



 , Joon Ha Park 2, Ji Hyeon Ahn 3, Myoung Cheol Shin 4, Joongbum Moon 4, Moo-Ho Won 4,*
, Joon Ha Park 2, Ji Hyeon Ahn 3, Myoung Cheol Shin 4, Joongbum Moon 4, Moo-Ho Won 4,* , Jun Hwi Cho 4,*
, Jun Hwi Cho 4,*1 Department of Medicine, Graduate School, Kangwon National University, 24341 Chuncheon, Gangwon, Republic of Korea
2 Department of Anatomy, College of Korean Medicine, Dongguk University, 38066 Gyeongju, Gyeongbuk, Republic Korea
3 Department of Physical Therapy, College of Health Science, Youngsan University, 50510 Yangsan, Gyeongnam, Republic of Korea
4 Department of Emergency Medicine, School of Medicine, Kangwon National University, 24341 Chuncheon, Gangwon, Republic of Korea
Abstract
Global cerebral ischemia remains a major cause of neurological morbidity and mortality, yet effective neuroprotective strategies have shown limited translational success. Experimental studies frequently rely on ischemic duration as a primary determinant of injury severity, implicitly assuming equivalence across global brain ischemia–reperfusion (IR) and cardiac arrest with return of spontaneous circulation (CA/ROSC) models. However, increasing experimental evidence indicates that identical ischemic durations can lead to substantially different neuronal outcomes depending on the physiological and systemic context of ischemia. In brain-restricted global IR models, partial preservation of systemic circulation allows residual metabolic activity, delayed stress responses, and region-specific neuronal vulnerability, most notably delayed neuronal death in the hippocampal cornu ammonis 1 region. By contrast, CA/ROSC is characterized by complete systemic circulatory arrest followed by a biologically hostile reperfusion phase that includes profound mitochondrial dysfunction, heterogeneous reperfusion, blood–brain barrier disruption, and amplification of systemic inflammatory responses. As a result, these qualitative differences shift ischemic injury thresholds toward earlier onset and broader neuronal damage in CA/ROSC, even when ischemic durations are nominally comparable. This review integrates experimental evidence from rat models to examine how energy failure, reperfusion biology, proteostasis disruption, and brain–body interactions collectively determine neuronal vulnerability beyond ischemic duration alone. Through direct comparison of global IR and CA/ROSC paradigms, we highlight limitations of duration-centric interpretations and outline implications for experimental design and translational neuroprotection. Recognition of context-dependent ischemic mechanisms is essential for improving model selection and advancing therapeutic strategies for global cerebral ischemia.
Keywords
- neuronal death
- mitochondrial dysfunction
- reperfusion injury
- oxidative stress
- autophagy
- apoptosis
- neuroinflammation
- brain ischemia
- animal models
Global cerebral ischemia remains a major cause of neurological morbidity and mortality in both experimental and clinical settings. Conditions such as cardiac arrest, severe hypotension, and asphyxia result in transient or sustained interruption of cerebral blood flow, leading to complex cascades of neuronal injury that evolve during ischemia and reperfusion [1, 2]. Despite decades of investigation, effective neuroprotective strategies that translate reliably from experimental models to clinical practice remain limited, particularly in the context of cardiac arrest–related brain injury [3]. Experimental research on global cerebral ischemia has relied heavily on rodent models to define ischemic thresholds, patterns of neuronal vulnerability, and mechanisms of delayed neuronal death. Early landmark studies established that neuronal injury does not occur uniformly across the brain but follows region-specific and time-dependent patterns, most prominently affecting hippocampal cornu ammonis 1 (CA1) pyramidal neurons after transient forebrain ischemia [4, 5]. These observations laid the foundation for the concept of selective neuronal vulnerability and shaped the interpretation of global IR injury for decades. A central assumption underlying much of this work has been that ischemic duration is a primary determinant of injury severity. Indeed, classic mapping studies demonstrated a graded relationship between the length of ischemia and the extent of neuronal damage in rat models of forebrain ischemia [6]. However, growing experimental evidence indicates that ischemic duration alone is insufficient to accurately predict neuronal outcome. Instead, neuronal fate is influenced by multiple interacting factors, including the depth of energy failure, the quality of reperfusion, regional metabolic demand, and systemic physiological disturbances [1, 2, 7].
This limitation becomes particularly evident when comparing brain-restricted global IR models with cardiac arrest followed by return of spontaneous circulation (CA/ROSC) models. Although both paradigms are frequently implemented using comparable ischemic durations—typically on the order of 5–10 minutes in rats—the resulting patterns of neuronal injury differ markedly. Global ischemia–reperfusion (IR) models often produce delayed, region-restricted neuronal death, whereas CA/ROSC models are associated with earlier, broader, and more heterogeneous brain injury that reflects the impact of complete systemic circulatory arrest and post-resuscitation pathophysiology [2, 7]. Despite these differences, the two model classes are frequently discussed interchangeably in the experimental literature, with ischemic duration used as a primary metric for cross-model comparison [6, 7]. This practice risks obscuring fundamental differences in ischemic quality, reperfusion biology, and brain–body interactions that shape neuronal outcomes. Consequently, neuroprotective strategies optimized in brain-restricted ischemia models have repeatedly failed to demonstrate efficacy in cardiac arrest settings, raising questions about the validity of duration-based equivalence across models.
In recent years, advances in molecular profiling, mitochondrial biology, and systemic inflammation research have further highlighted the complexity of global IR injury [8, 9, 10]. These findings support the view that neuronal injury thresholds are dynamic and context-dependent, rather than fixed values determined solely by ischemic duration. The aim of this review is therefore not to reassess ischemic duration per se, but to critically examine why identical ischemic durations can produce divergent neuronal outcomes in rat models of global cerebral ischemia. By comparing brain-restricted global IR and CA/ROSC paradigms, this review synthesizes evidence on ischemic thresholds, regional and temporal patterns of neuronal vulnerability, and mechanistic pathways involving energy failure, reperfusion biology, proteostasis disruption, and brain–body interactions. Through this integrative perspective, we aim to clarify the limitations of duration-centric interpretations and to emphasize implications for experimental design and translational neuroprotection.
This narrative review was designed to synthesize and critically integrate experimental findings that address why nominally identical ischemic durations result in divergent neuronal outcomes in rat models of global cerebral ischemia. Given the conceptual and mechanistic focus of the present work, we adopted a structured but non-systematic review framework, consistent with the aims of mechanistic integration rather than quantitative meta-analysis.
We performed a comprehensive literature search using the PubMed (https://pubmed.ncbi.nlm.nih.gov/), Web of Science (https://www.webofscience.com/), and Scopus (https://www.scopus.com/) databases. Search terms were combined using Boolean operators and included: “global cerebral ischemia”, “forebrain ischemia”, “four-vessel occlusion”, “cardiac arrest”, “return of spontaneous circulation”, “CA/ROSC”, “neuronal death”, “delayed neuronal death”, “ischemic duration”, “mitochondrial dysfunction”, “reperfusion injury”, and “rat model”. To ensure coverage of both foundational concepts and contemporary mechanistic advances, we considered publications spanning from the early 1980s through 2025. Seminal studies defining selective neuronal vulnerability and ischemic thresholds were intentionally included alongside recent experimental and translational reports addressing mitochondrial biology, proteostasis, reperfusion dynamics, and systemic inflammation. In general, studies were selected based on their relevance to mechanistic insights, experimental reproducibility, and their contribution to understanding context-dependent determinants of neuronal injury beyond ischemic duration alone.
Studies were included when they met the following criteria: (1) Experimental studies conducted in rats, focusing on global brain IR or CA/ROSC. (2) Use of defined ischemic durations, particularly within the commonly employed range of approximately 5–10 minutes, allows conceptual comparison across models. And (3) Reporting of histopathological, molecular, physiological, or functional outcomes related to neuronal injury or survival. Studies were excluded if they: (1) Primarily involved focal ischemia models (e.g., middle cerebral artery occlusion), (2) Used non-rodent species as the primary experimental system, or (3) Focused exclusively on anesthetic, pharmacokinetic, or technical outcomes without relevance to neuronal injury mechanisms. Model Stratification and Conceptual Framework: Included studies were stratified into two principal experimental paradigms: (1) brain-restricted global IR models, including four-vessel occlusion and related forebrain ischemia paradigms without complete systemic circulatory arrest; and (2) CA/ROSC models, encompassing asphyxial or ventricular fibrillation–induced cardiac arrest followed by standardized resuscitation protocols. Rather than comparing outcomes solely on the basis of ischemic duration, we evaluated studies within a conceptual framework that emphasizes ischemic quality, reperfusion biology, and systemic involvement. Particular attention was given to differences in energy failure depth, mitochondrial integrity, oxidative and calcium-mediated injury, proteostasis disruption, and brain–body interactions.
From each study, we extracted qualitative information regarding: (1) ischemic duration and experimental conditions, (2) regional and temporal patterns of neuronal injury, (3) molecular and cellular injury mechanisms, and (4) reported implications for neuroprotection or recovery. Rather than performing quantitative aggregation, we synthesized evidence thematically, integrating findings from both classical and contemporary literature to construct a mechanistic narrative explaining divergent neuronal outcomes despite comparable ischemic durations.
As a narrative review, this work does not include a formal quantitative assessment of publication bias or effect size. However, we placed particular emphasis on reproducible experimental paradigms, consistency of reported injury patterns, and convergence of mechanistic evidence across independent laboratories. By focusing on rat models exclusively, species-related variability was minimized, allowing clearer conceptual comparison between global IR and CA/ROSC paradigms.
Section 3 provides an integrative overview of experimental rat models used to study global cerebral ischemia, focusing on brain-restricted IR models and CA/ROSC models. Although these two paradigms are often implemented using comparable ischemic durations, they differ fundamentally in hemodynamic context, degree of systemic involvement, and the temporal and regional patterns of neuronal injury [5, 6, 7]. By outlining the methodological foundations, characteristic histopathological features, and evolving molecular insights of each model, this section establishes a conceptual framework for understanding why identical ischemic durations can produce divergent neuronal outcomes. This comparative perspective emphasizes the contribution of systemic factors, reperfusion biology, and brain–body interactions, thereby setting the stage for subsequent sections that examine ischemic thresholds, regional vulnerability, and mechanistic integration across models. These model-specific differences are schematically summarized in Fig. 1, illustrating how nominally similar ischemic durations can generate distinct neuronal injury trajectories depending on systemic context and reperfusion biology.
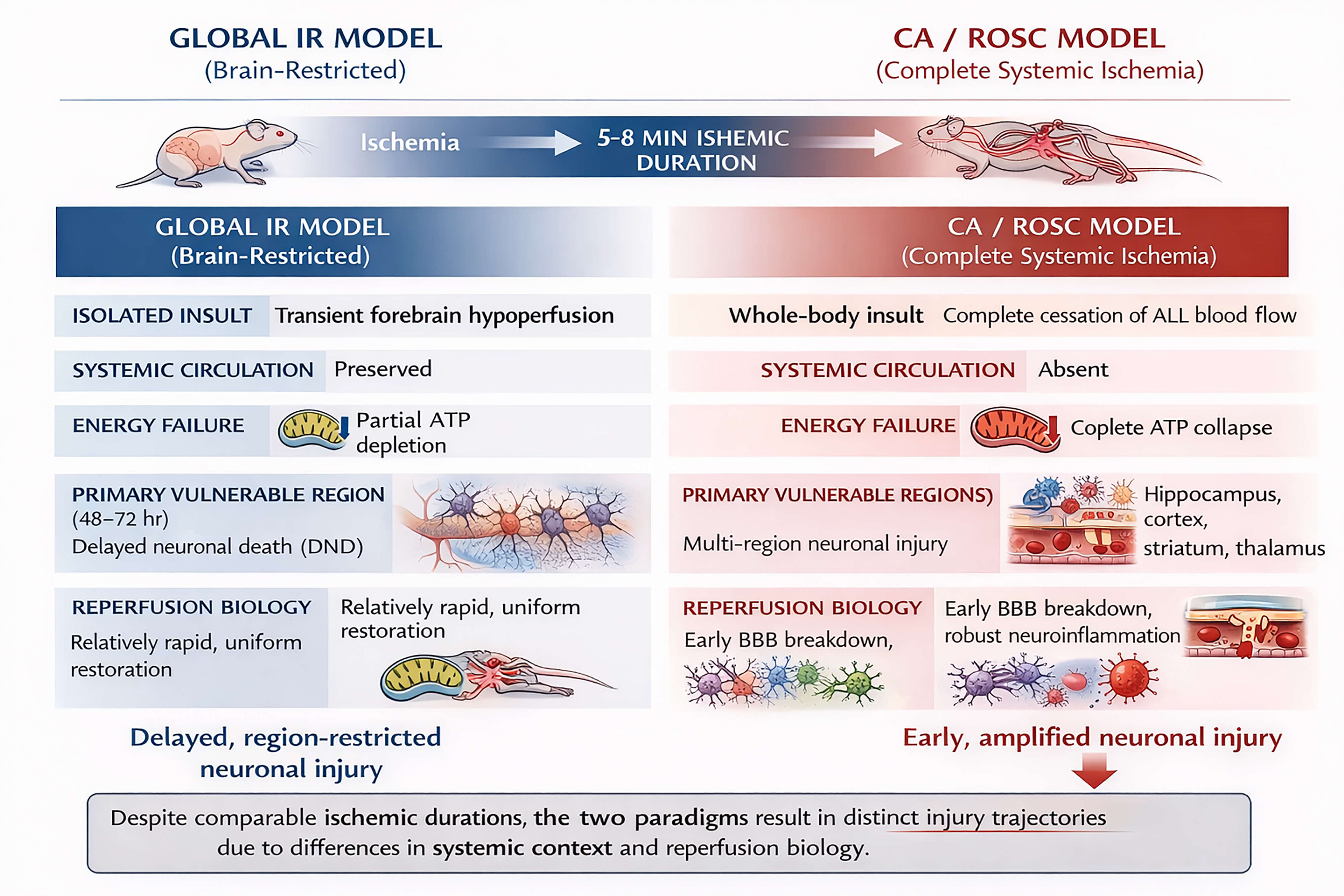 Fig. 1.
Fig. 1.
Divergent neuronal injury trajectories in rat models of brain-restricted global IR and CA/ROSC. This schematic compares experimental rat models of brain-restricted global IR (left) and CA/ROSC (right) implemented with comparable ischemic durations (typically 5–8 min). In global IR models, systemic circulation is preserved, resulting in partial energy failure, relatively uniform reperfusion, and delayed, region-restricted neuronal injury, most prominently delayed neuronal death in the hippocampal CA1 region. By contrast, CA/ROSC is characterized by complete systemic circulatory arrest, leading to abrupt global energy collapse, biologically hostile reperfusion, early blood–brain barrier disruption, systemic and central neuroinflammation, and spatially expanded neuronal injury across multiple brain regions. Overall, the figure illustrates that identical ischemic durations do not necessarily translate into equivalent injury severity or underlying mechanisms, underscoring the critical role of systemic context and reperfusion biology in shaping ischemic injury thresholds and translational interpretation. IR, ischemia–reperfusion; CA/ROSC, cardiac arrest with return of spontaneous circulation; ATP, adenosine triphosphate; BBB, blood–brain barrier; DND, delayed neuronal death; CA1, cornu ammonis 1.
Global brain IR models in rats have been extensively used to investigate the temporal thresholds and regional vulnerability of neurons under conditions of transient, brain-wide hypoperfusion without complete circulatory arrest [1, 5]. The most widely adopted paradigm is the four-vessel occlusion (4VO) model, originally introduced by Pulsinelli and Brierley [5]. This model involves permanent occlusion of the vertebral arteries followed by transient bilateral common carotid artery occlusion, resulting in forebrain ischemia while maintaining systemic circulation. Using this approach, early studies demonstrated that the duration of ischemia is a critical determinant of neuronal outcome, with approximately 5–10 minutes of ischemia sufficient to induce selective neuronal injury in vulnerable regions [5, 6]. Notably, neuronal damage in this model is not distributed uniformly across the brain. Instead, pyramidal neurons in the hippocampal CA1 region exhibit a characteristic pattern of delayed neuronal death, typically becoming evident 48–72 hours after reperfusion [4, 11]. The delayed nature of CA1 neuronal loss has been a defining feature of global IR models and has contributed substantially to the concept of selective neuronal vulnerability. Histopathologically, affected CA1 pyramidal neurons display cytoplasmic eosinophilia, nuclear pyknosis, and progressive neuronal loss over several days following reperfusion [12]. These pathological features occur in the relative absence of widespread necrosis in other brain regions, indicating that ischemic duration alone does not uniformly determine neuronal fate.
Histological and immunohistochemical analyses have further revealed that neuronal degeneration in global IR models is accompanied by robust glial responses. Astrocytic activation, reflected by increased expression of glial fibrillary acidic protein (GFAP), and microglial activation are consistently observed in the hippocampus during the delayed injury phase [13, 14]. In addition, white matter alterations and myelin loss have been reported, indicating that global IR engages both neuronal and non-neuronal compartments [13]. A study has extended classical histopathological observations by incorporating molecular and proteomic approaches. Proteome profiling of the hippocampus after global IR has shown that IR alters protein networks related to mitochondrial function, stress responses, and synaptic integrity [15]. Furthermore, experimental evidence suggests that delayed neuronal death in global IR involves overlapping cell death programs. Zakharova et al. [16] demonstrated that both autophagy- and apoptosis-related pathways contribute to neuronal loss in the hippocampal CA1 region, supporting the interpretation that delayed neuronal death reflects multifactorial disruption of cellular homeostasis rather than a single dominant death mechanism.
Collectively, global brain IR models provide a controlled framework for studying delayed neuronal injury and regional vulnerability under conditions of transient cerebral hypoperfusion. However, preservation of systemic circulation in these models limits the contribution of extracerebral physiological stressors, a distinction that becomes particularly relevant when comparing global IR with cardiac arrest–induced ischemia.
In contrast to brain-restricted ischemia, CA/ROSC models induce complete cessation of systemic blood flow, resulting in simultaneous ischemia of the brain and peripheral organs [7, 17]. In rats, CA is most commonly induced by asphyxia or ventricular fibrillation, followed by standardized cardiopulmonary resuscitation protocols to achieve ROSC [17, 18]. Typical ischemic durations range from 5 to 8 minutes, overlapping with those used in global IR models. Despite these nominal similarities in ischemic duration, neuronal outcomes in CA/ROSC models differ markedly from those observed in global IR. The abrupt loss of cerebral perfusion during CA leads to rapid depletion of adenosine triphosphate, severe metabolic acidosis, and early mitochondrial dysfunction. Consequently, neuronal injury in CA/ROSC models often emerges earlier and extends across broader brain regions, including the hippocampus, cerebral cortex, striatum, and thalamus [7, 19].
Experimental studies have shown that neuronal death after CA/ROSC is accompanied by pronounced blood–brain barrier disruption, cerebral edema, and neuroinflammatory responses during the early reperfusion period [20, 21]. These pathological features reflect the contribution of systemic ischemia–reperfusion injury, which is largely absent in brain-restricted global IR models [22]. Moreover, mitochondrial dysfunction and impaired energy recovery persist after ROSC, further exacerbating neuronal vulnerability [23]. Importantly, CA/ROSC models also underscore the central role of brain–body interactions in post-ischemic brain injury. Peripheral organ dysfunction, systemic inflammation, and metabolic disturbances can secondarily influence neuroinflammation and neuronal survival, emphasizing that ischemic duration cannot be interpreted independently of systemic context [22, 24].
Thus, while CA/ROSC models more closely approximate the clinical scenario of cardiac arrest, they introduce additional layers of systemic and physiological complexity that fundamentally distinguish them from global brain IR models, even when ischemic durations are matched.
Taken together, the experimental evidence reviewed in Sections 3.1 and 3.2 demonstrates that brain-restricted global IR and CA/ROSC models represent fundamentally distinct biological paradigms, despite frequent use of comparable ischemic durations. As summarized in Table 1 (Ref. [4, 5, 6, 7, 11, 13, 14, 16, 17, 18, 19, 20, 21, 22, 23, 24]), global IR models preserve systemic circulation and are characterized by delayed, region-restricted neuronal injury—most prominently delayed neuronal death in hippocampal CA1 pyramidal neurons—allowing mechanistic interrogation of selective vulnerability under relatively controlled reperfusion conditions. In contrast, CA/ROSC models involve complete systemic circulatory arrest followed by biologically hostile reperfusion, resulting in earlier and spatially expanded neuronal injury accompanied by blood–brain barrier disruption, neuroinflammation, and brain–body interactions that substantially modify post-ischemic outcomes. Collectively, these model-specific differences indicate that ischemic duration alone is insufficient to predict neuronal fate and must be interpreted within the broader context of hemodynamic conditions, systemic involvement, and reperfusion biology. Recognition of these distinctions provides a critical foundation for understanding divergent ischemic thresholds and mechanistic trajectories across models, which are explored in the subsequent sections.
| Category | Global brain IR models (4VO, 2VO) | CA/ROSC models | Key references |
| Ischemic mechanism | Transient forebrain hypoperfusion | Complete systemic circulatory arrest | [5, 6, 7] |
| Systemic circulation | Preserved | Absent | [5, 7, 17] |
| Typical ischemic duration | 5–10 min | 5–8 min | [5, 6, 17, 18] |
| Energy failure | Partial, region-dependent | Rapid and near-complete | [6, 7, 19, 23] |
| Reperfusion pattern | Rapid, relatively uniform | Heterogeneous, systemically disturbed | [7, 20, 21] |
| Primary vulnerable regions | Hippocampal CA1 | Hippocampus, cortex, striatum, thalamus | [4, 7, 11, 19] |
| Temporal profile of injury | Delayed (48–72 h) | Early + delayed | [4, 11, 19] |
| Dominant injury features | Delayed neuronal death, selective vulnerability | Early necrosis + apoptosis + inflammation | [7, 16, 20, 21] |
| Blood–brain barrier disruption | Mild or delayed | Early and pronounced | [20, 21] |
| Neuroinflammation | Predominantly intrinsic | Systemic + central | [14, 20, 22] |
| Glial responses | Delayed astro-/microgliosis | Early, amplified gliosis | [13, 14, 20] |
| Brain–body interaction | Minimal | Prominent (multi-organ IR) | [22, 24] |
| Translational relevance | Mechanistic, reductionist | Clinically representative | [7, 17, 22] |
Abbreviations: 4VO, four-vessel occlusion.
A central assumption in experimental cerebral ischemia research has been that the duration of ischemia is a primary determinant of neuronal injury severity. Nevertheless, accumulating evidence from rat models indicates that ischemic duration alone is insufficient to fully predict neuronal outcome, particularly when comparing brain-restricted IR models with CA/ROSC models. Instead, neuronal fate reflects the interaction between ischemic duration, hemodynamic context, systemic physiological disturbances, and reperfusion-associated processes. In this section, we examine how ischemic thresholds have been conceptualized and explain why nominally identical ischemic durations can generate divergent neuronal outcomes across experimental paradigms [1, 2, 7, 25, 26]. A comparative overview of ischemic duration–dependent threshold dynamics and neuronal injury characteristics in brain-restricted global IR and CA/ROSC models is summarized in Table 2 (Ref. [1, 2, 5, 6, 7, 11, 12, 17, 18, 19, 20, 21, 22, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35]).
| Category | Global brain IR models | CA/ROSC models | Key references |
| Primary determinant of injury | Interaction of ischemic duration with regional vulnerability | Systemic ischemia with compressed injury thresholds | [1, 2, 7, 25, 26] |
| Hemodynamic context | Brain-restricted hypoperfusion with preserved systemic circulation | Complete systemic circulatory arrest | [5, 7, 17] |
| Typical ischemic duration | 5–10 min | 5–8 min | [5, 6, 17, 18] |
| Energy failure | Partial, region-dependent ATP depletion | Rapid and near-complete ATP collapse | [2, 24, 31] |
| Threshold dynamics | Gradual, stepwise crossing of injury thresholds | Rapid exceedance of multiple thresholds | [1, 25, 29] |
| Temporal profile of neuronal injury | Delayed (48–72 h after reperfusion) | Early onset with overlapping delayed components | [11, 12, 19] |
| Spatial distribution of injury | Region-restricted (predominantly hippocampal CA1) | Spatially expanded (hippocampus, cortex, striatum, thalamus, brainstem) | [7, 19, 28] |
| Dominant cell death programs | Delayed apoptosis and proteostasis failure | Early necrosis plus apoptosis/autophagy | [27, 30, 33] |
| Role of reperfusion | Triggers delayed molecular stress responses | Amplifies injury via systemic and cerebral mechanisms | [20, 21, 32] |
| Blood–brain barrier disruption | Mild or delayed | Early and pronounced | [20, 21] |
| Neuroinflammatory contribution | Predominantly intrinsic | Systemic + central (brain–body interaction) | [22, 34] |
| Translational implication | Ischemic duration defines susceptibility | Ischemic duration insufficient without systemic context | [29, 35] |
The concept of ischemic thresholds emerged from early experimental studies demonstrating that neuronal injury evolves in a stepwise manner as cerebral blood flow and energy availability decline. Thresholds have been proposed for reversible electrical failure, loss of ionic homeostasis, and irreversible structural injury [1, 25, 36]. Importantly, these thresholds should not be regarded as fixed values, but instead vary according to multiple factors, including baseline metabolic demand, temperature, and the speed and completeness of reperfusion. At the cellular level, ischemic neuronal death reflects progressive failure of bioenergetic homeostasis, excitotoxic signaling, and mitochondrial integrity rather than a simple function of ischemic duration alone [26]. Recent experimental work further supports this dynamic framework by demonstrating that molecular stress responses and mitochondrial resilience can shift effective injury thresholds even under nominally comparable ischemic durations [9].
In forebrain ischemia models, partial reductions in cerebral perfusion may permit short-term neuronal survival despite suppressed synaptic activity, whereas more severe or prolonged ischemia leads to delayed or immediate neuronal death [37, 38]. This framework has been particularly influential in interpreting delayed neuronal death in the hippocampal CA1 region, where neurons survive the initial ischemic insult but succumb during the reperfusion phase [4, 11, 39]. More recent evidence indicates that delayed neuronal death reflects progressive failure of cellular homeostasis during reperfusion, including disturbances in proteostasis and mitochondrial quality control, rather than representing a direct consequence of ischemic duration alone [27]. Thus, ischemic duration should be viewed as one component within a broader pathophysiological continuum rather than as an isolated determinant.
In global brain IR models, ischemic durations of approximately 5–10 minutes are sufficient to induce selective neuronal injury, most prominently in the hippocampal CA1 region [5, 6, 39]. Despite comparable durations, widespread cortical or subcortical necrosis is generally absent, and neuronal injury evolves gradually over 48–72 hours after reperfusion [12]. Notably, recent analyses further confirm that even under standardized ischemic durations, neuronal injury remains spatially restricted in global IR models, reinforcing the concept of region-specific vulnerability rather than uniform ischemic damage [28]. This delayed and region-restricted pattern of injury reflects several features of global IR models [11, 29]. First, partial preservation of systemic circulation limits the severity of metabolic derangements during ischemia. Second, reperfusion restores oxygen and glucose delivery relatively rapidly, thereby shifting the dominant injury process toward delayed mechanisms such as oxidative stress, impaired protein homeostasis, and dysregulated calcium signaling. At the cellular level, delayed neuronal death in CA1 pyramidal neurons has been shown to involve apoptotic pathways rather than immediate necrosis, highlighting the prolonged vulnerability of these neurons after transient ischemia [30]. Proteomic and molecular studies further demonstrate that reperfusion in global IR preferentially activates stress-response pathways, including proteasomal dysfunction and endoplasmic reticulum stress, rather than immediate necrotic cascades [27]. Histopathological findings—including cytoplasmic eosinophilia, nuclear pyknosis, and progressive neuronal loss—are therefore interpreted as consequences of post-ischemic cellular dysfunction rather than immediate energy collapse [11, 40]. Taken together, these observations indicate that in global IR models, ischemic duration defines susceptibility rather than inevitability, allowing delayed neuronal death to serve as a window for mechanistic investigation.
In CA/ROSC models, ischemic durations often overlap with those used in global IR experiments, typically ranging from 5 to 8 minutes [17, 18]. Nevertheless, despite these nominal similarities, the temporal onset, spatial distribution, and mechanistic drivers of neuronal injury differ markedly from those observed in brain-restricted IR models. Neuronal injury in CA/ROSC models frequently manifests earlier and involves broader brain regions, including the hippocampus, cerebral cortex, striatum, thalamus, and brainstem [7, 19]. A defining feature of CA/ROSC models is the complete cessation of systemic circulation, resulting in abrupt depletion of cerebral adenosine triphosphate, rapid intracellular acidosis, loss of mitochondrial membrane potential, and collapse of ion homeostasis across multiple brain regions [2, 24]. Unlike global IR, where residual perfusion and rapid reperfusion limit the depth of energy failure, CA produces a more uniform and severe ischemic insult at the whole-body level, a concept emphasized early in cerebral resuscitation research [31]. The reperfusion phase following ROSC further amplifies neuronal injury through systemic mechanisms. Experimental studies consistently demonstrate early blood–brain barrier disruption, cerebral edema, and robust neuroinflammatory activation within hours after ROSC [20, 21]. These processes promote secondary neuronal injury by facilitating immune cell infiltration, cytokine release, and oxidative stress, thereby accelerating neuronal loss beyond the delayed paradigm typically observed in global IR models [32]. At the cellular level, neuronal death in CA/ROSC models reflects a convergence of injury pathways rather than a predominantly delayed program. Early necrotic injury driven by profound energy failure coexists with delayed apoptotic and autophagic processes during reperfusion, as evidenced by caspase activation, mitochondrial dysfunction, and dysregulated autophagy signaling in vulnerable regions [23, 33]. This convergence of injury mechanisms shortens the temporal interval between ischemia and irreversible neuronal damage, effectively shifting ischemic thresholds toward earlier time points. Importantly, CA/ROSC models highlight the critical role of brain–body interactions in post-ischemic neuronal injury. Systemic inflammatory responses, metabolic derangements, and multi-organ dysfunction contribute secondarily to neuroinflammation and impaired recovery, reinforcing the interpretation that neuronal fate cannot be dissociated from extracerebral physiology in this setting [22, 34].
Taken together, findings from global IR and CA/ROSC models indicate that ischemic thresholds are model-dependent and context-sensitive. In brain-restricted IR models, thresholds for irreversible neuronal injury are reached gradually and selectively, permitting delayed neuronal death. By contrast, CA/ROSC models rapidly exceed multiple injury thresholds as a result of systemic ischemia and reperfusion-associated insults, resulting in accelerated and spatially expanded neuronal damage. These divergent threshold dynamics are consistent with contemporary interpretations that distinguish localized cerebral ischemia from whole-body ischemia–reperfusion as fundamentally different pathophysiological entities [29]. These distinctions carry important implications for experimental interpretation. Matching ischemic duration across models does not ensure equivalence of injury severity or mechanism. Instead, ischemic duration must be interpreted in conjunction with hemodynamic conditions, systemic responses, and reperfusion biology. Failure to account for these contextual factors may contribute to inconsistencies in experimental outcomes and limit the translational relevance of neuroprotective strategies, particularly when extrapolating from global IR paradigms to clinical cardiac arrest settings, where prolonged post-resuscitation care and temperature management further modulate injury trajectories [35].
Neuronal vulnerability to ischemia is neither spatially uniform nor temporally synchronized across the brain. Experimental rat models of global brain IR and CA/ROSC consistently demonstrate that specific brain regions exhibit distinct ischemic thresholds, injury kinetics, and dominant modes of neuronal death. Notably, these regional and temporal patterns diverge substantially between brain-restricted IR and systemic ischemia induced by CA, even when ischemic durations are nominally comparable [1, 2].
The hippocampus, particularly pyramidal neurons in the CA1 region, represents the most extensively characterized example of selective neuronal vulnerability following transient global ischemia. In global IR models, CA1 neurons typically survive the ischemic episode itself but undergo delayed neuronal death that becomes histologically apparent 48–72 h after reperfusion [4, 11]. Histopathologically, CA1 injury is characterized by cytoplasmic eosinophilia, nuclear pyknosis, and progressive neuronal loss, often accompanied by pronounced astrocytic reactivity marked by increased GFAP expression [12, 41]. These changes occur despite relative preservation of adjacent hippocampal subfields, reinforcing the concept of region-specific vulnerability. More recent studies have further refined this paradigm by indicating that CA1 vulnerability reflects the accumulation of post-ischemic disturbances, including mitochondrial dysfunction, impaired proteostasis, endoplasmic reticulum stress, and dysregulated calcium signaling [27, 40]. Notably, accumulating evidence suggests that CA3 neurons, although more resistant, are not uniformly protected and may exhibit injury under more severe or prolonged ischemic conditions, highlighting a graded rather than absolute vulnerability gradient within the hippocampus [28].
Mechanistic analyses additionally demonstrate that delayed CA1 neuronal death involves overlapping caspase-dependent and caspase-independent pathways together with impaired protein homeostasis, rather than a single dominant process [27, 40]. Consistent with this gradient concept, CA3 pyramidal neurons—while relatively resistant—can undergo injury under conditions of increased ischemic severity or altered reperfusion dynamics [28]. In CA/ROSC models, hippocampal injury often manifests earlier and is less restricted to CA1. Experimental evidence indicates that CA1 neuronal loss may occur sooner after reperfusion and frequently coexists with injury to CA3 and dentate regions, reflecting the deeper metabolic collapse associated with systemic ischemia [7, 19].
Cortical vulnerability differs markedly between global IR and CA/ROSC models. In classical global IR paradigms, neocortical neurons are relatively spared following short ischemic durations, with neuronal injury largely confined to selectively vulnerable regions such as the hippocampal CA1 sector [6]. When cortical injury occurs under these conditions, it is typically delayed and regionally heterogeneous, reflecting partial preservation of cerebral perfusion and rapid restoration of metabolic substrates during reperfusion.
By contrast, CA/ROSC models more frequently produce early cortical neuronal injury, particularly within frontal and parietal cortices. This early cortical involvement reflects the abrupt global energy failure associated with complete circulatory arrest, followed by impaired reperfusion and systemic inflammatory amplification after ROSC [7, 19]. Experimental studies have demonstrated that cortical edema and neuronal degeneration can be detected within days after resuscitation, indicating that cortical ischemic thresholds are exceeded more rapidly in CA/ROSC than in brain-restricted global IR paradigms [19, 20, 22].
Taken together, these observations indicate that cortical vulnerability is strongly shaped by the hemodynamic and systemic context of ischemia, rather than ischemic duration alone.
Subcortical structures, particularly the striatum and thalamus, also show clear model-dependent differences in vulnerability. In brain-restricted global IR paradigms, injury is often concentrated in selectively vulnerable regions such as the hippocampal CA1 sector, whereas striatal and thalamic neuronal loss is comparatively limited or absent at commonly used ischemic durations. This pattern is consistent with classic mapping studies demonstrating non-uniform regional injury distributions after brief forebrain ischemia in rats [6].
In contrast, following CA/ROSC, subcortical injury becomes more prominent, reflecting the combined impact of systemic no-flow ischemia, post-resuscitation hypoperfusion, metabolic acidosis, and reperfusion-associated inflammation. In a multimodal rat CA model, cortical and striatal edema were detectable within several days after resuscitation, followed by evolving white-matter injury over 1–2 weeks, supporting the presence of a broader spatiotemporal injury footprint than that typically observed in brain-restricted global IR models [42]. Moreover, experimental CA paradigms have reported early thalamic injury accompanied by microglial activation within 24 h, particularly under severe asphyxial conditions, indicating that thalamic vulnerability can emerge rapidly when systemic and reperfusion-related stressors are substantial [43]. Contemporary syntheses of preclinical post–cardiac arrest brain injury further emphasize that degenerating neurons can be detected in both cortex and striatum in rat CA models, often in association with blood–brain barrier disruption and inflammatory signatures [44].
Taken together, these findings indicate that striatal and thalamic vulnerability is not simply determined by ischemic duration, but instead reflects the quality of ischemia (brain-restricted versus systemic), the post-resuscitation hemodynamic milieu, and reperfusion biology, which together shift regional injury thresholds in CA/ROSC relative to global IR.
The brainstem represents a critical but often underappreciated target of ischemic injury. In global brain IR models, brainstem nuclei are typically preserved, consistent with partial maintenance of systemic circulation and relatively rapid reperfusion, even when selectively vulnerable forebrain regions such as the hippocampal CA1 sector undergo delayed neuronal death [4, 6]. Recent comparative analyses of experimental ischemia models further support that brainstem structures remain largely resistant to injury in brain-restricted global IR paradigms, unless ischemia is prolonged or compounded by systemic instability [28]. Under these experimental conditions, ischemic thresholds in the brainstem are rarely exceeded at commonly used ischemic durations, and overt histopathological damage is uncommon.
In contrast, CA/ROSC models consistently demonstrate vulnerability of brainstem structures, including respiratory and autonomic nuclei, even after relatively brief ischemic durations [7, 24]. This heightened susceptibility likely reflects the high metabolic demand of brainstem nuclei, their continuous role in cardiorespiratory regulation, and their limited tolerance for systemic hypoxia, hypercapnia, and metabolic acidosis during no-flow ischemia. Experimental studies have shown that CA/ROSC is associated with persistent impairment of brainstem-mediated functions, including blunted hypoxic ventilatory responses and disrupted autonomic regulation, which correlate with reduced long-term survival despite restoration of spontaneous circulation [24, 45]. These observations indicate that brainstem dysfunction is not merely a secondary consequence of supratentorial injury, but instead represents a primary and clinically relevant component of post–cardiac arrest brain injury [2, 34].
The cerebellum represents another region in which vulnerability is strongly influenced by ischemic context. In global IR models, cerebellar neurons—particularly Purkinje cells—are relatively resistant to brief forebrain ischemia, and overt degeneration is uncommon unless ischemic durations are prolonged or compounded by additional stressors such as hyperthermia or hypotension [1, 6]. However, following CA/ROSC, cerebellar injury has been reported in several experimental paradigms, often emerging in a delayed fashion during the post-resuscitation period. Recent studies suggest that cerebellar vulnerability after CA/ROSC is linked to impaired microcirculatory reperfusion, mitochondrial dysfunction, and neuroinflammatory amplification rather than ischemic duration alone [22, 34]. Although cerebellar injury is less consistent than hippocampal or brainstem damage, its occurrence highlights that systemic ischemia and reperfusion-associated disturbances can extend neuronal injury beyond classically defined vulnerable regions, particularly under conditions of severe or prolonged circulatory arrest.
Taken together, these observations indicate that the brainstem and cerebellum are preferentially affected by systemic ischemia and reperfusion stress rather than brain-restricted hypoperfusion, reinforcing the importance of ischemic quality and systemic context in shaping regional patterns of neuronal vulnerability.
Across brain regions, temporal patterns of neuronal injury differ substantially between global IR and CA/ROSC models. Global IR is characterized by delayed and region-restricted neuronal death, with hippocampal CA1 injury typically emerging over several days after reperfusion and remaining the dominant lesion during the subacute phase [4, 11, 12]. This delayed temporal profile reflects neuronal survival through the ischemic episode itself, followed by progressive disruption of cellular homeostasis during reperfusion, rather than immediate energy collapse.
By contrast, CA/ROSC models exhibit a more compressed temporal profile, with early neuronal injury, rapid propagation across multiple brain regions, and overlapping necrotic, apoptotic, and autophagic processes during reperfusion [2, 34]. Experimental CA studies consistently demonstrate that neuronal degeneration, blood–brain barrier disruption, and neuroinflammatory activation can be detected within hours to days after resuscitation, indicating that ischemic thresholds are exceeded earlier and across a broader spatial extent than in brain-restricted global IR paradigms [7, 19].
Taken together, these observations indicate that ischemic thresholds are dynamically shifted by systemic factors and reperfusion biology. Regions that tolerate brief ischemia under brain-restricted conditions may exceed injury thresholds rapidly when systemic ischemia, metabolic derangement, and inflammatory amplification are superimposed. Accordingly, the temporal evolution of injury should be regarded as a model-defining feature rather than a simple function of ischemic duration, with important implications for interpreting experimental outcomes and designing time-sensitive neuroprotective interventions.
Overall, regional and temporal patterns of neuronal vulnerability indicate that ischemic injury cannot be predicted solely by ischemic duration. Instead, neuronal fate reflects a dynamic interplay between region-specific metabolic demands, network connectivity, and intrinsic cellular resilience, as well as the broader physiological context in which ischemia and reperfusion occur. Comparisons between brain-restricted global IR and cardiac arrest–induced ischemia further demonstrate that identical ischemic durations can yield fundamentally different injury phenotypes depending on systemic involvement and reperfusion biology (Table 3, Ref. [1, 2, 4, 6, 7, 11, 19, 20, 22, 24, 28, 34, 42, 43, 44, 45]). Recognition of these spatiotemporal and model-dependent patterns is essential for interpreting experimental outcomes and for designing neuroprotective strategies that are appropriately matched to the underlying ischemic context. If these distinctions are not adequately considered, key mechanisms of selective vulnerability may be obscured and the translational relevance of preclinical neuroprotection studies may be reduced.
| Brain region | Global brain IR models | CA/ROSC models | Temporal pattern of injury | Dominant injury features | References |
| Hippocampus (CA1) | Highly vulnerable; selective delayed neuronal death | Vulnerable with earlier onset; CA1 plus CA3 and dentate gyrus often involved | IR: delayed (48–72 h); CA/ROSC: early–subacute | Mitochondrial dysfunction, impaired proteostasis, apoptosis/autophagy | [4, 7, 11, 19] |
| Hippocampus (CA3, DG) | Relatively resistant; injury under severe or prolonged IR | Frequently involved together with CA1 | IR: late/conditional; CA/ROSC: early | Energy failure, reperfusion stress, inflammatory amplification | [19, 28] |
| Cerebral Cortex | Largely spared after brief IR; heterogeneous delayed injury | Early and prominent cortical injury (frontal/parietal regions) | IR: delayed/heterogeneous; CA/ROSC: early | Cerebral edema, neuronal degeneration, BBB disruption | [7, 19, 20, 22] |
| Striatum | Minimal injury at common IR durations | Frequent injury after CA/ROSC | IR: absent or late; CA/ROSC: early–subacute | Metabolic collapse, inflammatory signaling | [7, 42, 44] |
| Thalamus | Generally preserved | Early vulnerability under severe CA or asphyxial arrest | IR: rare; CA/ROSC: early | Microglial activation, neuronal degeneration | [43, 44] |
| Brainstem | Largely resistant | Highly vulnerable, including autonomic and respiratory nuclei | IR: resistant; CA/ROSC: early and persistent | Energy failure, autonomic dysfunction | [2, 24, 34, 45] |
| Cerebellum (Purkinje cells) | Relatively resistant; injury with prolonged or severe IR | Delayed vulnerability reported after CA/ROSC | IR: resistant; CA/ROSC: delayed/subacute | Microcirculatory failure, mitochondrial dysfunction | [1, 6, 22, 34] |
| Overall Pattern | Region-restricted, delayed neuronal injury | Spatially expanded, systemically amplified injury | IR: delayed; CA/ROSC: temporally compressed | Systemic ischemia–reperfusion, inflammation | [1, 2, 7, 19, 34] |
Abbreviations: DG, dentate gyrus.
Despite the frequent use of comparable ischemic durations in experimental studies, neuronal outcomes differ markedly between global IR models and CA/ROSC models. These mechanistic distinctions, together with their distinct temporal relationships to ischemic thresholds, are summarized schematically in Fig. 2. This divergence reflects not merely quantitative differences in ischemic severity, but rather qualitative differences in energy failure, reperfusion biology, cellular stress handling, and systemic–central nervous system interaction [1, 2, 46]. In the following subsections, we integrate these mechanisms to explain why nominally identical ischemic durations can exceed neuronal injury thresholds in CA/ROSC but not in brain-restricted global IR.
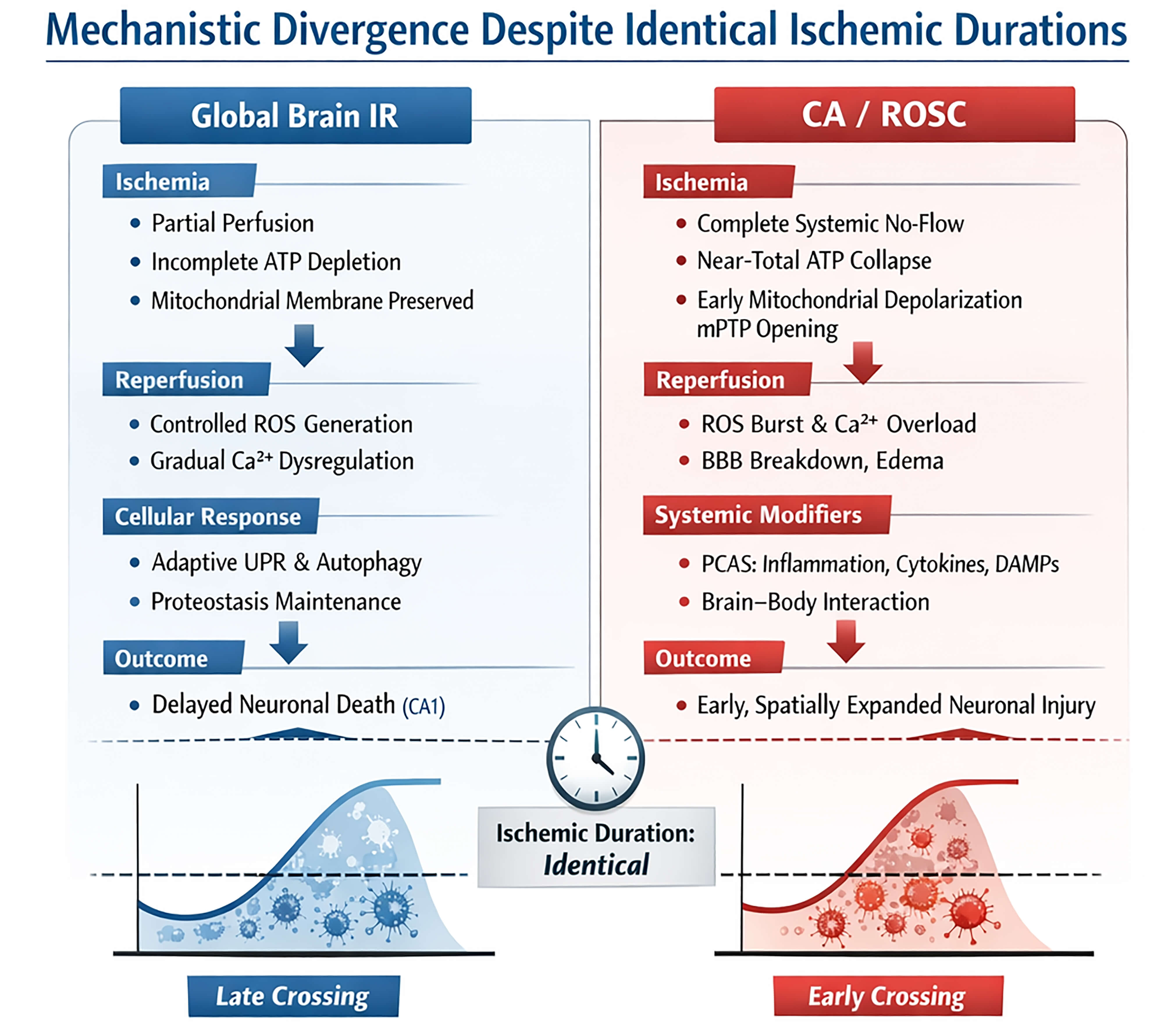 Fig. 2.
Fig. 2.
Mechanistic divergence between global brain ischemia–reperfusion (IR) and cardiac arrest with return of spontaneous circulation (CA/ROSC) despite identical ischemic durations. Schematic comparison illustrating why equivalent ischemic durations result in fundamentally different neuronal outcomes across experimental models. In global brain IR (left), partial preservation of systemic circulation limits energy failure, allowing incomplete ATP depletion and relative mitochondrial integrity during ischemia. Reperfusion is characterized by controlled ROS generation, gradual Ca2+ dysregulation, and activation of adaptive stress responses, including unfolded protein response (UPR) signaling and autophagy, which together culminate in delayed and region-restricted neuronal death, most prominently in hippocampal CA1 neurons. By contrast, CA/ROSC (right) is associated with abrupt and complete systemic no-flow ischemia, leading to near-total ATP collapse, early mitochondrial depolarization, and opening of the mitochondrial permeability transition pore (mPTP). Reperfusion is biologically hostile, with exaggerated ROS bursts, Ca2+ overload, BBB disruption, cerebral edema, and systemic inflammatory amplification as part of post–cardiac arrest syndrome (PCAS). These combined processes promote early and spatially expanded neuronal injury that rapidly crosses injury thresholds despite identical ischemic durations. The lower panels schematically illustrate the differential timing of injury-threshold crossing in global IR (late crossing) versus CA/ROSC (early crossing). ROS, reactive oxygen species; DAMPs, damage-associated molecular patterns.
A fundamental distinction between global IR and CA/ROSC models lies in the depth and completeness of energy failure during ischemia. In brain-restricted global IR, partial preservation of systemic circulation allows residual oxygen and substrate delivery, resulting in profound suppression of synaptic activity but incomplete depletion of adenosine triphosphate (ATP) in many brain regions [1]. Recent experimental work further suggests that, under these conditions, mitochondrial respiration and membrane potential can remain partially preserved during ischemia, enabling neurons to engage delayed recovery or stress-adaptive responses during reperfusion [9, 38].
By contrast, CA/ROSC is associated with abrupt and complete cessation of systemic circulation, leading to rapid and near-total ATP collapse across the brain. This profound energy failure promotes early mitochondrial depolarization and increases the likelihood of mitochondrial permeability transition pore opening, irreversibly compromising oxidative phosphorylation and priming neurons for early death [2]. Experimental studies further demonstrate that mitochondrial functional recovery during reperfusion is markedly impaired after CA/ROSC, even when cerebral blood flow is restored, indicating that ischemic duration alone does not adequately reflect the depth of mitochondrial injury [23, 47, 48].
In global IR models, reperfusion typically restores oxygen and glucose delivery relatively rapidly and with limited systemic disturbance. Under these conditions, neurons transition from ischemic suppression to delayed injury pathways in which ROS generation and calcium dysregulation evolve over hours to days rather than minutes [12]. Recent evidence further suggests that reperfusion after brain-restricted ischemia preferentially activates mitochondrial and cytosolic oxidative stress pathways without immediately overwhelming endogenous antioxidant defenses, permitting delayed neuronal injury rather than rapid necrosis [15, 27].
By contrast, reperfusion following CA/ROSC is frequently heterogeneous and biologically hostile. Systemic inflammatory activation, endothelial dysfunction, and impaired microcirculatory flow amplify the early ROS burst and exacerbate calcium influx through compromised membrane integrity and excitotoxic signaling [7]. Excessive ROS generation and calcium overload destabilize mitochondria and activate calcium-dependent proteases, accelerating neuronal injury [20, 49].
Notably, clinical trials examining post–cardiac arrest temperature management indicate that modulation of reperfusion-associated metabolic stress can influence neurological outcome, underscoring the biological severity of early reperfusion injury in CA/ROSC [50, 51]. Consistent with these observations, blood–brain barrier (BBB) disruption, cerebral edema, and oxidative damage emerge early after CA/ROSC, indicating that reperfusion-associated stress rapidly exceeds neuronal injury thresholds [20, 21].
In global IR models, delayed neuronal death is closely linked to progressive failure of cellular proteostasis rather than immediate structural collapse. Following transient ischemia, neurons activate adaptive stress responses, including the unfolded protein response (UPR), proteasomal degradation, and autophagy, to manage ischemia-induced protein misfolding and organelle damage [9]. In selectively vulnerable populations, such as hippocampal CA1 pyramidal neurons, these protective mechanisms initially preserve cellular integrity but can become maladaptive under sustained stress, leading to delayed neuronal death [4, 27, 52].
By contrast, CA/ROSC imposes a qualitatively different proteostatic burden on neurons. Profound energy depletion rapidly impairs ATP-dependent protein quality control, resulting in early collapse of proteostasis during reperfusion. Experimental CA studies demonstrate robust engagement of apoptotic cascades alongside necrotic features, reflecting the limited capacity of neurons to sustain controlled stress responses under conditions of severe metabolic failure [16, 40]. Autophagy, which may serve adaptive roles in global IR, appears to be insufficiently activated or dysregulated after CA/ROSC, contributing to mixed necrotic–apoptotic neuronal loss.
In brain-restricted global IR models, neuronal injury develops largely within the central nervous system. Although transient ischemia activates local inflammatory and glial responses, systemic physiology remains relatively preserved, limiting extracerebral contributions to post-ischemic brain injury [14]. Under these experimental conditions, neuroinflammation is driven predominantly by intrinsic mechanisms within selectively vulnerable regions, such as the hippocampal CA1 [28].
By contrast, CA/ROSC induces a systemic ischemia–reperfusion syndrome that profoundly reshapes the cerebral injury landscape. Complete circulatory arrest results in simultaneous ischemia of the brain and peripheral organs, followed by reperfusion accompanied by systemic inflammation and immune activation—collectively described as post–cardiac arrest syndrome [2, 46]. Peripheral organ injury leads to the release of circulating cytokines and damage-associated molecular patterns that amplify neuroinflammation and endothelial dysfunction after ROSC [22, 34].
Beyond inflammatory signaling, persistent systemic hypoperfusion and metabolic instability can further compromise cerebral energy recovery, even after apparent restoration of cerebral blood flow. Experimental and clinical evidence indicate that such systemic disturbances contribute to delayed neurological deterioration and heterogeneous recovery trajectories after CA/ROSC [24, 53]. This biological heterogeneity has, in turn, motivated the development of multimodal neuroprognostication strategies incorporating electrophysiology and neuroimaging to improve outcome prediction [54, 55, 56].
Overall, these mechanisms indicate that ischemic duration is a necessary but insufficient predictor of neuronal outcome. Global IR models primarily engage delayed, region-restricted injury governed by partial energy failure, adaptive stress responses, and controlled reperfusion. By contrast, CA/ROSC is characterized by complete systemic ischemia, severe mitochondrial collapse, intensified reperfusion injury, proteostasis failure, and systemic inflammatory amplification. As a result, nominally identical ischemic durations occupy fundamentally different positions along the injury continuum across experimental models [46].
This section emphasizes why global IR models and CA/ROSC should not be treated as interchangeable paradigms in translational neuroprotection. Although “ischemic duration” is often used as a matching variable, converging experimental and clinical evidence indicates that these models occupy different positions on the injury continuum because systemic physiology, reperfusion biology, and brain–body interactions diverge profoundly after CA/ROSC. As a result, methodological choices, including model selection, endpoint definition, and the timing of intervention and prognostication, can strongly influence conclusions regarding efficacy and translational relevance and therefore must be aligned with the dominant mechanisms operating in each experimental paradigm [1, 2].
A major challenge in translational neuroprotection research is the persistent extrapolation of findings from brain-restricted global IR models to CA/ROSC. Although both paradigms are often implemented using comparable ischemic durations, accumulating experimental and clinical evidence indicates that they occupy fundamentally different positions along the ischemic injury continuum [1, 2]. Recent international guidelines and consensus statements increasingly frame post–cardiac arrest brain injury as a dynamic, multisystem syndrome rather than a simple extension of transient global ischemia, underscoring why translation from global IR models often fails [57, 58, 59]. One major explanation for this translational gap lies in qualitative differences in injury biology, rather than ischemic duration alone. In global IR models, partial preservation of systemic circulation allows neurons to retain residual metabolic capacity, engage delayed stress responses, and progress toward injury over hours to days. Under these conditions, therapeutic interventions targeting oxidative stress, excitotoxicity, or delayed apoptotic signaling can meaningfully influence neuronal fate when administered within an extended post-ischemic window [1, 12].
By contrast, CA/ROSC is characterized by abrupt and complete systemic ischemia followed by a biologically hostile reperfusion phase, marked by profound mitochondrial dysfunction, systemic inflammation, and microcirculatory impairment—features collectively conceptualized as post–cardiac arrest syndrome (PCAS) [60, 61, 62]. These systemic modifiers fundamentally reshape the temporal and mechanistic landscape of neuronal injury compared with brain-restricted ischemia.
As a result, neuroprotective agents optimized in global IR paradigms may be administered outside their effective temporal or mechanistic window when tested in CA/ROSC models or in clinical cardiac arrest settings. Experimental and clinical studies demonstrate that mitochondrial failure, blood–brain barrier disruption, and inflammatory amplification occur early after resuscitation, often preceding the therapeutic windows inferred from brain-restricted ischemia models [7, 21, 63]. Accordingly, translational failure should be interpreted primarily as a mismatch between model-specific injury mechanisms and therapeutic targeting, rather than as evidence that candidate interventions are intrinsically ineffective.
The distinction between global IR and CA/ROSC has direct implications for experimental design and interpretation. First, simply matching ischemic duration across models does not ensure equivalence of injury severity or underlying mechanism. Identical ischemic durations can produce delayed, region-restricted neuronal death in global IR but early and widespread neuronal dysfunction in CA/ROSC, reflecting differences in systemic involvement and reperfusion biology [1, 7, 62].
Second, the selection of outcome measures critically shapes translational relevance. Global IR studies have traditionally emphasized histopathological endpoints—particularly delayed neuronal death in the hippocampal CA1 region—as primary indicators of injury. While such measures are well suited for studying selective vulnerability, they may fail to capture early, diffuse, or functionally relevant impairments that dominate CA/ROSC pathology [4, 6]. In CA/ROSC models and clinical cohorts, functional outcomes, brainstem integrity, cerebral perfusion, and systemic recovery parameters are increasingly recognized as more clinically meaningful endpoints than delayed forebrain histology alone [24, 64].
Third, the temporal alignment between injury mechanisms and therapeutic intervention windows requires careful reconsideration. In global IR, delayed neuronal death provides an extended window for mechanistic interrogation and therapeutic intervention. In CA/ROSC, by contrast, injury mechanisms unfold rapidly and overlap during early reperfusion, resulting in compressed and partially overlapping therapeutic windows [2]. Clinical trials of targeted temperature management illustrate this principle, demonstrating that modulation of systemic physiology and prevention of secondary insults may be more critical than precise temperature targets per se [65, 66]. Moreover, clinical studies conducted within the framework of targeted temperature management indicate that physiological instability, hemodynamic requirements, and the timing of prognostic assessment can substantially influence neurological outcome interpretation, underscoring the limited applicability of delayed-injury paradigms derived from global IR [67, 68].
Overall, these considerations argue for a shift toward integrative experimental strategies that explicitly align model selection with mechanistic targets. Rather than treating global IR and CA/ROSC as interchangeable models distinguished only by ischemic duration, future studies should instead recognize them as complementary paradigms that interrogate distinct dimensions of ischemic brain injury.
Global IR models remain indispensable for dissecting delayed neuronal death, selective vulnerability, and intrinsic neuronal stress-response pathways under controlled conditions [69, 70]. By contrast, CA/ROSC models are essential for capturing the impact of systemic ischemia–reperfusion, brain–body interactions, cerebral hypoperfusion, and early reperfusion-associated injury, features that dominate clinical cardiac arrest [62, 64].
From a translational perspective, this integrative framework is consistent with contemporary clinical paradigms emphasizing early hemodynamic optimization, mitigation of systemic inflammation, and multimodal neuroprognostication rather than exclusive focus on delayed neuronal death pathways [71, 72, 73, 74]. Effective neuroprotection after cardiac arrest is therefore likely to require early mitochondrial stabilization, modulation of reperfusion biology, and control of systemic inflammatory responses, rather than delayed interventions extrapolated from brain-restricted ischemia alone [22, 23, 48]. In addition, translational success should be evaluated across the continuum of recovery, extending from early coma assessment to long-term neurological and functional outcomes, which are increasingly recognized as critical determinants of meaningful survival after cardiac arrest [75].
While most neuroprognostication frameworks have been developed in adult populations, recent evidence indicates that integrated electrophysiological and neuroimaging approaches may further improve prognostic accuracy in pediatric cardiac arrest, where etiologies, injury patterns, and recovery trajectories differ substantially from adults [76]. These observations highlight the need for age- and context-specific prognostication strategies when translating experimental findings or adult-derived paradigms to broader clinical populations.
Finally, it should be recognized that early post-ROSC management decisions, including airway control, circulatory stabilization, and metabolic correction, can substantially influence subsequent neurological injury trajectories. Emergency medicine–oriented analyses emphasize that these early systemic interventions frequently precede any opportunity for neuroprotective targeting derived from experimental ischemia models, thereby highlighting a critical translational gap between controlled laboratory paradigms and real-world cardiac arrest care [77].
Aging represents a critical biological modifier of ischemic vulnerability that is often underrepresented in experimental global ischemia studies. Experimental evidence indicates that aged animals exhibit exacerbated mitochondrial dysfunction and reduced cerebrovascular and metabolic resilience following IR, resulting in accelerated neuronal injury and diminished recovery capacity [78, 79, 80]. Age-related vascular stiffening, endothelial dysfunction, and baseline inflammatory priming further shift ischemic injury thresholds toward earlier failure, particularly in CA/ROSC paradigms where systemic circulatory collapse is superimposed on pre-existing vascular vulnerability [81]. These observations highlight that ischemic duration equivalence across age groups cannot be assumed and underscore the importance of incorporating age as a critical stratification variable in translational ischemia research.
Sex-dependent differences in ischemic vulnerability have been consistently reported in both experimental and clinical studies. Female animals, particularly during reproductive age, often demonstrate relative resistance to ischemic injury compared with males, an effect attributed in part to estrogen-mediated suppression of mitochondrial oxidative stress and modulation of cerebrovascular and inflammatory responses [82, 83, 84]. Conversely, loss of endogenous sex hormone protection following ovariectomy or aging abolishes this protective phenotype and shifts ischemic thresholds toward increased vulnerability [82]. These findings suggest that sex and hormonal status represent biologically meaningful modifiers of ischemic outcome that interact with systemic physiology and reperfusion biology, further complicating duration-based injury prediction across experimental models.
From a contemporary perspective, sex differences in ischemic brain injury are increasingly recognized as reflecting not only differences in injury magnitude but also divergence in dominant injury pathways [85]. Recent studies indicate that male and female brains exhibit distinct mitochondrial stress responses, microglial activation profiles, and inflammatory resolution dynamics following global ischemia and cardiac arrest, which may differentially influence reperfusion injury and delayed neuronal death [86, 87]. These sex-specific biological responses suggest that uniform neuroprotective strategies may obscure clinically relevant heterogeneity and contribute to translational failure when sex is not explicitly incorporated into experimental design and data interpretation.
Beyond classical inflammatory mediators, endogenous neurotoxic metabolites such as homocysteine have emerged as additional contributors to ischemic vulnerability. Elevated homocysteine levels are associated with endothelial dysfunction and excitotoxic signaling and have been linked to oxidative stress–related pathways, all of which may exacerbate IR injury [88, 89]. Experimental and clinical evidence further suggests that hyperhomocysteinemia may lower neuronal injury thresholds and worsen post-ischemic neurological outcomes, with potential relevance to systemic IR settings [89, 90]. These biochemical modifiers further emphasize that ischemic outcome reflects a complex interaction between systemic metabolic status and cerebral vulnerability rather than ischemic duration alone.
From a contemporary viewpoint, endogenous neurotoxic metabolites such as homocysteine should be considered dynamic systemic modifiers that may lower effective ischemic injury thresholds, rather than static vascular risk markers [91, 92]. In the context of CA/ROSC, where systemic metabolic derangement and reperfusion stress coexist, such metabolites may amplify mitochondrial dysfunction, endothelial injury, and neuroinflammatory cascades, thereby accelerating neuronal injury even under comparable ischemic durations [93, 94].
Experimental studies of global cerebral ischemia have long relied on ischemic duration as a central determinant of neuronal injury severity. However, the evidence synthesized in this review indicates that ischemic duration alone is insufficient to reliably predict neuronal outcome, particularly when comparing brain-restricted global IR models with CA/ROSC models. Instead, neuronal fate reflects an integrated interplay among energy failure, reperfusion biology, cellular stress handling, and systemic physiological context. Global IR models are characterized by partial energy preservation, controlled reperfusion, and delayed, region-restricted neuronal death, most prominently affecting selectively vulnerable populations such as hippocampal CA1 pyramidal neurons. These models have provided invaluable insights into delayed neuronal death, selective vulnerability, and intrinsic neuroprotective stress responses. By contrast, CA/ROSC models impose complete systemic ischemia followed by a biologically hostile reperfusion environment, marked by mitochondrial collapse, inflammatory amplification, and microcirculatory dysfunction. As a consequence, nominally identical ischemic durations can exceed multiple injury thresholds more rapidly in CA/ROSC, producing earlier, broader, and mechanistically heterogeneous neuronal injury. This distinction carries important translational implications. The repeated failure of neuroprotective strategies in cardiac arrest settings should not be interpreted solely as a lack of therapeutic promise, but rather as a consequence of misalignment between experimental model mechanisms and the timing of therapeutic intervention. Interventions optimized in global IR paradigms often target delayed injury mechanisms that are already overwhelmed or bypassed in CA/ROSC. Recognizing these differences is therefore critical for improving preclinical study design and aligning experimental outcomes with clinical realities.
With respect to future research directions, several priorities can be identified. Importantly, biological modifiers such as age, sex-related hormonal status, and endogenous neurotoxic metabolites further shape ischemic vulnerability and injury thresholds, reinforcing the need for biologically stratified experimental designs in translational neuroprotection research. From a contemporary perspective, these biological modifiers should not be regarded as secondary covariates but as integral determinants of ischemic injury thresholds that interact with systemic physiology and reperfusion biology. Failure to incorporate age-, sex-, and metabolically stratified frameworks may contribute to persistent translational failure, particularly in cardiac arrest–related neuroprotection where biological heterogeneity is pronounced. Future experimental designs should therefore move beyond duration-centric and biologically uniform models toward stratified, mechanism-aligned paradigms that more accurately reflect clinical complexity. First, experimental model selection should be explicitly guided by the mechanistic question under investigation, rather than by ischemic duration alone. Global IR and CA/ROSC should be treated as complementary, not interchangeable, paradigms that probe distinct dimensions of ischemic brain injury. Second, greater emphasis should be placed on early reperfusion biology and systemic modifiers of neuronal injury, particularly in CA/ROSC models. Approaches targeting mitochondrial stabilization, microcirculatory recovery, and systemic inflammatory control may hold greater translational relevance than strategies focused exclusively on delayed neuronal death pathways. Third, outcome measures should extend beyond delayed forebrain histopathology to include functional assessments, brainstem integrity, and indices of whole-body recovery, especially in CA/ROSC paradigms. Such multidimensional endpoints are more likely to capture clinically meaningful injury and recovery trajectories.
Finally, integrative frameworks that incorporate brain–body interactions and dynamic ischemic thresholds are required to bridge experimental and clinical perspectives. By reframing ischemic duration as one component within a broader pathophysiological continuum, future studies may achieve more accurate mechanistic insight and improved translational success.
In summary, understanding why identical ischemic durations yield divergent neuronal outcomes requires moving beyond duration-centric paradigms toward a systems-level view of IR injury. Adoption of this integrative perspective may ultimately guide the development of more effective and context-appropriate neuroprotective strategies for global cerebral ischemia and cardiac arrest.
Future translational efforts will benefit from experimental frameworks that integrate systemic physiology, biological heterogeneity, and reperfusion biology, thereby enabling more accurate prediction of therapeutic efficacy across preclinical and clinical settings.
4VO, four-vessel occlusion; ATP, adenosine triphosphate; BBB, blood–brain barrier; CA1, cornu ammonis 1; CA/ROSC, cardiac arrest with return of spontaneous circulation; DND, delayed neuronal death; IR, ischemia–reperfusion; GFAP, glial fibrillary acidic protein; UPR, unfolded protein response; mPTP, mitochondrial permeability transition pore; PCAS, post–cardiac arrest syndrome; ROS, reactive oxygen species.
All data reported in this paper will also be shared by the lead contact upon request.
JSY, M-HW, and JHC designed the research study. JSY, JHP, JHA, MCS, JM, M-HW and JHC wrote the manuscript. JHP and JHA contributed to figure preparation. MCS and JM contributed to the reference organization. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Moo-Ho Won is serving as one of the Editorial Board members of this journal. We declare that Moo-Ho Won had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Bettina Platt.
During the preparation of this work the authors used ̵ChatGPT-5.2 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.