







 , Yan Sun 2,†, Minyuan Zhang 1, Xiangying Gao 1, Junlu Wang 1, Qinxue Dai 1,*
, Yan Sun 2,†, Minyuan Zhang 1, Xiangying Gao 1, Junlu Wang 1, Qinxue Dai 1,*
1 Department of Anesthesia, The First Affiliated Hospital of Wenzhou Medical University, 325000 Wenzhou, Zhejiang, China
2 The Second Clinical Medical College, Wenzhou Medical University, 325000 Wenzhou, Zhejiang, China
†These authors contributed equally.
Abstract
Cerebral ischemia-reperfusion injury (CIRI) is a severe neurological condition where restoring neuronal mitochondrial function critically impacts prognosis. While electroacupuncture (EA) has demonstrated neuroprotective effects by improving mitochondrial function, the precise underlying mechanisms remain unclear. Emerging evidence suggests that astrocyte-to-neuron mitochondrial transfer, facilitated by mitochondrial Rho-GTPase 1 (Miro1), serves as a vital neuroprotective pathway. Therefore, this study investigates whether astrocytic Miro1 participates in the neuroprotective effects of EA against CIRI in mice by regulating the expression of the mitochondrial marker translocase of the outer mitochondrial membrane 40 (TOM40) and adenosine triphosphate (ATP) levels in damaged neurons.
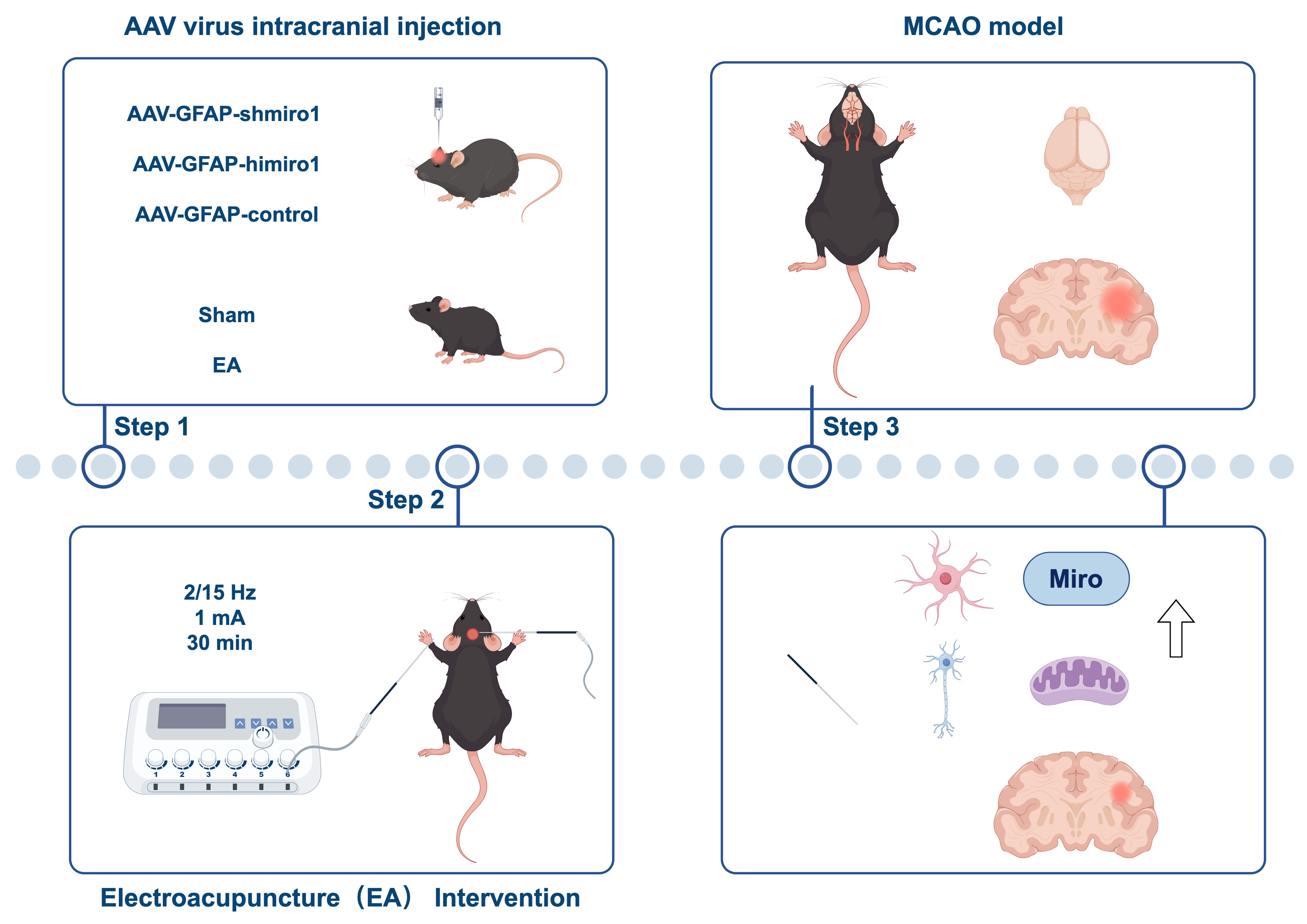
126 C57BL/6 mice were randomly allocated into seven experimental groups (n = 18 per group): Sham-operated (Sham), middle cerebral artery occlusion (MCAO) model, EA, sham electroacupuncture (SEA), EA combined with astrocyte-specific Miro1 knockdown (GFAP: glial fibrillary acidic protein, EA+AAV-GFAP-shMiro1), astrocyte-specific Miro1 over-expression (AAV-GFAP-hiMiro1), and adenoviral empty vector control (AAV-GFAP-control). The CIRI model was induced using MCAO. Prior to model induction, the EA group received pretreatment with EA at the Baihui (GV20) acupoint. The SEA group underwent identical procedures to the EA group except for electrical stimulation. For the EA+AAV-GFAP-shMiro1, AAV-GFAP-hiMiro1, and AAV-GFAP-control groups, mice received intracerebroventricular injections of AAV-GFAP-shMiro1, AAV-GFAP-hiMiro1, or AAV-GFAP-control, respectively, 48 hours prior to EA treatment, with other procedures matching the EA group. At 24 hours post-reperfusion, neurological deficit scores, cerebral infarct volume, and neuronal survival in the peri-infarct penumbra were assessed. Astrocytes and neurons from the peri-infarct penumbra were isolated to measure ATP levels and expression of the mitochondrial-specific protein TOM40 in neurons, as well as ATP levels, TOM40, and Miro1 protein expression in astrocytes.
Relative to the Sham group, the MCAO group displayed a significant increase in cerebral infarct volume and neurological deficit scores, accompanied by a marked reduction in neuronal viability, TOM40 expression, and ATP levels (p < 0.01). In contrast to the MCAO and SEA groups, the EA and AAV-GFAP-hiMiro1 groups demonstrated improved neurological scores, reduced infarct volume, enhanced neuronal viability, elevated neuronal ATP levels and TOM40 expression, as well as decreased astrocytic ATP and TOM40 levels, but significantly increased Miro1 expression in astrocytes (p < 0.01). When compared to the EA group, the EA+AAV-GFAP-shMiro1 group exhibited a reversal of all the aforementioned improvements (p < 0.01), while the AAV-GFAP-hiMiro1 group showed no significant changes (p > 0.05).
EA exerts neuroprotective effects in MCAO mice by upregulating Miro1 protein expression in astrocytes and upregulating the mitochondrial marker TOM40 alongside ATP levels in neurons. Silencing Miro1 abolished the neuroprotective effects of EA and reduced neuronal TOM40 expression, while Miro1 overexpression increased this mitochondrial marker and mimicked EA-mediated neuroprotection. These findings identify Miro1 as a key effector of EA-induced neuroprotection, although the upstream signaling pathways linking EA to Miro1 upregulation require further investigation.
Graphical Abstract
Keywords
- electroacupuncture
- mitochondrial transfer
- GTP phosphohydrolases
- reperfusion injury
- brain ischemia
- neuroprotection
Cerebral infarction represents a common neurological disorder affecting the central nervous system. It is distinguished by its elevated occurrence frequency, significant fatality risk, and substantial impairment potential, thereby constituting a serious global health concern [1]. Mitochondria are critical organelles that regulate cellular homeostasis and function through energy metabolism, calcium signaling, cellular metabolism, and apoptosis [2]. During cerebral ischemia-reperfusion injury (CIRI), the restoration of mitochondrial function in neurons profoundly impacts stroke prognosis [3, 4]. During electroacupuncture (EA) intervention, a sparse-dense wave at a frequency of 2/15 Hz was applied for stimulation at the Baihui (GV20) acupoint. The choice of this parameter is based on the classical “frequency-specific effect” theory, which posits that different electrical stimulation frequencies can activate distinct endogenous neurochemical pathways [5]. Specifically, the 2/15 Hz pattern is designed to integrate the potential benefits of low-frequency (2 Hz) and medium-to-high-frequency (15 Hz) stimulation, thereby exerting synergistic neuroprotective effects through multi-target mechanisms [6]. Studies have shown that EA improves mitochondrial function in neurons of rats with middle cerebral artery occlusion (MCAO), contributing to its neuroprotective effects [7, 8]. However, the underlying mechanisms remain unclear.
Hayakawa et al. [9] utilized transgenic mice with fluorescently labeled astrocytes to establish an MCAO model, observing fluorescently tagged mitochondria within damaged neurons in the peri-infarct penumbra, suggesting that astrocytes transfer their mitochondria to injured neurons, thus implicating mitochondrial transfer as a neuroprotective mechanism. Furthermore, Tseng et al. [10] demonstrated that the upregulation of mitochondrial Rho-GTPase 1 (Miro1) expression in mesenchymal stem cells enhances the intercellular transfer of mitochondria to oxidant-damaged neurons, thereby improving neuronal metabolism and survival. Conversely, decreasing Miro1 expression was found to impair this mitochondrial transfer capacity and diminish the neuroprotective benefits. Building on these insights, it is hypothesized that EA at Baihui may promote astrocyte-to-neuron mitochondrial transfer via astrocytic Miro1, thereby mediating neuroprotection.
To investigate this hypothesis, this study employed an MCAO mouse model combined with adeno-associated viruses carrying glial fibrillary acidic protein (GFAP) specific promoters to either over-express or silence Miro1 in astrocytes. The effects of EA on mitochondrial quantity and function were evaluated in neurons and astrocytes. The aim was to elucidate novel mechanisms underlying EA-induced neuroprotection.
Male C57BL/6 mice (20–25 g) were procured from Beijing Vital River Laboratory Animal Technology (China). These animals were maintained, with food and water ad libitum, in controlled laboratory environments. Prior to surgical procedures, a 24-hour fasting period, withdrawal of both food and water, was enforced. The entire experimental protocol and animal care procedures followed the ethical standards established by the First Affiliated Hospital of Wenzhou Medical University.
1% 2,3,5-triphenyltetrazolium chloride (TTC) staining reagent (Cat# C0651, Beyotime Biotechnology Co., Ltd.,
Shanghai, China); Isoflurane (Cat# R510-22-2, RWD Life Science Co., Ltd.,
Shenzhen, Guangdong, China); Brain matrix (Cat# 68707, RWD Life Science Co.,
Ltd.); AAV-GFAP-hiMiro1 (Genewiz Biotech Co., Ltd.,
Guangzhou, Guangdong, China); AAV-GFAP-control (Genewiz Biotech Co., Ltd.); AAV-GFAP-shMiro1 (Genewiz Biotech Co., Ltd.); Acupuncture needles (Size: 0.18
Anesthesia was induced via inhalation of 5% isoflurane and maintained with 2%
isoflurane. Following hair removal and sterilization of the cervical region, a
midline incision was performed in the neck area using microsurgical techniques.
The internal carotid artery (ICA), external carotid artery, and right common
carotid artery were isolated. A blunt-tipped monofilament suture was then
introduced into the right ICA and carefully advanced until reaching the middle
cerebral artery origin to induce vascular occlusion. Following a two-hour
ischemic period, the suture was removed to restore cerebral blood flow, after
which the surgical wound was closed. Neurological function was evaluated using
standardized scoring criteria after recovery from anesthesia, with animals
scoring below one point being excluded from subsequent analysis. Core body
temperature was maintained at 37.0
126 C57BL/6 mice were randomly divided into seven groups (n = 18 per group) using a randomized block design, including: Sham operation group (Sham), model group (MCAO), EA, sham EA group (SEA), EA + astrocytic Miro1 knockdown group (EA+AAV-GFAP-shMiro1), astrocytic Miro1 overexpression group (AAV-GFAP-hiMiro1) and adenoviral empty vector control group (AAV-GFAP-control).
The Baihui acupoint (GV20) was located at the intersection of the line connecting the two ear tips and the sagittal midline in mice [12]. After localization, an acupuncture needle was inserted approximately 1 mm into the Baihui acupoint. Another needle was inserted into the left forelimb (non-acupoint site) as the return electrode to complete the circuit. The Han’s EA device was employed to administer electrical stimulation under controlled conditions, with specific settings including a dual-frequency mode (2/15 Hz), current strength set at 1 mA, and a treatment period lasting 30 minutes.
For the respective experimental groups, ICV injections of AAV-GFAP-shMiro1, AAV-GFAP-hiMiro1, or AAV-GFAP-control (1 µL per mouse) were performed 48 h prior to EA treatment. The infusion procedure was conducted at a constant flow rate of 0.2 µL per minute, with the entire administration process completed within five minutes. For the intracerebroventricular (ICV) delivery, the stereotactic positioning parameters were established at 0.4 mm caudal to bregma, with a 1 mm lateral deviation from the midline and a penetration depth of 2 mm [13].
Following a 24-hour period after CIRI, mice were deeply anesthetized via inhalation of 3% isoflurane in an oxygen/air mixture and subsequently euthanized by decapitation. The penumbral region
surrounding the infarct area was isolated and mechanically dissociated in chilled
phosphate-buffered saline (PBS) utilizing pipette techniques. Tissue digestion
was performed using a 0.25% trypsin solution, after which the enzymatic reaction
was halted by introducing a stop solution. Subsequent centrifugation at
300
A suspension of 1
To prepare the cell suspension, 1
Neurological function was evaluated at 24 h post-reperfusion using a standardized scoring system, the Longa 5-point scale [11]. 0: No neurological deficits, 1: Failure to fully extend contralateral forelimb; 2: Circling toward the contralateral side; 3: Falling to the contralateral side; 4: Loss of spontaneous movement with impaired consciousness. All assessments were performed by an experimenter blinded to the group allocations.
24 hours after reperfusion, mice were deeply anesthetized via inhalation of 3% isoflurane in an oxygen mixture prior to decapitation for brain tissue collection. The harvested cerebral tissues were
rapidly frozen at –20 °C for 20 minutes, followed by coronal sectioning into four
sequential 2 mm thick slices utilizing a specialized brain matrix apparatus.
These tissue sections were subsequently immersed in a 1% solution of TTC
staining reagent and maintained at 37 °C for precisely 20 minutes during the
incubation process. Viable tissue stained red, while infarcted areas remained
pale. Images were scanned and analyzed using a LUZEX-F image analysis system
(Nireco Corporation, Tokyo, Japan). Infarct volume was calculated using the
Swanson method to correct for brain edema. Briefly, the areas of the
contralateral hemisphere and the non-infarcted ipsilateral hemisphere were
measured on each slice. Infarct volume percentage was calculated as:
(contralateral hemisphere volume – ipsilateral non-infarcted volume) /
contralateral hemisphere volume
24 hours after CIRI, mice underwent anesthesia
and were subjected to transcardial perfusion using 4% paraformaldehyde solution.
Harvested brain tissues were subsequently embedded in paraffin blocks for
histological preparation. After deparaffinization, coronal sections 3.5 µm
thick were treated with 3% hydrogen peroxide solution for 10 minutes to inhibit
endogenous peroxidase activity. For antigen retrieval, tissue sections were
heated in 10 mmol/L citrate buffer (pH 6.0) for 20 minutes, followed by gradual
cooling to ambient temperature. Prior to primary antibody incubation, sections
were blocked with PBS containing 5% bovine serum albumin for one hour at room
temperature. The primary antibody employed was rabbit anti-MAP2 (1:500 dilution,
ab221693, Abcam), which was applied overnight at 4 °C. Following three PBS washes,
sections were exposed to Alexa Fluor 594-conjugated secondary antibody (1:500
dilution, ab150068, Abcam) for one hour at room temperature. Nuclear
counterstaining was performed using 4′,6-diamidino-2-phenylindole (DAPI, 1
µg/mL concentration) for five minutes. After final PBS rinses, prepared
slides were coverslipped and examined under an Olympus BX51 fluorescence
microscope (Olympus Corporation, Tokyo, Japan) with standardized imaging
parameters (including gain, threshold, and black level settings), maintained
throughout all experimental procedures. Quantification of viable neurons was
based on the identification of cells exhibiting a MAP2+ (red fluorescence)
signal surrounded by DAPI+ (blue) nuclear staining. For statistical
analysis, five randomly selected microscopic fields (
The isolated neurons and astrocytes underwent lysis, after which intracellular ATP concentrations were measured utilizing a commercially available ATP detection system (ATP Bioluminescence Assay Kit CLS II, Roche) based on the luciferin-luciferase reaction. In this procedure, cellular lysates were combined with the enzyme substrate solution, followed by immediate luminescence detection performed with a microplate luminometer. To standardize the measurements, ATP quantities were adjusted according to the total protein levels quantified through the BCA protein assay, with results presented in units of nmol per milligram of protein.
To prepare protein samples, neuronal or astrocytic cells were solubilized using 1% Triton X-100 lysis buffer, followed by protein concentration determination through BCA protein assay. For electrophoretic analysis, identical protein quantities (5 µg) were loaded onto SDS-polyacrylamide gel wells. After electrophoretic separation, proteins were transferred onto nitrocellulose membranes using wet transfer methodology. Membrane blocking was performed with 5% skim milk dissolved in tris-buffered saline (TBST) containing 0.1% Tween-20 for 60 minutes at ambient temperature. Subsequently, membranes were probed with primary antibodies targeting TOM40 (dilution 1:5000) and Miro1 (dilution 1:1000) overnight at 4 °C. Following TBST washes, membranes were exposed to HRP-conjugated secondary antibodies (1:5000 dilution) for one hour at room temperature. Protein detection was achieved through enhanced chemiluminescence reaction, with signal capture on Kodak X-Omat AR radiographic film (Eastman Kodak Company, Rochester, NY, USA). Quantitative analysis of band intensities was conducted using ImageJ software (Version 1.54g, National Institutes of Health, Bethesda, MD, USA). Representative Western blot images are shown in the main figures, and the corresponding original, uncropped blots are provided in the Supplementary Material.
All statistical analyses and graph construction were performed using GraphPad
Prism software (Version 10.1.0, GraphPad Software, San Diego, CA, USA).
Continuous variables with normal distribution were presented as mean
As shown in Fig. 1, the neurological deficit scores were significantly elevated
in the MCAO group when compared with the Sham group (p
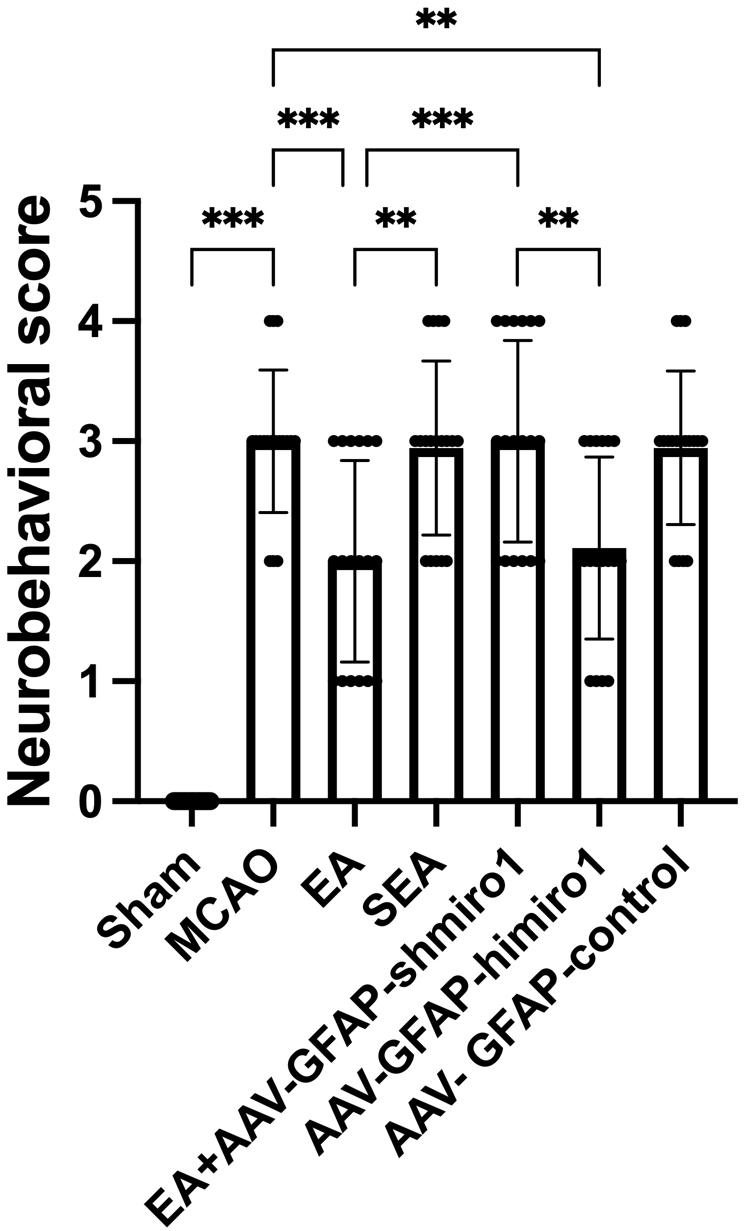 Fig. 1.
Fig. 1.
Comparison of neurological deficit scores among experimental
groups (n = 18 per group). Data are presented as mean
As shown in Fig. 2, the infarct size was significantly greater in the MCAO group
when compared with the Sham-operated controls (p
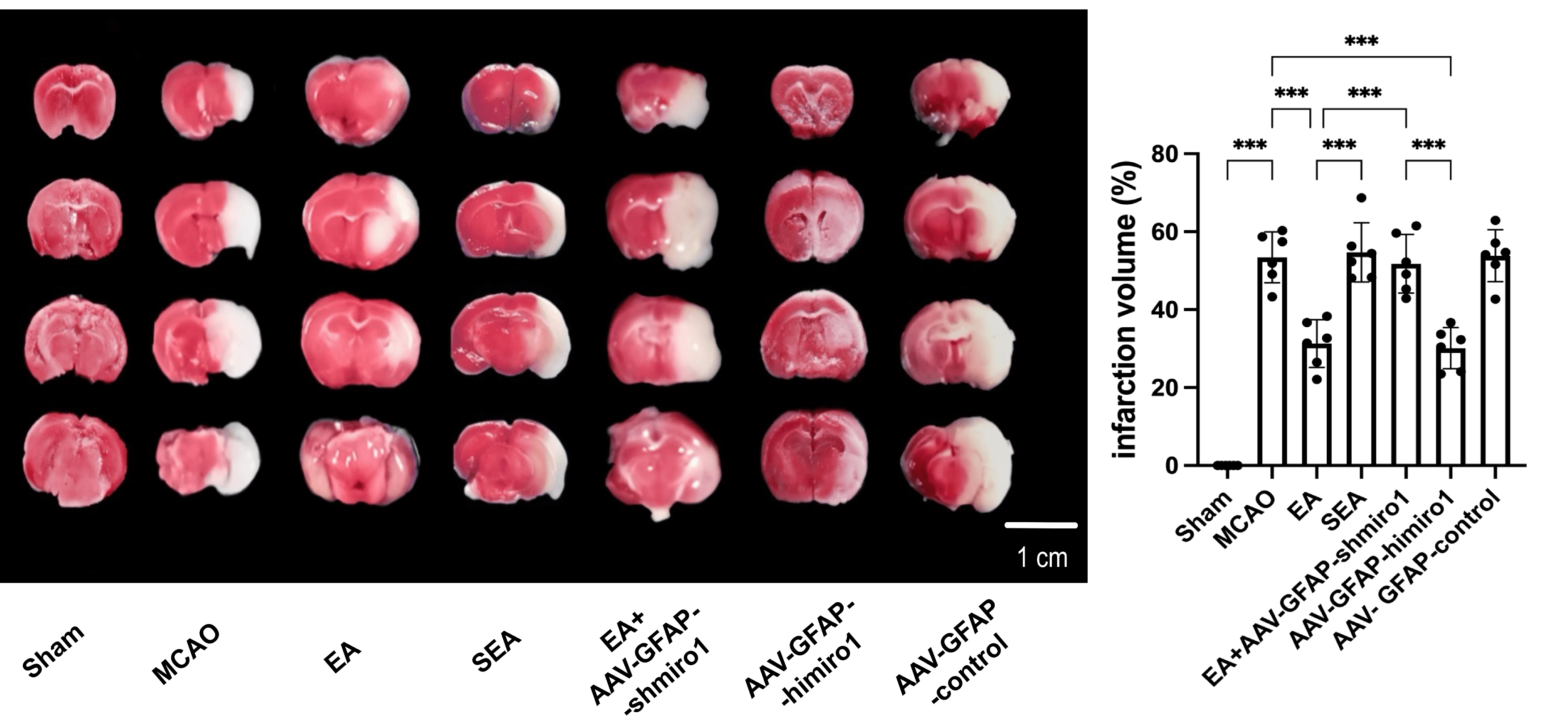 Fig. 2.
Fig. 2.
Comparison of cerebral infarct volume measured by 2,3,5-triphenyltetrazolium chloride (TTC) staining
across experimental groups (n = 6, randomly selected from 18 per group). Data are presented as
mean
As shown in Fig. 3, neuronal survival rates in the peri-infarct penumbra were
significantly lower in the MCAO group when compared with the Sham operated
controls (p
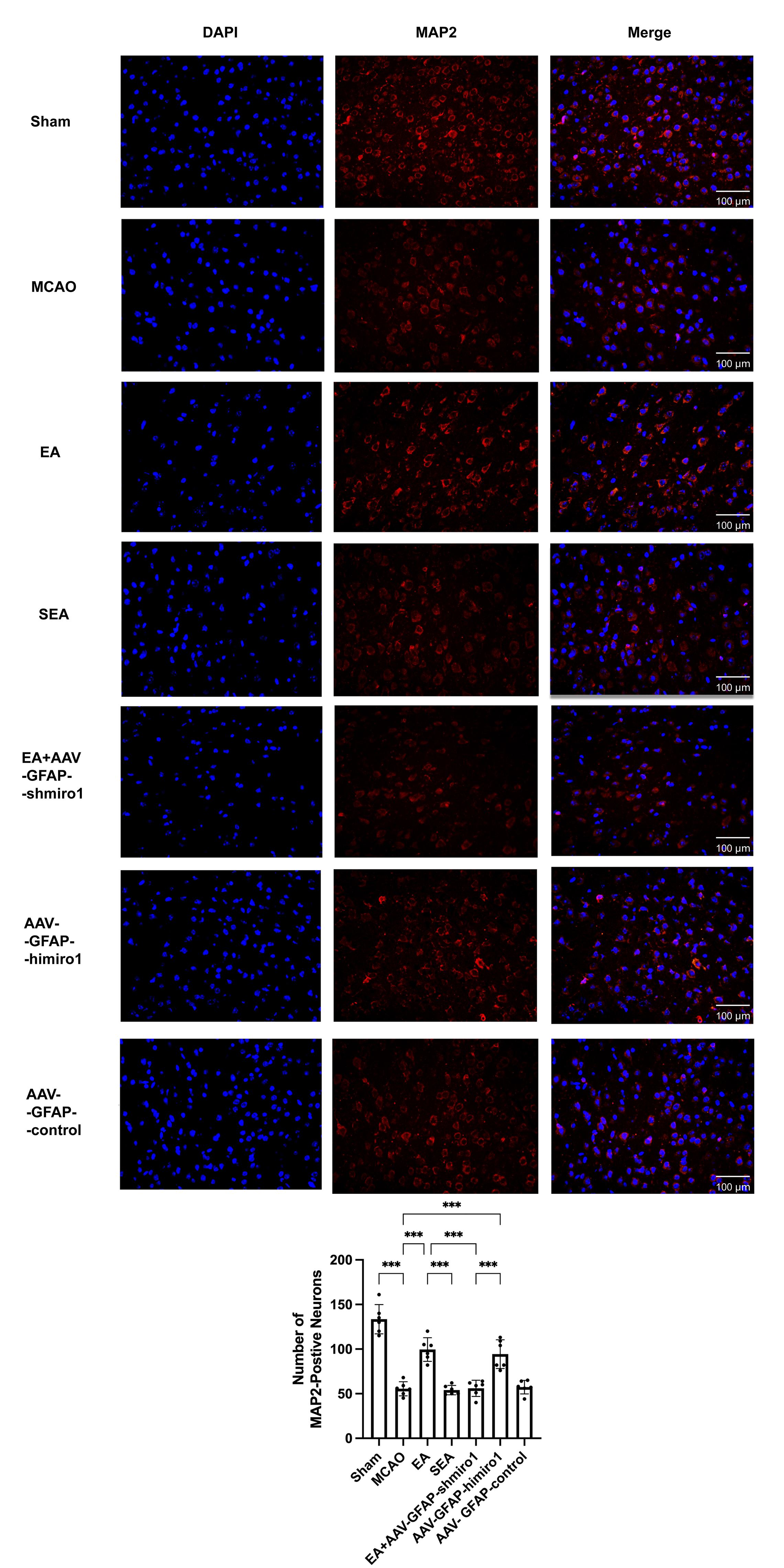 Fig. 3.
Fig. 3.
Comparison of neuronal viability in the peri-infarct penumbra
across experimental groups (immunofluorescence staining,
As shown in Fig. 4, neuronal ATP concentrations in the peri-infarct penumbra
were substantially lower in the MCAO group than in the Sham group (p
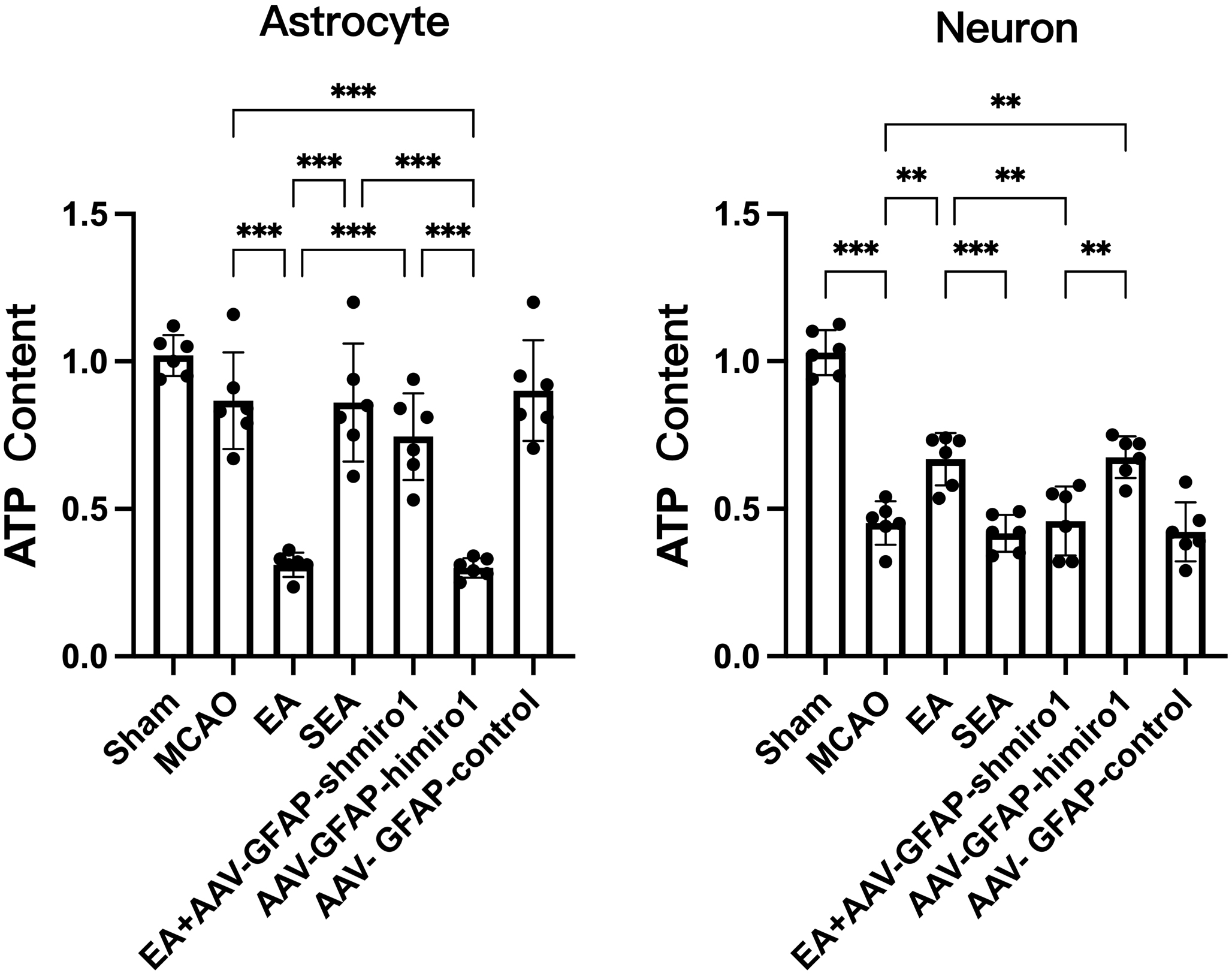 Fig. 4.
Fig. 4.
Comparison of ATP levels in neurons and astrocytes across
experimental groups (bioluminescence assay; n = 6, randomly selected from 18 per group). Data are
presented as mean
As illustrated in Fig. 5, the MCAO group showed no significant changes in TOM40
or Miro1 protein expression in astrocytes of the peri-infarct penumbra compared
to the Sham group. In contrast, the EA group exhibited significantly increased
Miro1 expression and decreased TOM40 levels in astrocytes compared to both the
MCAO and SEA groups (p
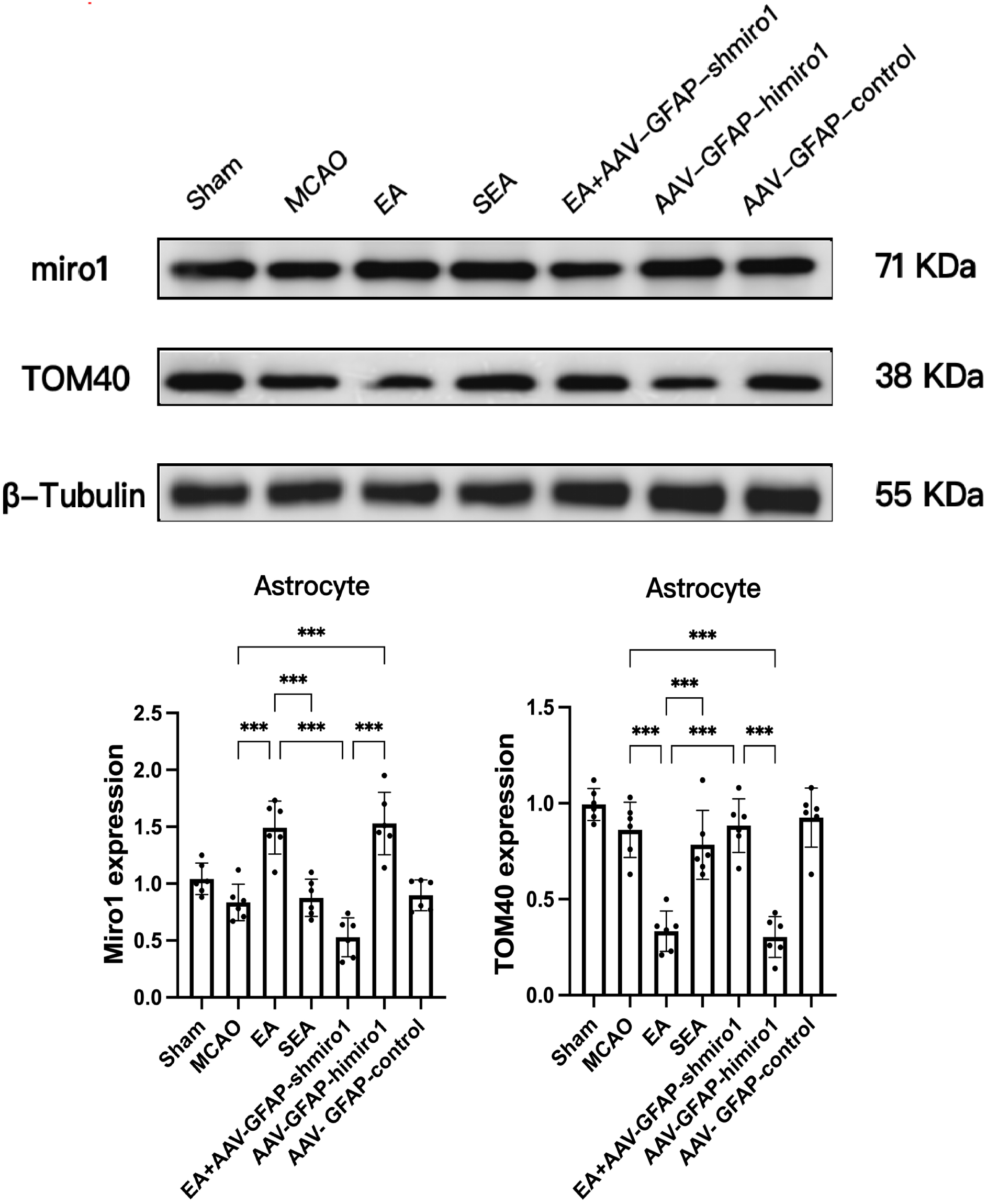 Fig. 5.
Fig. 5.
Comparison of TOM40 and Miro1 protein expression in astrocytes
across experimental groups (Western blot analysis; n = 6, randomly selected from 18 per group).
Data are presented as mean
As illustrated in Fig. 6, the MCAO group exhibited significantly reduced TOM40
expression in neurons of the peri-infarct penumbra compared to the Sham group
(p
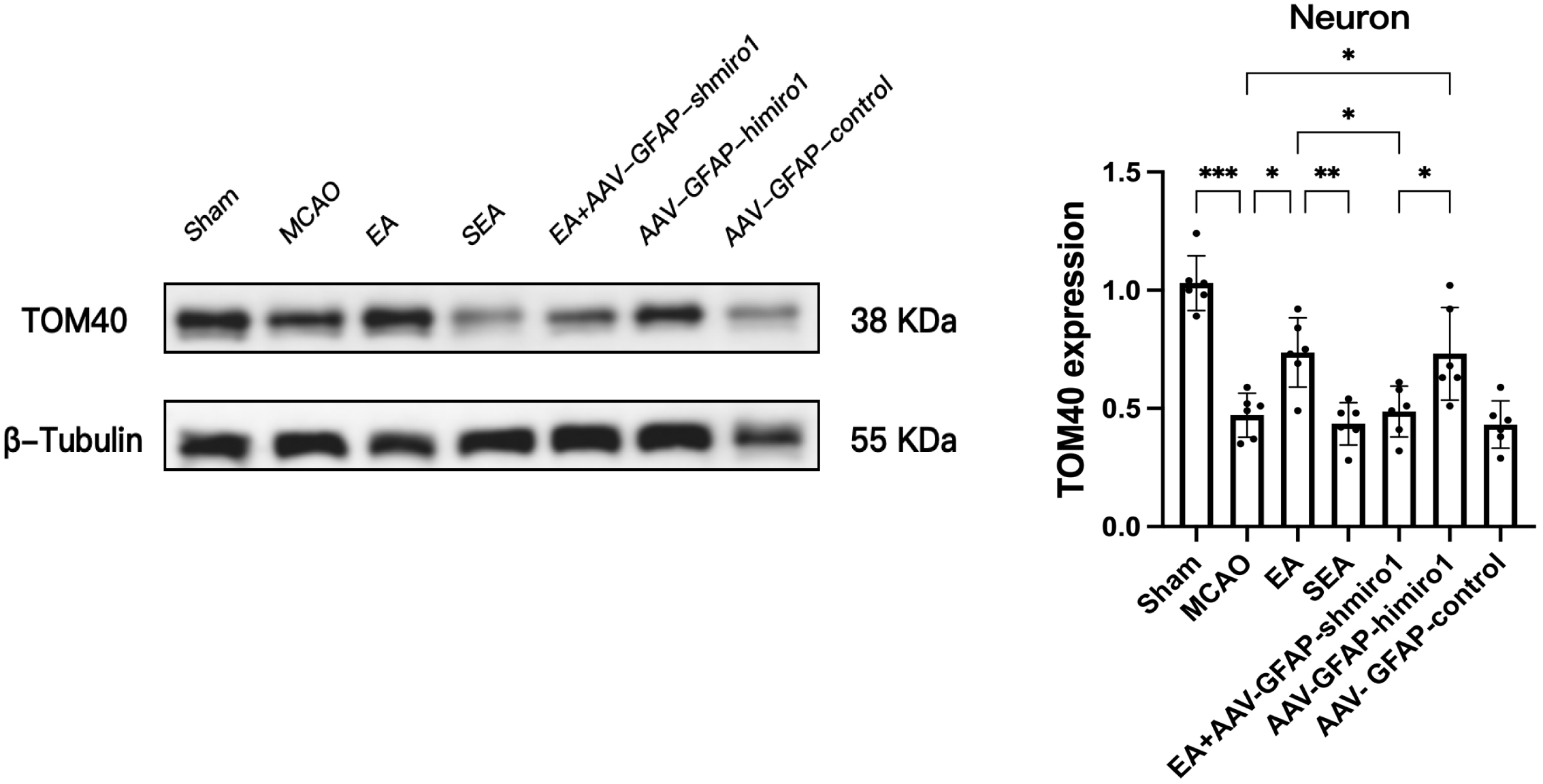 Fig. 6.
Fig. 6.
Comparison of TOM40 expression in neurons across experimental
groups (Western blot analysis; n = 6, randomly selected from 18 per group). Data are presented as
mean
As the predominant glial cell population in the central nervous system, astrocytes perform essential functions in modulating synaptic activity, upholding blood-brain barrier integrity, facilitating synaptic plasticity, and ensuring neuronal stability [14]. During cerebral ischemic events, these cells exhibit reactive changes marked by cellular hypertrophy and elevated production of GFAP, phenomena strongly correlated with ischemic neuronal injury [15]. Following ischemic stroke, astrocytes exert multifaceted neuroprotective effects [16]. For instance, astrocytic scar formation around ischemic lesions isolates damaged areas from healthy tissue, shielding surviving neurons from harmful substances released by the infarct core [17]. Thus, astrocytes are pivotal for post-stroke recovery.
The mechanisms governing mitochondrial transfer remain incompletely understood. Key players include the motor protein KIF5 and the adaptor protein Miro1/2, which mediate mitochondrial transport [18]. Miro1, a calcium-sensitive protein anchored to the mitochondrial outer membrane, binds KIF5 to form a transport complex [18, 19]. This complex facilitates mitochondrial transfer between cells via tunneling nanotubes (TNTs) [19, 20], which are extensively distributed between astrocytes and neurons [21] with their connectivity intensifying under ischemic conditions and enhancing intercellular communication and tissue repair [22]. In neurons, Miro1 governs mitochondrial trafficking; its deficiency disrupts mitochondrial distribution and motility, contributing to neurological disorders [23]. Notably, studies suggest that peri-infarct astrocytes transfer functional mitochondria to ischemic neurons, promoting neuronal survival [9]. It is hypothesized here that during CIRI, astrocytes export mitochondria to damaged neurons via Miro1-mediated TNTs, thereby exerting neuroprotection.
This study investigated whether EA enhances astrocyte-to-neuron mitochondrial transfer via Miro1 to mitigate CIRI. Compared to the Sham group, MCAO mice exhibited enlarged infarct volumes, worsened neurological deficits, and reduced neuronal ATP/TOM40 levels, while astrocytic ATP/TOM40 remained unchanged. This indicates severe mitochondrial dysfunction in neurons but preserved mitochondrial integrity in astrocytes, likely due to astrocytic glycogen storage and superior hypoxia tolerance [16]. EA significantly increased astrocytic Miro1 expression while reducing astrocytic ATP/TOM40 and elevating neuronal ATP/TOM40, correlating with improved neurological outcomes and reduced infarct volume. These findings suggest EA promotes mitochondrial transfer from astrocytes to neurons.
Mechanistically, astrocyte-specific Miro1 knockdown (AAV-GFAP-shMiro1) abolished EA-induced increases in neuronal ATP/TOM40 and reversed EA’s neuroprotective effects. Conversely, astrocytic Miro1 overexpression (AAV-GFAP-hiMiro1) mimicked EA’s benefits, confirming Miro1’s essential role. Critically, viral controls (AAV-GFAP-control) did not alter outcomes, excluding nonspecific viral effects. These results strongly support that EA upregulates astrocytic Miro1 to enhance mitochondrial export to neurons via TNTs, restoring neuronal ATP levels and the expression of mitochondrial biogenesis markers.
We fully acknowledge that this study has several limitations. First, although our findings strongly suggest that EA promotes astrocyte-to-neuron mitochondrial transfer via Miro1, direct evidence using TNT inhibitors is lacking, and the proposed mechanism remains hypothetical. Second, the specific transduction pathways linking the physical stimulus (EA) to the molecular signal (Miro1 upregulation) have not been elucidated. This represents a core gap in the current mechanistic framework. Based on existing literature and preliminary clues, we propose the following testable hypotheses: (1) EA may induce Ca2+ transients in astrocytes, activating calcineurin-nuclear factor of activated T-cells (NFAT) or calcium/calmodulin-dependent protein kinase (CaMK)-cAMP response element-binding protein (CREB) pathways to promote Miro1 transcription [24, 25]; (2) EA stimulation at Baihui (GV20) may trigger neuronal glutamate release, activating astrocytic metabotropic glutamate receptors 3/5 (mGluR3/5) receptors and downstream signaling cascades [26, 27]; (3) EA-induced adaptive oxidative stress may regulate Miro1 expression via the nuclear factor erythroid 2-related factor 2-antioxidant response element (Nrf2-ARE) pathway [28]. All of these hypotheses require rigorous experimental validation in future studies. We have planned subsequent investigations using in vitro co-culture systems combined with pathway-specific inhibitors, chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR), and other techniques to systematically dissect the molecular mechanisms by which EA regulates Miro1.
Furthermore, regarding the assessment of mitochondria, we used TOM40 protein expression alongside ATP levels to evaluate neuronal mitochondrial status. It should be noted that changes in TOM40 levels may primarily reflect mitochondrial biogenesis or protein assembly processes, rather than serving as a direct equivalent of total mitochondrial mass or content. To definitively confirm an increase in absolute mitochondrial content, future studies employing direct methods such as mitochondrial DNA quantification or electron microscopic morphometry are warranted.
This study demonstrates that EA may protect against CIRI by upregulating astrocytic Miro1 to boost mitochondrial transfer to neurons, thereby contributing to the restoration of neuronal mitochondrial function and biogenesis markers. These findings unveil a novel astrocyte-dependent mechanism of EA-mediated neuroprotection, offering potential therapeutic targets for ischemic stroke.
The datasets used and analysed during the current study available from the corresponding author on reasonable request.
HL: Writing – original draft, Validation, Conceptualization. YS: Validation, Formal analysis, Writing – original draft. MZ: Writing – original draft, Validation, Conceptualization. XG: Investigation, Data curation, Writing – review & editing. JW: Conceptualization, Formal analysis, Writing – review & editing, Resources. QD: Writing – review & editing, Supervision, Conceptualization, Project administration. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal care and experiments were conducted according to the Institutional Animal Care and Use Committee of Wenzhou Medical University, and all experiments were designed to minimize animal suffering. The study protocol was approved by the Animal Experiment Committee of Wenzhou Medical University (WYYY-AEC-2021-295).
We would like to express our gratitude to all those who helped us during the writing of this manuscript. Thanks to all the peer reviewers for their opinions and suggestions.
This study was supported by the Natural Science Foundation of China (Nos.81704180).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN48953.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.