320033, Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS, 10099141, Gibco), the cell suspension was passed through a sterile filter and seeded into culture flasks that had been pre-equilibrated at 37 °C with 5% CO2. The cells were subsequently expanded for 3–4 additional passages under standard conditions. Finally, at least 95% positive cells of glial fibrillary acidic protein, as determined by the immunofluorescent assay, were applied to the experiments in vitro. Astrocytes were serum-starved for 12 h in FBS-free medium before IL-17 stimulation. All primary cells were validated for their identity by surface marker analysis and tested negative for mycoplasma.
Based on the drug manufacturers’ instructions (MCE), previous studies [28, 29, 30, 31], and our preliminary experiments, drug treatments are as follows:
Animal: Met 100 mg/kg/day and Pio 15 mg/kg/day were suspended in DMSO/Saline solution, respectively, and delivered individually via intraperitoneal injection, or combined and administered in a single injection of 100 µL total volume. The drug administration was performed from day 0 to day 19 post-immunization. Female C57BL/6 mice aged 6–8 weeks were randomly assigned to the following groups: normal control (NC) group, EAE group, DMSO + EAE group, Met + EAE group, Pio + EAE group, and Met + Pio + EAE group (15 mice per group).
Astrocytes: 10 mM Met is administered for 2 h, followed by stimulation with IL-17, and 10 µM Pio is administered for 1 h before IL-17 stimulation in astrocytes.
Cells or spinal cord tissues were processed for RNA extraction using TRIzol reagent (15596-026, Invitrogen). The first-strand cDNAs were developed using the Prime-Script TM RT reagent kit (RR037A, TaKaRa, Kusatsu, Shiga, Japan) from (1 µg RNA), and real-time PCR was carried out on a Roche LightCycler® 480 system (Roche Diagnostics, Basel, Switzerland) with SYBR Green qPCR Master Mix (04887352001, Roche Diagnostics). The sequences of all primers were listed in Supplementary Table 1. The specificity of each primer pair was confirmed by melting curve analysis, and the amplification efficiency was validated using a standard curve, with efficiencies ranging from 95% to 105% and R2 values greater than 0.999. The gene transcription levels were quantified by the 2-ΔΔCT method.
ELISA was performed in accordance with the kit manufacturer’s protocol. In
brief, the sera obtained from mouse blood or conditioned medium from primary mouse
cultured astrocytes were harvested to quantify the secreted level of IL-6,
TNF-
As described previously [27], the total protein was extracted from primary
astrocytes and spinal cord tissues of mice, respectively. Equivalent protein
extracted from the mouse brain tissue (50 µg) or primary astrocytes (20
µg) was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), then electro-transferred onto polyvinylidene
fluoride (PVDF) membrane. After blocking with 5% non-fat milk for 2 h at room
temperature, the membrane was probed with primary antibody overnight at 4
°C. Primary antibodies were applied as follows: p-mTOR (1:1000), mTOR
(1:2000), p-AKT (1:1000), AKT (1:2000), p-STAT3 (1:500), STAT3 (1:1000) and
Immunofluorescent staining of primary astrocytes was performed as described previously [7]. Briefly, frozen sections (10 µm) from the tissue of the spinal cord were incubated at room temperature for 15 minutes with 10% goat serum for blocking, followed by overnight incubation with the primary antibody at 4 °C. The primary antibodies used are as follows: anti-C3 (1:50), anti-p-STAT3 (1:100) and anti-glial fibrillary acidic protein (1:200). After a rinse, the sections were incubated at 37 °C for 1 h with Alexa488-conjugated donkey anti-mouse IgG (A21202, Life Technologies) and Alexa594-conjugated anti-Rabbit IgG (A21207, Life Technologies). Ultimately, the sections were counterstained using 4′,6-diamidino-2-phenylindole (DAPI) (D9542, Sigma-Aldrich) and subsequently imaged with an Olympus IX51 (Olympus Corporation, Tokyo, Japan) or BX51 microscope (Olympus Corporation) (exposure times of 30 ms-100 ms). Quantitative fluorescence analysis was performed using ImageJ software (version 1.53, National Institutes of Health, Bethesda, MD, USA), where the mean fluorescence intensity is calculated by dividing the Integrated Density by the Area of the selected region.
Mice were anesthetized and perfused with 4% paraformaldehyde (P6148, Sigma-Aldrich) in 0.1 M sodium phosphate buffer (pH 7.4). Spinal cords were then rapidly extracted and fixed with the same fixation solution overnight at 4 °C. To evaluate inflammation and demyelination of EAE mice, 4 µm paraffin-embedded spinal cord sections were performed by hematoxylin and eosin (H&E) and luxol fast blue (LFB) staining, respectively [7]. Moreover, the ultrastructure of spinal cords was examined using an electron microscope (EM, HT7800, Hitachi High-Technologies, Tokyo, Japan).
Data are given as mean
Studies on MS patients have confirmed that Th17 cells are abundant in peripheral
blood, cerebrospinal fluid and focal lesions. Furthermore, the number of Th17
cells and the levels of Th17 cells-associated inflammatory mediators are
significantly increased during the relapse phase of MS. Therefore, IL-17, as the
key cytokine predominantly secreted by effector Th17 cells, plays an important
role in the process of MS [31, 32]. We firstly detected the activation of
neurotoxic astrocytes and the production of inflammatory cytokines in mouse
primary astrocytes stimulated by IL-17 in vitro. The results of real-time PCR showed that the mRNA transcription levels of the related markers of
A1-like astrocytes, including UDP glucuronosyltransferase 1 family, polypeptide a
(Ugt1a), Histocompatibility 2, D region locus 1 (H2D1), Serpin peptidase
inhibitor, clade G, member 1 (Serping1), Histocompatibility 2, T region locus
23 (H2T23), serglycin (Srgn) and C3, were
significantly increased at 6 h by IL-17 stimulation and persisted until 12 h,
compared with 0 h group (Fig. 1A). Meanwhile, the mRNA level of inflammatory
cytokines and chemokines, including IL-6, TNF-
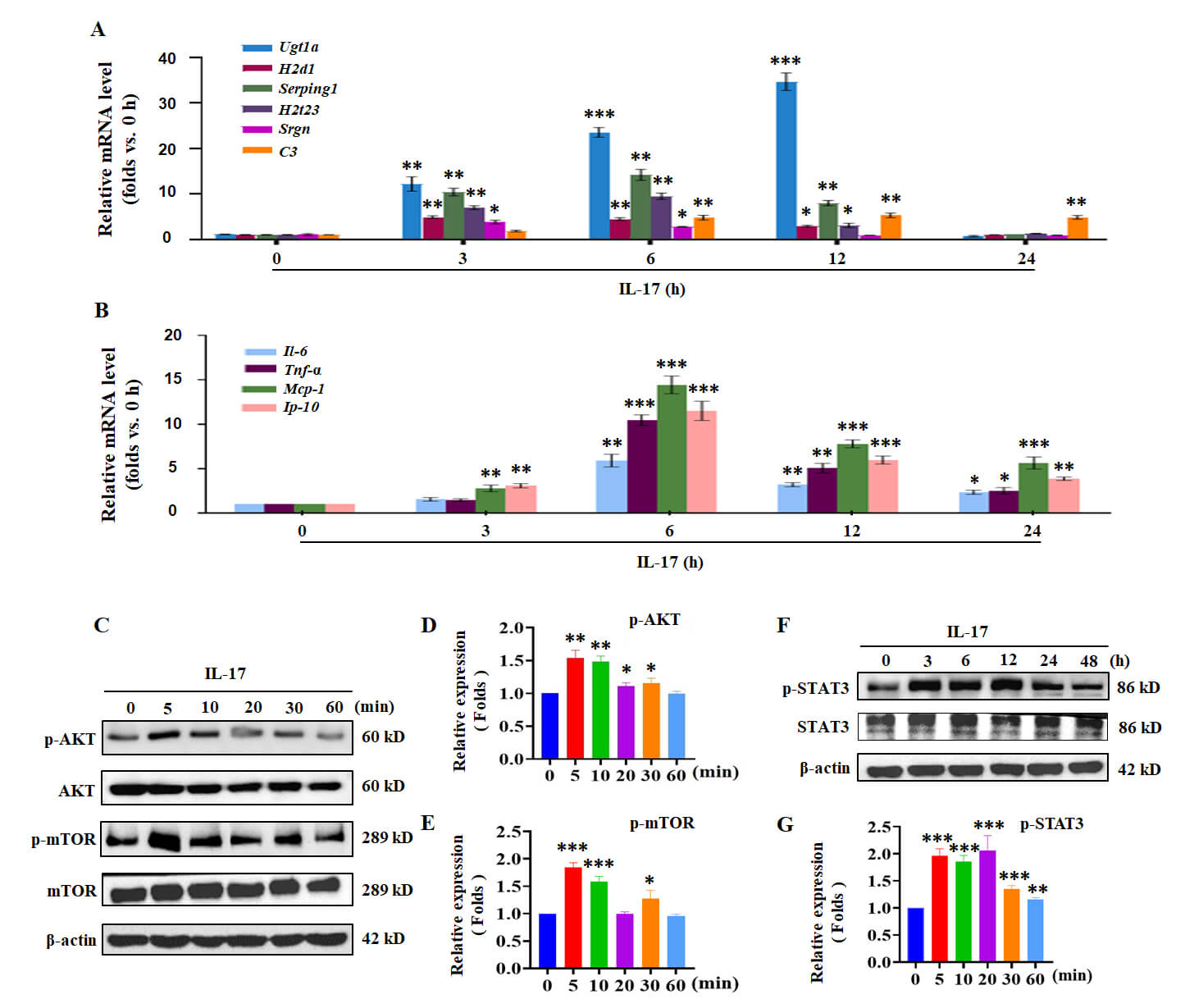 Fig. 1.
Fig. 1.
IL-17 induces the activation of A1-like reactive astrocytes and
AKT/mTOR/STAT3 signals in vitro. (A) The mRNA transcription
levels of the related markers of A1 astrocytes (Ugt1a, H2D1, Serping1, H2T23,
Srgn and C3) and (B) of inflammatory cytokines and chemokines (IL-6,
TNF-
Due to the association of Akt/mTOR/STAT3 pathways with Th17 response and inflammation [23, 25, 33], we further explored the possible molecular mechanisms of IL-17-induced activation of A1-like astrocytes. Western blotting assay was used to detect the activation of the AKT/mTOR/STAT3 pathway. The results showed that the phosphorylation of AKT and mTOR reached their peak at 5 min after IL-17 stimulation (Fig. 1C–E; The original Western blotting images are provided in the Supplementary Material-WB image). Meanwhile, the level of STAT3 phosphorylation (p-STAT3) was increased at 3 h and maintained until 12 h (Fig. 1F,G). Overall, the results suggested that IL-17 induced the activation of neurotoxic astrocytes and the AKT/mTOR/STAT3 signaling pathway.
To determine the effect of Met and Pio on the activation of A1-like astrocytes, primary mouse astrocytes were pre-treated with Met and Pio prior to being stimulated by IL-17. As shown in Fig. 2A, the mRNA transcription levels of the related markers of A1-like astrocytes, including Ugt1a, H2D1, Serping1, H2T23, Srgn and C3, were significantly decreased by Met and Pio, compared with IL-17 stimulation. C3 is one of the most important markers of A1-like astrocytes [9]. Furthermore, the results of immunofluorescence saining sssay (IFA) showed that the protein expression of C3 was decreased in astrocytes treated with Met and Pio prior to IL-17 stimulation (Fig. 2B,C). These results suggested that Met and Pio may downregulate the activation of A1-like reactive astrocytes.
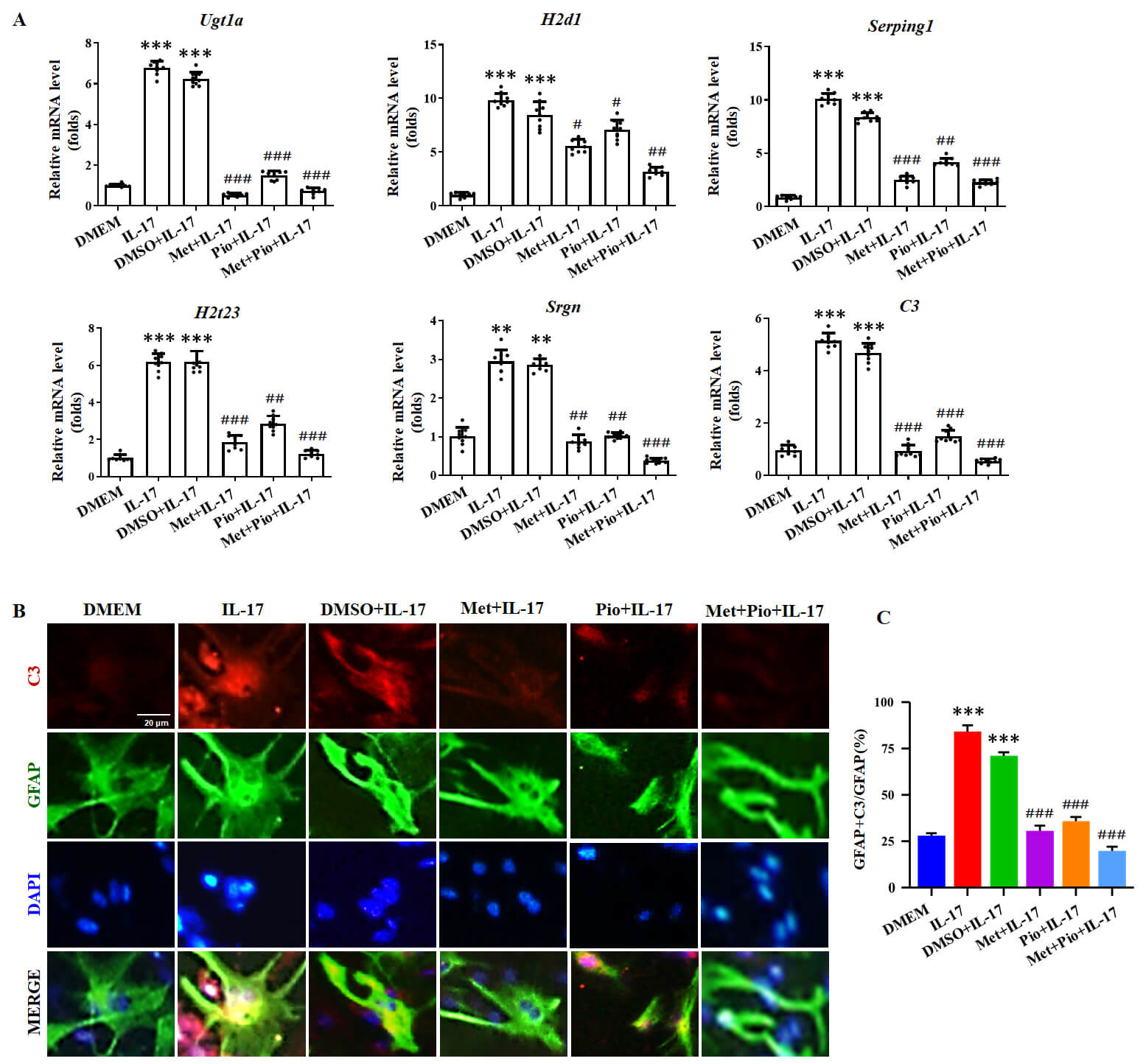 Fig. 2.
Fig. 2.
Met and Pio suppress the A1-like phenotype of astrocytes induced
by IL-17. Primary mouse astrocytes were pretreated with Metformin (Met) and
Pioglitazone (Pio), prior to being stimulated with IL-17 for 6 h. (A) The mRNA
transcription levels of the related markers of A1 astrocytes were detected by
real-time PCR assay. (B) Immunofluorescent staining for glial fibrillary acidic
protein (GFAP), C3 and nuclear staining of
4′,6-diamidino-2-phenylindole (DAPI) in primary astrocytes treated with IL-17.
Scale bars, 20 µm. (C) The number of C3-positive cells was analyzed.
Data are derived from three independent experiments and are presented as the
means
To explore the possible mechanism of Met and Pio in downregulating A1-like astrocytes activation, the activation of AKT/mTOR/STAT3 pathway was measured in primary mouse astrocytes treated with IL-17, prior to pre-treatment with Met and Pio. The results presented that the phosphorylation level of AKT and mTOR was suppressed by Met and Pio, compared to the IL-17 + DMSO treatment group (Fig. 3A–C; The original Western blotting images are provided in the Supplementary Material-WB image). And then, IFA was employed to observe the phosphorylation level of STAT3. The results displayed that the level of p-STAT3 was dramatically decreased and the nuclear translocation was significantly reduced in pre-treatment of primary mouse astrocytes with Met and Pio, compared with the IL-17 + DMSO treatment group (Fig. 3D,E).
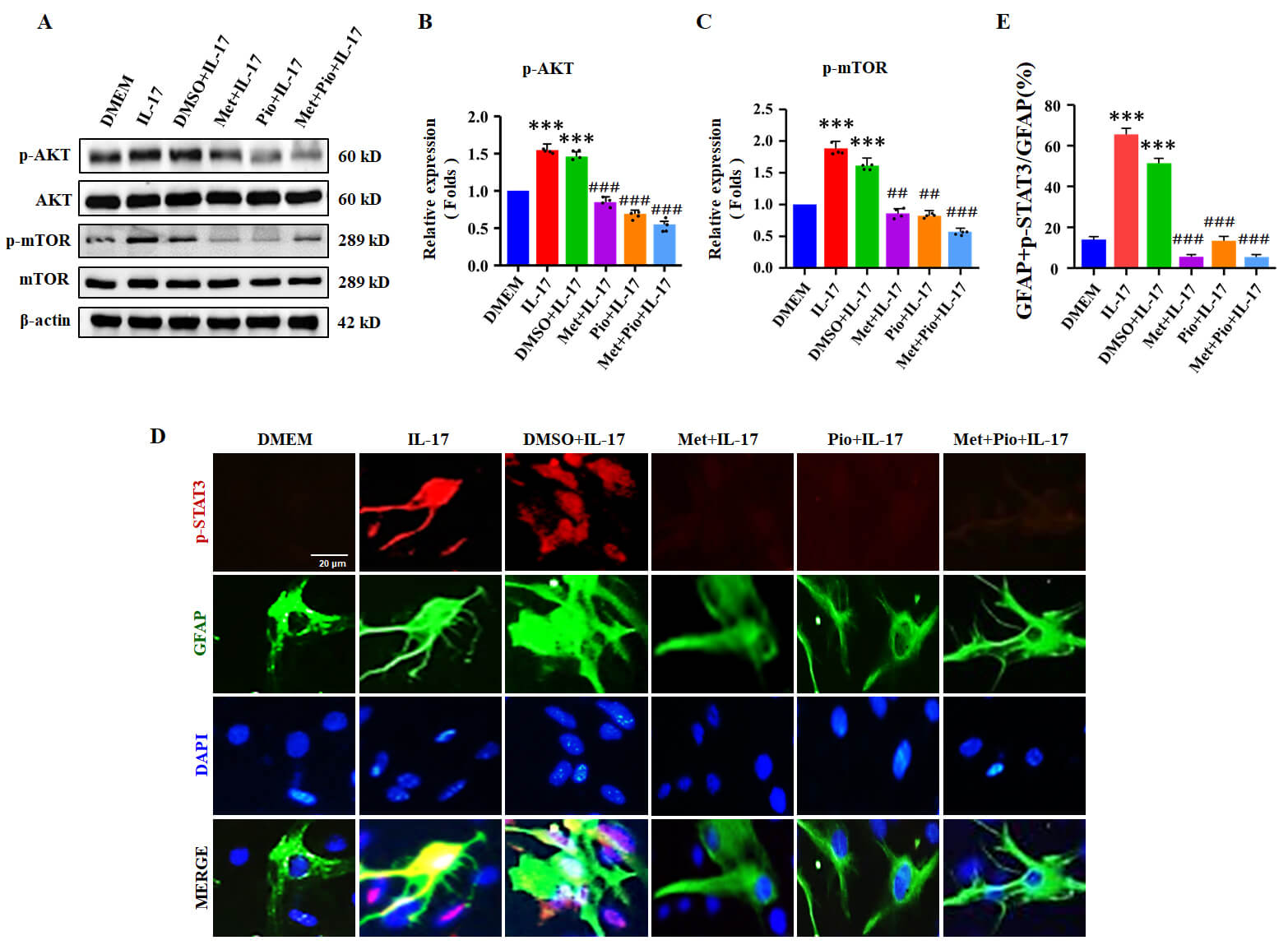 Fig. 3.
Fig. 3.
Met and Pio downregulate AKT/mTOR/STAT3 signals in astrocytes
activated by IL-17. Primary mouse astrocytes were pretreated with Met and Pio,
prior to being stimulated by IL-17. (A) The levels of p-AKT and p-mTOR were
evaluated by Western blotting assay in primary mouse astrocytes stimulated by
IL-17 for 5 min and 3 h, respectively. (B,C) Relative density was employed to
evaluate the protein expression of p-AKT and p-mTOR. (D) Immunofluorescent
staining for GFAP (green), p-STAT3 (red) and nuclear staining of DAPI (blue) in
primary astrocytes treated with IL-17 for 6 h. Scale bars, 20 µm.
(E) The number of p-STAT3-positive cells was analyzed. The data are derived from
three separate experiments and are presented as the means
Furthermore, we asked whether Met and Pio disturbed Akt/mTOR/STAT3 pathways to
regulate the inflammatory reaction. Thus, the mRNA transcription and secretion
levels of inflammatory cytokines and chemokines (IL-6, TNF-
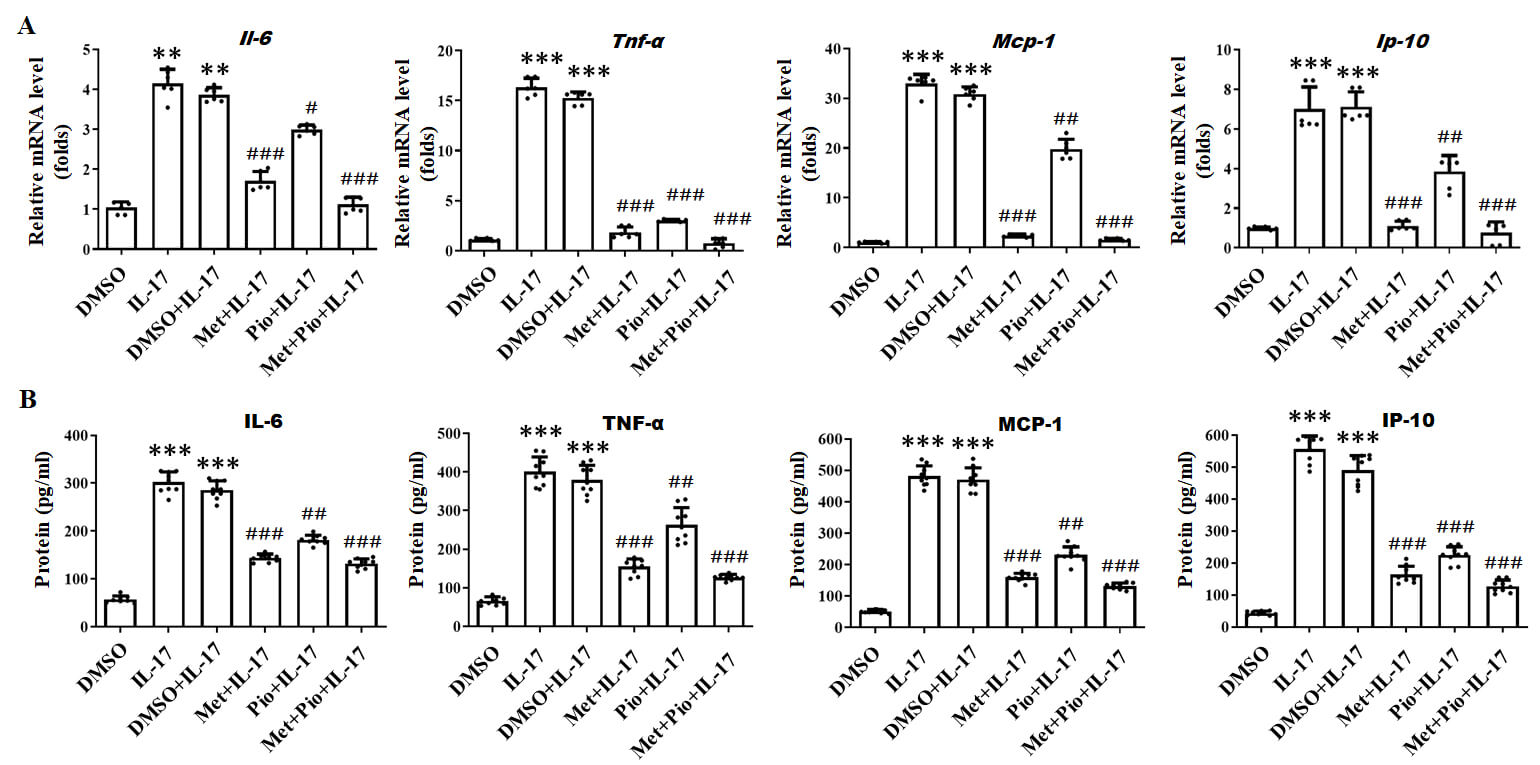 Fig. 4.
Fig. 4.
Met and Pio decrease the production of pro-inflammatory
cytokines in astrocytes treated with IL-17. (A) The mRNA transcription
levels and (B) the secreted levels of inflammatory cytokines and
chemokines (IL-6, TNF-
To determine whether to affect the activation of A1-like astrocytes in
vivo, Met and Pio were injected into mice constituted by MOG35-55on day 0
post immunization (dpi) till 19th dpi. Firstly, the clinical score revealed that
Met and Pio not only delayed the onset time of EAE but also alleviated
pathogenesis (Fig. 5A, Supplementary Fig. 1). Meanwhile, the production
and release of IL-6, TNF-
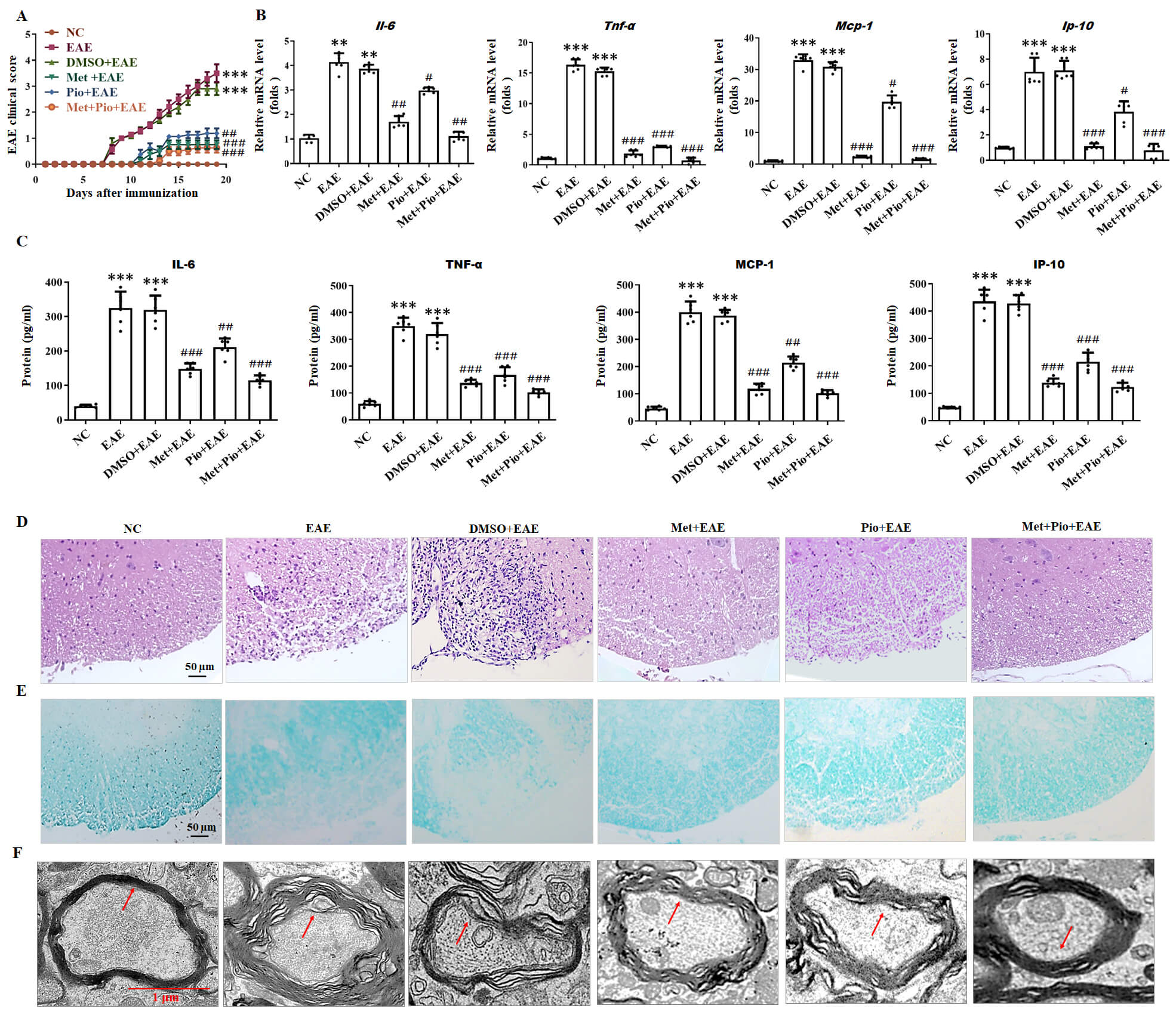 Fig. 5.
Fig. 5.
Met and Pio alleviate experimental autoimmune
encephalomyelitis (EAE) pathogenesis and reduce the production of inflammatory
cytokine in mice. (A) The EAE clinical scores were assessed daily on 15 mice per
group. (n = 15 mice per group). (B,C) The concentrations of IL-6,
TNF-
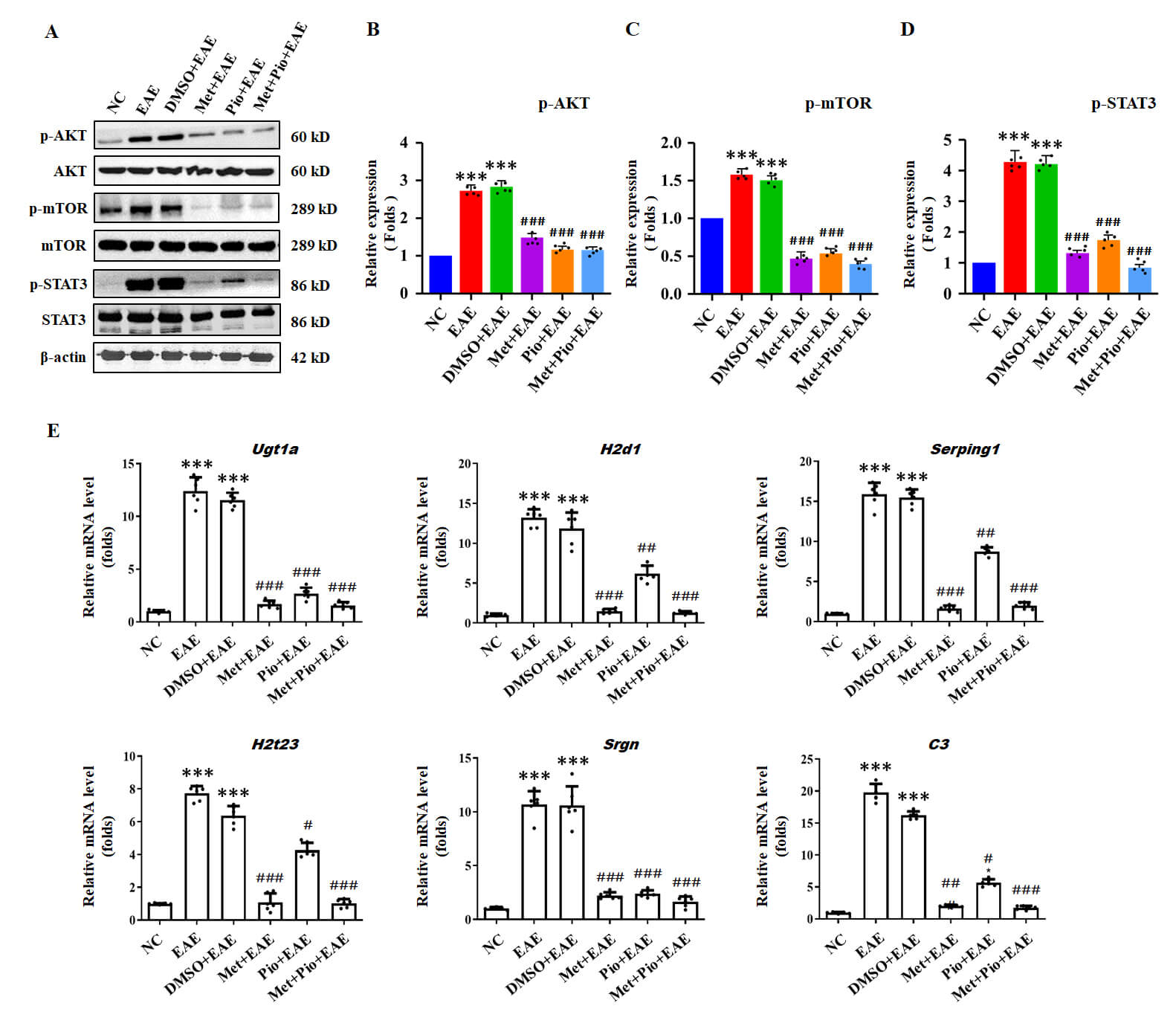 Fig. 6.
Fig. 6.
Met and Pio inhibit AKT/mTOR/STAT3 signals and A1
astrocytes activity in EAE mice. (A) The levels of p-AKT, p-mTOR and p-STAT3
were evaluated by Western blotting assay. (B–D) Relative density was used to
analyze the protein expression of p-AKT, p-mTOR and p-STAT3. (E) The mRNA
transcription levels of the related markers of A1 astrocytes (Ugt1a, H2D1,
Serping1, H2T23, Srgn and C3) were detected by real-time PCR assay. ***p
Reactive astrocytes play a vital role in recruiting inflammatory cells to the lesion sites, which participates in the positive-feedback loop to drive the progression of MS/EAE pathogenesis [8, 34]. Persistent and excessive pro-inflammatory cytokines and neurotoxic mediators from reactive astrocytes exacerbate BBB dysfunction and the massive infiltration of immune cells into the CNS [34]. Therefore, these combined effects of reactive astrocytes produce an inflammatory environment in the MS lesions to enlarge the process of illness. Herein, we find that Met and Pio, two antidiabetic drugs, alleviate the pathological process of EAE mice by downregulating A1-like astrocytes and inflammatory response via inhibiting AKT/mTOR/STAT3 signals.
It is well known that T cells, B cells, glial cells and peripheral inflammation are all implicated in the disease progression of MS/EAE. Among these pathogenic mediators, Th17 cells play a pivotal role in the pathological process of MS/EAE. Th17 cells promote inflammatory responses by secreting IL-17, thereby exacerbating the onset and progression of EAE/MS. Previous studies have shown that IL-17 can activate astrocytes, which in turn triggers the secretion of large amounts of pro-inflammatory cytokines [10, 11, 12, 13, 14]. Beyond amplifying the inflammatory cascades, IL-17-mediated astrocyte activation further enhances microglial activation and contributes to oligodendrocyte damage, thus aggravating the pathological process of MS/EAE. In this study, we found that IL-17 enhances the secretion of pro-inflammatory cytokines in mouse primary astrocytes, manifesting a neurotoxic astrocyte phenotype.
Neurotoxic reactive astrocytes, also termed A1 phenotype astrocytes, participate in various CNS diseases, such as neurodegenerative and demyelinating diseases [9, 35, 36, 37]. A1 astrocytes induced by activated microglia are impaired in supporting neuronal survival, neurite outgrowth, synaptogenesis and phagocytosis, in turn promoting the death of neurons and oligodendrocytes [9]. Inhibiting the conversion of astrocytes to the neurotoxic A1 phenotype exerts neuroprotective properties in some neurodegenerative disorders and neurologic injuries [35, 36]. Our results and Li et al. data [16] showed that IL-17 enhanced the expression levels of specific markers of A1-like astrocytes, such as Ugt1a, H2D1, Serping1, H2T23, Srgn and C3, which suggests that IL-17 may induce the activation of A1 reactive astrocytes.
Emerging evidence indicates that the PI3K/Akt/mTOR and AKT/STAT3 pathways are
closely implicated in the activation of A1-like astrocytes [38, 39, 40, 41]. A recent
study shows that the downregulation of the PI3K/AKT pathway alters the conversion
of the A1 phenotype [38]. Moreover, IL-10, an anti-inflammatory cytokine,
inhibits A1 phenotype through suppressing the STAT3 pathway [39]. Here, IL-17
activated AKT/mTOR/STAT3 signal pathways, and enhanced the production of key
inflammatory cytokines and chemokines: IL-6, TNF-
Met is a popular oral glucose-lowering drug, widely used as therapy for patients with type 2 diabetes mellitus (T2DM) [42]. Mechanistically, Met interferes with the AMPK and mammalian target of rapamycin complex 1 (mTORC1) signaling pathways, thereby exerting effects on mitochondrial function and antioxidant activity [43, 44, 45]. And Pio is another drug to be approved for the treatment of T2DM, and also possesses potent anti-inflammation activity [46, 47]. Pio downregulates mTOR signaling in the inflammatory response of astrocytes [25]. Our data demonstrated that Met and Pio alleviated EAE pathogenesis in mice by suppressing the A1 phenotype astrocyte, reducing pro-inflammatory cytokine production, and downregulating AKT/mTOR/STAT3 signal pathways. However, the mechanisms by which Met and Pio regulate the neurotoxicity mediated by activated astrocytes require further investigation.
So far, our study still has several limitations. Firstly, our data indicate that
IL-17 drives A1-like astrocyte polarization, as evidenced by the upregulated mRNA
transcription levels of established A1-like markers (including Ugt1a, H2D1,
Serping1, H2T23, Srgn, and C3), but the expression levels of canonical
A1-polarizing cytokines (IL-1
In summary, our current findings display that two antidiabetic drugs, Met and Pio decrease IL-17 activated neurotoxic astrocytes, downregulate AKT/mTOR/STAT3 signal pathways, and repress the production of inflammatory cytokines and chemokines in astrocytes, in turn eventually lessening the development of EAE pathogenesis. Therefore, these findings emphasize that Met and Pio may play critical roles in reducing astrocyte-driven neuroinflammation and MS pathogenesis.
The paper is listed as, “Roles of Metformin and Pioglitazone in Regulating A1-like Astrocyte Activation in EAE Mice” as a preprint on Research Square at: https://www.researchsquare.com/article/rs-5406730/v1.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
XML and FZ designed the experiments; SPQ, JJG, BHY, XJZ, TXZ, and YD performed the experiment; DXS, SWW, YD, TXZ, and XTW analyzed the data, and SPQ wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was conducted with the approval of the Animal Ethics Committee of Xuzhou Medical University (Ethics Approval Number: 202209S050). All procedures were performed in accordance with the ARRIVE guidelines and the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Not applicable.
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20231347 to Liu), Jiangsu Provincial Department of Education (20KJA320004 to Zhou), and the Technology Innovation Foundation of Xuzhou City (KC23242 to Qin).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN47364.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.