




 , Hongguo Li 1, Yuchen Zhu 1, Siqi Song 1, Liang Kang 3,*
, Hongguo Li 1, Yuchen Zhu 1, Siqi Song 1, Liang Kang 3,* , Yushi Hu 1,2,*
, Yushi Hu 1,2,*1 School of Sports Medicine and Health, Chengdu Sport University, 610041 Chengdu, Sichuan, China
2 Sports Medicine Key Laboratory of Sichuan Province & Key Laboratory of Sports Medicine, General Administration of Sport of China, 641418 Chengdu, Sichuan, China
3 Sichuan Province Orthopedic Hospital, 610041 Chengdu, Sichuan, China
Abstract
Circadian rhythms and emotional health are fundamentally interconnected. Circadian rhythms are characterized by self-sustaining oscillations within biological systems that synchronize sleep-wake cycles and related physiological and biochemical processes with periodic environmental changes. Clinical observations have demonstrated the significant role of the biological clock in the regulation of mood and anxiety, indicating that disruptions in circadian pacemaker control may contribute to the development of mood disorders and psychopathology. Conversely, therapeutic interventions aimed at correcting circadian misalignment have been shown to alleviate symptoms of mood disorders. The mechanistic involvement of astrocytes in mood disorders has been well-established. Recent research indicates that astrocytes possess the ability to autonomously regulate circadian rhythms, independent of pacemaker neurons. In addition to modulating rhythmicity in the central pacemaker, the suprachiasmatic nucleus (SCN), astrocytes are involved in circadian regulation within emotion-related brain regions, such as the nucleus accumbens (NAc) and amygdala. Nonetheless, the precise mechanisms by which astrocytes influence mood through circadian pathways, and the potential for their rhythmic alterations to serve as therapeutic targets for mood disorders, remain incompletely understood. In this review we synthesizes contemporary evidence regarding the role of astrocytes as pivotal regulators of circadian rhythms, focusing on how their intrinsic transcriptional–translational feedback loop (TTFL) oscillations influence mood disorders. Additionally, we investigate the role of astrocytic neurotransmitters, such as glutamate, γ-aminobutyric acid (GABA), and purinergic signaling, in the circadian-mediated amelioration of mood pathologies. These insights offer novel perspectives for identifying chronotherapeutic intervention targets for mood disorders.
Keywords
- astrocytes
- circadian rhythms
- mood disorders
- glutamate
- chronotherapeutic
Mood disorders, characterized by significant disruptions in emotional regulation, pose a global health challenge in modern society [1, 2]. According to data from the World Health Organization (WHO), over 300 million individuals suffer from depression, which is the leading cause of non-fatal health loss worldwide [3]. These disorders impose considerable personal and societal burdens, yet our understanding of their etiology and therapeutic targets remains limited [4]. From this standpoint, we argue that a closer integration of psychiatry with sleep and circadian science will enhance the understanding and treatment of mental illnesses [5]. Clinical research consistently indicates that disruptions in sleep-circadian rhythms are prevalent across all psychiatric disorder diagnostic categories [6]. Shift work, which poses challenges to the sleep-circadian system, is associated with an increased risk of developing depression and anxiety disorders [7]. Diurnal mood variations and sleep disturbances are commonly observed in major depressive disorder (MDD), while cyclical mood episodes and alterations in sleep patterns are fundamental clinical features of bipolar disorder (BD) [8]. Importantly, this relationship is bidirectional: both acute and chronic stress can disrupt circadian rhythms, and such disruptions can, in turn, influence emotional states. For example, exposure to light or manipulations of sleep that affect the circadian clock have a direct impact on mood [9]. Pharmacological treatments for MDD and BD, such as paroxetine and lithium, also have effects on the biological clock [10, 11]. Furthermore, even in healthy individuals, the circadian clock plays a role in regulating emotions, as evidenced by distinct circadian rhythms in affective states and reward motivation under controlled conditions [12, 13].
Astrocytes, which comprise approximately 30% of brain cells, are distributed
throughout the central nervous system and engage in close interactions with
neurons to maintain neuronal homeostasis. These glial cells support neurons
through various mechanisms, including neurotransmitter uptake, ion buffering,
metabolic support, and the secretion of neurotrophic factors [14, 15, 16]. Dysfunction in
astrocytes impairs synaptic activity, and accumulating evidence suggests that
astrocytes modulate neuronal circuits and influence behavior [17, 18]. Recent
research indicates that astrocytes play a role in modulating a range of
behavioral domains, including sleep, circadian rhythms, perception, memory, and
emotional states such as anxiety [19, 20, 21]. Postmortem histopathological studies
of individuals with depression consistently report reduced glial cell densities,
suggesting that aberrant astrocyte function may contribute to the pathophysiology
of mood disorders [22, 23, 24]. Structural and functional deficits in astrocytes
are correlated with maladaptive behaviors, positioning these cells as promising
therapeutic targets for psychiatric and mood disorders. Traditionally, neurons
were considered the sole timekeepers in the suprachiasmatic nucleus (SCN), with
glial cells playing only supportive roles [25, 26]. Recent discoveries indicate
that astrocytes play an active role in the timing of the SCN network
[25, 27, 28, 29, 30, 31]. These glial cells possess cell-autonomous clocks that
regulate extracellular concentrations of glutamate and
This review synthesizes current evidence on the bidirectional relationship between circadian rhythms and mood disorders, with a particular focus on the involvement of astrocytes. Our aim is to consolidate diverse perspectives on astrocyte-mediated circadian modulation of mood pathology, identify existing research gaps, and propose mechanistic investigations for future astrocyte-targeted interventions in mood disorders.
Circadian rhythms encompass the rhythmic physiological, biochemical, and
behavioral changes that organisms undergo to adapt to approximately 24-hour
light-dark cycles [25]. In mammals, circadian rhythms represent a hierarchical
system that includes a central clock located within the SCN of the hypothalamus,
as well as peripheral clocks distributed across various tissues throughout the
body [43, 44]. The SCN is anatomically and functionally divided into two
subregions: the ventrolateral “core” and the dorsomedial “shell”, which are
distinguished by their neuropeptide content. The core subregion contains neurons
that express vasoactive intestinal polypeptide (VIP) and gastrin-releasing
peptide (GRP), whereas the shell subregion contains neurons that express arginine
vasopressin (AVP) [45]. These neuronal populations work synergistically to
facilitate photic entrainment. The core subregion of the SCN receives photic
input via the retinohypothalamic tract (RHT), which is mediated by glutamate and
pituitary adenylate cyclase-activating polypeptide (PACAP) [46, 47]. This photic
information is then relayed to the shell subregion through neurotransmitters such
as GABA, VIP, and GRP [48]. The mammalian circadian clock is primarily composed
of a core feedback loop and a stabilizing loop. Within the core circadian loop,
the heterodimer composed of circadian locomotor output cycles kaput
(CLOCK) and brain and muscle ARNT-like 1 (Bmal1) facilitates
the transcription of Period (Per) and Cryptochrome (Cry) genes through
E-box enhancer sequences located in their promoters. The subsequent accumulation
of the Period-Cryptochrome (PER-CRY) protein complex in the nucleus exerts
negative feedback, thereby inhibiting the activity of the CLOCK-BMAL1
heterodimer. As the PER-CRY complex undergoes degradation, a new cycle is
initiated approximately every 24 hours. In the stabilizing loop, the
CLOCK-BMAL1 heterodimer regulates the nuclear receptors
retinoic-acid-related orphan receptors alpha (Ror
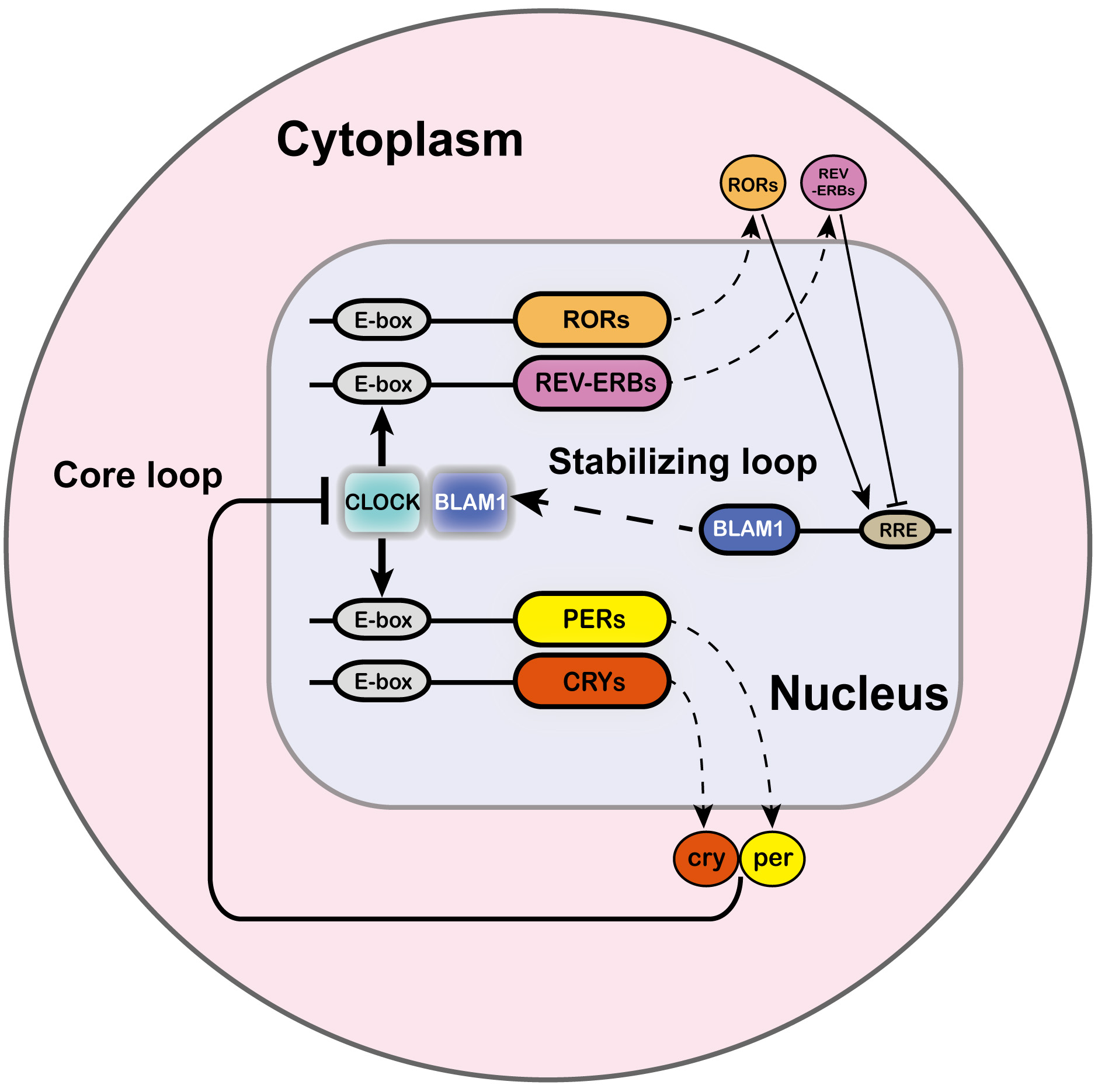 Fig. 1.
Fig. 1.
The mechanism of TTFLs in circadian rhythms. The TTFL is
intricate and involves multiple regulatory components. In mammals, the circadian
rhythm primarily consists of the core loop and the stabilizing loop. Within the
core loop, the heterodimer formed by CLOCK and BMAL1 proteins initiates
the expression of Per and Cry genes through enhancer box
(E-box) regulatory sequences located in their promoters. The PER-CRY protein
complexes subsequently accumulate in the nucleus, where they exert a negative
feedback effect by inhibiting the activity of the CLOCK-BMAL1
heterodimer. As the PER-CRY complexes degrade, a new cycle is permitted to
commence approximately every 24 hours. In the stabilizing loop, the
CLOCK-BMAL1 heterodimer drives the expression of the nuclear receptors
Ror
The central circadian pacemaker regulates the organism’s circadian rhythms through these mechanisms. Under typical conditions, both central and peripheral rhythms are synchronized, thereby preparing the body for expected daily behavioral and environmental change [50]. However, circadian disruption occurs when there is a misalignment between the phase of entrainment and the external environment, potentially resulting in mood and behavioral disorders [51]. Mood disorders, in addition, can also affect circadian rhythms, and possibly accompanied by structural and functional brain alterations [52, 53]. Neuroimaging studies have revealed volumetric changes in multiple brain regions in individuals with mood disorders, including the amygdala [54, 55] and hippocampus [56, 57]. Localized atrophy in the anterior-middle region of the right hippocampus has been found to correlate with later chronotypes in patients experiencing acute depression [58]. Supporting evidence from animal study indicates that unpredictable chronic mild stress (UCMS) induces anxiety- and depression-like behaviors in mice, significantly diminishes the amplitude of circadian locomotor activity (paralleling observations in patients with major depressive disorder), and simultaneously reduces molecular rhythm amplitude in the SCN while increasing rhythm amplitude in the NAc [59]. Chronotype refers to an individual’s preference for, or actual timing of, behavioral activities, and is influenced by genetic factors, age, sex, timing of light exposure, and social factors such as work and family schedules [50]. Chronotype also has significant implications for mood disorders, with late chronotypes and individuals experiencing social jet lag being at an increased risk for psychiatric disorders [60, 61]. Notably, a late chronotype is well-recognized to be associated with a higher prevalence of depressive symptoms in both individuals with major depressive disorder and healthy controls [60, 62, 63]. A diffusion tensor imaging study has revealed differences in white matter integrity within the frontal and temporal lobes, cingulate gyrus, and corpus callosum among individuals with an evening chronotype, as compared to those with morning and intermediate chronotypes [64]. Furthermore, the evening chronotype is associated with increased bilateral amygdala sensitivity to negative emotional facial expressions and reduced functional connectivity between the dorsal anterior cingulate cortex (dACC) and amygdala, suggesting potential disruptions in emotion regulation circuitry [65]. Notably, Sex differences play a crucial role in the regulation of circadian rhythms and the manifestation of mood disorders. During adolescence, boys exhibit a more significant shift towards a later chronotype relative to girls [66]. Additionally, adult males typically demonstrate a later chronotype than their female counterparts [67]. Moreover, factors such as sex hormones [68] and variations in brain structure and function [69] contribute to the increased vulnerability of women to mood disorders, such as anxiety, compared to men. Consequently, incorporating sex as a biological variable is essential for the development of more personalized, rhythm-based intervention strategies for individuals with mood disorders.
Additionally, clock gene polymorphisms have been associated with mood disorders,
such as major depressive disorder and anxiety [70, 71, 72, 73]. Research involving
BD patients has identified that the CLOCK rs1801260 polymorphism, linked to the
evening chronotype, correlates with discrepancies between subjective and
objective assessments of depressive symptom severity. Similarly, the efficacy of
antidepressants in MDD patients may have been influenced by variations in the
Clock gene [73]. In addition to its association with depression and anxiety,
human CRY1
SCN receive numerous synaptic inputs, most notably from the RHT, which transmits
light-responsive signals directly from the retina to synchronize biological
rhythms via this zeitgeber [78, 79]. Remarkably, astrocytes within the SCN exhibit
an increase in cellular FBJ osteosarcoma oncogene (cFOS) expression in response
to photic stimulation, indicating their potential role in light response and
possibly in circadian entrainment [80, 81]. Emerging evidence suggests the
existence of an SCN-independent pathway, mediated by intrinsically photosensitive
retinal ganglion cells (ipRGCs), which provide excitatory synaptic input to
neurons in the perihabenular nucleus (PHb) of the dorsal thalamus and play a role
in mood regulation [82]. Research has confirmed that abnormal exposure to
nighttime light significantly influences mood and may substantially elevate the
risk of developing mood disorders, such as depression [83]. Mice exposed to
constant light or light during the night has been shown to significantly
exacerbate depression-like behaviors, such as decreased sucrose preference and
increased immobility time, while also diminishing the amplitude of circadian
rest-activity rhythms and reducing nighttime activity levels [84, 85].
Additionally, dim light at night interferes with Per1 gene expression in
the hippocampus of postpartum mice, with a potential link to depression through
the TGF-
Although extensive clinical evidence and foundational research substantiate the therapeutic efficacy and underlying mechanisms of circadian rhythm interventions for mood disorders, current intervention strategies predominantly target neurons. Astrocytes, which engage in bidirectional interactions with neurons to dynamically modulate cellular functions, are pivotal glial cells in the context of mood disorder interventions [100]. The role of astrocytes in circadian regulation is gaining recognition, necessitating further investigation into their potential as key therapeutic targets for modulating circadian-related mood disorders.
The SCN is predominantly comprised of neurons and astrocytes. Neurons within the SCN are essential for sustaining circadian rhythms, as they autonomously regulate clock gene expression to initiate rhythmic patterns and synchronize molecular clocks in peripheral tissues [25, 26]. Increasing evidence suggests that astrocytes, which are neuronally active and possess cell-autonomous TTFL clocks, are vital components of the SCN’s timekeeping system [27, 28]. Astrocytes display pronounced intrinsic rhythmicity, as evidenced by the 24-hour oscillations of glial fibrillary acidic protein (GFAP), a specific marker for astrocytes, under both light-dark (LD) cycles and constant darkness (DD) conditions [101]. The [Ca2+]i in astrocytes, crucial for neuroglial interactions, exhibits rhythmic fluctuations, peaking at night and reaching troughs during the day [29]. Notably, these astrocytic [Ca2+]i oscillations are antiphasic to those observed in neurons [27, 29]. The observed phase difference between SCN astrocytes and neurons indicates a mutually antagonistic coupling mechanism. In this model, daytime signals emitted by electrically active neurons inhibit astrocytic functions, whereas the nocturnal suppression of circadian neurons and their associated neuropeptidergic networks permits astrocytic activation, which may in turn suppress neuronal activity [29].
Astrocytes have been demonstrated to participate in the modulation of circadian
rhythms in the SCN. The conditional knockout of Bmal1 in SCN astrocytes,
identified by the Aldehyde dehydrogenase 1 family member L1 (Aldh1l1) promoter,
exhibit an extension of the circadian period of the SCN and alterations in
locomotor activity rhythms in mice [30]. Similarly, targeting astrocytes via the
Glutamate aspartate transporter (GLAST) promoter in Bmal1 conditional
knockout mice, exhibit a delayed onset of the active phase of locomotor activity
[102]. These findings collectively underscore the correlation of clock genes
within astrocytes in regulating the organism’s circadian rhythms. Additionally,
the circadian period is extended in Csnk1
Research has established the critical role of astrocytes in circadian
regulation. Furthermore, astrocytic dysfunction may disrupt normal brain function
and potentially contribute to the development of mood disorders [105]. Research
on astrocyte morphology and astrocyte-specific biomarkers has identified
hypertrophy of astrocyte cell bodies and processes in the anterior cingulate
cortex (ACC) of individuals who have died by suicide with depression [106].
Additionally, studies have reported a reduction in astrocyte density and
decreased expression of the classical astrocyte marker GFAP in various brain
regions, such as the amygdala and ACC, in patients with MDD [106, 107]. In
contrast, in depressed suicide victims, a downregulation of GFAP mRNA and protein
expression has been observed in the dorsomedial thalamus and caudate nucleus,
despite the presence of enlarged astrocyte cell bodies and processes [108].
Moreover, increased serum levels of the calcium-binding protein S100
Furthermore, studies on astrocyte rhythm-regulating proteins have demonstrated downregulated expression of astrocyte-associated proteins, including glutamine synthetase (GS), glutamate transporters, and connexins, in patients with mood disorders [116, 117]. Glutamate serves as a critical gliotransmitter through which astrocytes modulate circadian rhythms to sustain the TTFL in the SCN. Inhibition of astrocytic glutamate transport has been shown to disrupt the TTFL in the SCN [29]. Empirical evidence indicates that the mRNA expression of GS, indicative of astrocytic glutamate handling capacity, is diminished in the dorsolateral prefrontal cortex (dlPFC), premotor cortex, and amygdala of individuals with depression [116]. Similarly, analyses of specific cortical regions in patients with MDD reveal downregulation of genes encoding the high-affinity astrocytic glutamate transporters solute carrier family 1 member 2 (SLC1A2) and solute carrier family 1 member 3 (SLC1A3) [118]. Connexin 30 (Cx30) and connexin 43 (Cx43) are essential proteins that facilitate calcium wave propagation and inter-astrocytic communication. Targeted inhibition of glutamate release via Cx43 hemichannels can influence the expression of circadian clock genes in SCN neurons [31]. Research has demonstrated reduced expression of Cx30 and Cx43 in the dlPFC of individuals with MDD who have died by suicide [119]. Similar findings have been reported for aquaporin-4 (AQP4), with decreased expression levels observed in MDD patients [105]. Glutamate transporters serve as a principal mechanism through which astrocytes regulate glutamate concentrations. Additionally, in astrocytes and hippocampal slices, glutamate release is predominantly facilitated by Cx43 hemichannels [120]. This observation implies that the downregulation of glutamate transporters and connexins in patients with mood disorders corresponds with the observed trend of diminished rhythmic expression in the SCN and may constitute a mechanism contributing to circadian disruption. Nonetheless, research concerning the regulation of astrocytic glutamate rhythms has predominantly concentrated on the SCN. It remains uncertain whether the circadian disruption symptoms associated with mood disorders originate from intrinsic rhythm alterations within mood-related brain regions or are contingent upon glutamate projections from the SCN. Human-based studies, particularly those involving post-mortem analyses, possess substantial limitations that obscure the generalizability of these findings. Consequently, further mechanistic studies employing animal models are necessary for validation.
A review of genetic evidence from various animal models suggests that astrocytic networks within the brain modulate behavioral changes, including those related to sleep, circadian rhythms, and depression-like behaviors [20]. Moreover, exposure to acute or chronic stress stimuli induces both functional and structural alterations in astrocytes, potentially resulting in deficits in emotion-related behaviors [121]. The SCN functions as the central circadian pacemaker of the brain. The rhythmic expression of TTFL genes and circadian behaviors are directly modulated by chemogenetic or optogenetic activation of SCN neurons [78, 79]. Additionally, the amplitude of molecular rhythms within the SCN diminishes following stress intervention with UCMS [59]. Recent studies have revealed that astrocytes also exhibit cell-autonomous TTFL rhythms, a finding corroborated by ex vivo SCN slices expressing astrocyte-specific TTFL genes [31, 104].
In addition to its established role in modulating reward-related rhythms, the NAc may also serve as a circadian oscillator, integrating sensory and circadian inputs to temporally regulate motivated behaviors [122]. Electrophysiological study has demonstrated that medium spiny neurons (MSNs) within the NAc exhibit circadian fluctuations in glutamatergic synaptic transmission, peaking during the excitatory phase that aligns with the active/dark period in mice [123]. Moreover, dopamine levels and its metabolites in the NAc display pronounced circadian rhythms [124, 125, 126], and the transcription of genes such as tyrosine hydroxylase, dopamine transporter, monoamine oxidase A, and dopamine receptor D1 (Drd1), D2 (Drd2), D3 (Drd3) is directly influenced by the molecular circadian clock [127, 128, 129, 130, 131, 132]. Research indicates that UCMS not only reduces the amplitude of circadian locomotor activity and molecular rhythms in the SCN of depressed mice but also increases the amplitude of molecular rhythms in the NAc brain region. Furthermore, there is a negative correlation between the time spent swimming in the forced swim test (FST) and the time spent in the open arms of the elevated plus maze (EPM) with the Per2::luc rhythm amplitude in the NAc [59]. Recent research has demonstrated that NAc astrocytes not only express robust rhythms in all canonical clock genes, but approximately 43% of the entire NAc astrocyte transcriptome exhibits a significant diurnal rhythm [34]. In contrast, only 6% of the entire NAc (encompassing all cell types) was found to be rhythmic [133], the astrocyte-specific rhythm highlights the complexity of circadian regulation within the NAc. Disruption of the molecular clock specifically within NAc astrocytes showed alterations in exploratory and emotional behaviors during the daytime, but not at night, suggesting significant circadian variation [34] (Table 1, Ref. [32, 34, 35, 89, 134, 135, 136, 137, 138]). The NAc is a critical brain region involved in emotion regulation, receiving projections from PFC neurons and modulating mood and stress responses [139]. Collectively, these studies suggest the presence of an intrinsic, functional circadian clock system within the NAc that orchestrates its functions across different times of the day. Furthermore, these rhythms may be entrained by rewarding stimuli and/or indirectly influenced by innervation from the SCN [122, 140, 141, 142, 143]. Although a direct neural pathway from the SCN to the NAc has yet to be identified, it is plausible that the NAc receives circadian information indirectly from the SCN via the paraventricular nucleus of the thalamus (PVT) [122]. However, the detailed mechanisms underlying these processes remain to be elucidated. Furthermore, the neuronal composition of the SCN and NAc is notably similar, as both regions are predominantly composed of GABAergic neurons [144, 145, 146]. Importantly, the use of UCMS models selectively altered rhythm amplitudes specifically in the SCN and NAc, while rhythms in other regions typically associated with anxiety and depression remained relatively unaffected [59]. Consequently, astrocyte rhythms within the NAc might play a pivotal role in the regulation of mood disorders.
| Affected region | Manipulation | Astrocyte phenotype | Emotional behavior | References |
| NAc | Bmal1 deletion | GFAP expression ↑ | Circadian day: locomotor response to novelty ↑ | [34] |
| AMPA (Gria1/2/3/4) expression ↑ | Circadian day: the number of center entries (OFT) ↑ | |||
| NMDA (Grin2a/2d/3a) expression ↑ | Circadian day: time in the lighted (dark/light box) ↑ | |||
| mGluRs (Grm2) expression ↑ | ||||
| GLT-1 expression ↑ | ||||
| AMPA/NMDA EPSC ratio during the day ↓ | ||||
| GCre+ Per2fl/fl | GAT-2/Slc6a13 and Drd3 expression ↓ | Immobile time (FST) ↓ | [35] | |
| Glutamate levels ↓ | Time in the open section (O-maze) ↑ | |||
| Per2 expression ↓ | ||||
| Per1/2 deletion | N/A | Time in the lighted (dark/light box) ↓ | [134] | |
| Locomotor activity in the light (dark/light box) ↓ | ||||
| Time in the open arms (EPM) ↓ | ||||
| Per2 deletion | Glutamate levels ↓ | Despair and anxiety ↓ | [135, 136] | |
| GAT-2/Slc6a13 and Drd3 expression ↓ | ||||
| SCN | Bmal1 deletion | N/A | Escape latencies and number ↑ | [89] |
| Cry1-null slices | Circadian day: GAT-3 ↑ [GABA]e ↓ | N/A | [32] | |
| Circadian night: GAT-3 ↓ [GABA]e ↑ | ||||
| VTA | Clock-Δ19 | N/A | Anxiety-related behavior ↑ | [137, 138] |
PM, Elevated plus maze; EPSC, excitatory postsynaptic currents; FST, forced swim
test; GABA,
Neurons in the mPFC receive inputs from the cortex, thalamus, and limbic system,
and project to multiple brain regions, including the amygdala, NAc, dorsal raphe
nucleus (DRN), and lateral habenula (LHb), to regulate emotion and stress
responses, establishing the mPFC as a vital region for emotional control [139].
Clinical investigations into the mPFC and the striatum—brain regions associated
with positive affect—indicate that individuals with an evening chronotype
exhibit diurnal patterns of positive affect characterized by a phase delay and
reduced amplitude, alongside diminished overall metabolic activity in these
regions during both morning and evening wakefulness. Collectively, these
preliminary findings may represent a potential mechanism underlying the
heightened risk of mood disorders among late chronotype [42]. This suggests a
possible link between the emotional regulatory functions of the mPFC and the
circadian clock. Previous studies have demonstrated that the ablation of mPFC
astrocytes via L-
| Affected region | Manipulation | Change in astrocytes | Rhythmic molecular phenotype | Emotional behavior | Reference |
| SCN | GFAP-Cre mice; | CB1/2Rs ↑ | Per2::Luc rhythms ↑ | N/A | [153] |
| WIN 55,212-2 treatment | ADO ↑ | ||||
| Vgat-Cre; ChR2 mice | N/A | The homecage activity period or tau ↓ | Time in the center (OFT) ↓ | [150] | |
| The amplitude of homecage activity rhythms ↓ | Open arm entries and open arm time (EPM) ↓ | ||||
| mPFC | Cx43-shRNA mice | Cx43 ↓ | ATP levels ↓ | Sucrose preference (SPT) ↓ | [40] |
| Immobility (FST) ↑ | |||||
| Time in the central area (OFT) ↓ | |||||
| Time in the open arms (EPM) ↓ | |||||
| Probability of entering the open arms (EPM) ↓ | |||||
| WT mice; | Cx43 ↑ | ATP levels ↑ | Sucrose preference (SPT) ↑ | [40] | |
| AAV2/5-GfaABC1D-Cx43-mCherry treatment | Immobility (FST) ↓ | ||||
| Time in the central area (OFT) ↑ | |||||
| Time in the open arms (EPM) ↑ | |||||
| Probability of entering the open arms (EPM) ↑ | |||||
| WT mice; | EAAT2 ↓ | Typical waking EEG | Latency to begin drinking sucrose ↑ | [147] | |
| DHK treatment | Intracranial self-stimulation thresholds ↑ | ||||
| WT mice; | Astracyte ↓ | N/A | Sucrose preference (SPT) ↓ | [37] | |
| L-AAA treatment | Latency to feed in novelty suppressed feeding test ↑ | ||||
| Immobility duration (FST) ↑ | |||||
| Escape latency (AAT) ↑ | |||||
| WT mice; | N/A | N/A | CSDS-induced social avoidance (SIT) ↓ | [152] | |
| ATP or ATP- |
CSDS-induced immobility (FST) ↓ | ||||
| CMS-induced fur condition ↓ | |||||
| Post-CMS sucrose preference (SPT) ↑ | |||||
| ZI | WT mice; | Ca2+ ↓ | GABAergic neuronesactivation ↑ | Anxiety-like behaviour ↓ | [151] |
| hPMCA2w/b treatment | GAT-3 expression ↑ | ||||
| BLA | LDN-212320 treatment | EAAT2 ↓ | N/A | Entries and time spent in the center (OFT) ↓ | [149] |
| Exploration of the open arms (EPM) ↓ | |||||
| LHb | WT mice; | EAAT2 ↓ | N/A | Entries and time spent in the center (OFT) ↑ | [149] |
| DHK treatment | Exploration of the open arms (EPM) ↑ | ||||
| GLT-1flox/flox mice; | EAAT2 ↓ | LHb neurons ↑ | Immobility duration (TST) ↑ | [148] | |
| DHK treatment | REM sleep ↑ | Latency to the first immobility episode ↓ | |||
| Latency to the onset of REM ↓ | |||||
| Unimodal distributions for all three sleep stages | |||||
| CeM | WT mice; | Ca2+ ↑ | N/A | Freezing response ↓ | [149] |
| AAV8-GFAP-hM3D-mCherry treatment | Fear response ↓ | ||||
| No anxiety behavior | |||||
| N/A | Per2::Luc transgenic mice; | N/A | Per1 and Per2 expression ↑ | N/A | [154] |
| IB-MECA treatment | Per1 and Per2 rhythms period length ↑ | ||||
| THP-1 (TIB-202) cells; CGS21680 treatment | N/A | Clock and Bmal mRNA ↑ | N/A | [155] | |
| Per2 mRNA ↓ | |||||
| Rev-erb |
AAT, active avoidance test; ATP-
The amygdala, a downstream target of mPFC neuronal regulation, plays a crucial role in the modulation of emotions, including fear and anxiety [156]. The basolateral amygdala (BLA) functions as the primary input structure of the amygdala, receiving a wide array of sensory information from the thalamus and cortex [157]. Previous research has demonstrated that chronic unpredictable mild stress intervention leads to a reduction in reactive astrocyte activity within the BLA. In contrast, chronic (21-day) optogenetic stimulation of Channelrhodopsin-2 (ChR2) in BLA astrocytes help to mitigate anxiety-like behaviors in mice exposed to UCMS [109]. Notably, investigations into neuronal involvement have revealed that activation of BLA astrocytes contribute to alleviates anxiety, whereas activation of BLA glutamatergic neurons does not reduce anxiety-like behaviors in stressed mice [158]. Furthermore, clinical studies indicate that the observed reduction in amygdala volume in patients with mood disorders may primarily result from a decrease in astrocyte density rather than neuronal loss [159, 160]. Collectively, these findings suggest that the function of amygdala astrocytes may operate independently of neuronal activity. Recent clinical research suggests a potential association between the emotional regulatory function of the amygdala and the circadian clock, as evidenced by increased bilateral amygdala sensitivity to negative emotional facial expressions in patients with an evening chronotype [65]. This finding may elucidate, from a neuroregulatory perspective, the frequent comorbidity of mood disorders and circadian rhythm disruptions. Conversely, animal study has demonstrated that while UCMS diminishes the amplitude of circadian locomotor activity and molecular rhythms in the SCN of depressed mice, the rhythmicity of Per2 expression in the frontal cortex and amygdala remains largely unaffected [59]. It is hypothesized that astrocytes within the amygdala may play a pivotal role in rhythm regulation. However, further research is needed to elucidate how these astrocytes modulate rhythmic changes in the amygdala under conditions of mood disorders.
Integrating the aforementioned evidence, astrocytes appear to be integral components of the anxiety regulatory circuitry, extending from the mPFC to downstream regions such as the amygdala and NAc. In light of the increased sensitivity to negative emotional expression observed in individuals with an evening chronotype, the regulation by the mPFC, amygdala, and NAc demonstrates a distinct temporal bias. Research indicates that astrocytic [Ca2+]i undergoes rhythmic fluctuations, characterized by a peak during the night and a trough during the day [29]. Neuronal signals released during the daytime suppress astrocytic function, whereas the nocturnal silencing of neuronal networks permits astrocytic activation [29, 130], a phenomenon consistent with chronotype trends identified in clinical studies. Nonetheless, these studies predominantly focus on specific brain regions and primarily describe effects. Therefore, the precise mechanistic role of astrocytic rhythmic alterations in mood disorders necessitates further investigation.
Rhythmic neuronal activity and neurotransmitter expression within the SCN are regulated by the Clock gene. Consequently, alterations in circadian clock gene expression may lead to disruptions in neuronal physiology, ultimately impacting peripheral biological systems governed by the SCN [25, 43, 44]. Remarkably, astrocytes in other brain regions beyond the SCN were also found to have significant rhythmicity [122].
Studies have demonstrated significant rhythmicity in the circadian molecular
clock function of the NAc. Sustained bioluminescent rhythms have been observed in
the NAc ex vivo using the PER2:LUC mouse model [59, 161],
corroborating gene expression analyses in mice that reveal significant diurnal
variation in molecular clock mRNA levels within the NAc [59, 133, 142, 162, 163, 164].
This phenomenon is corroborated by RNA-sequencing analyses of human post-mortem
NAc tissue, which reveal pronounced rhythmic expression of canonical clock genes,
such as Bmal1, Npas2, Period, Cryptochrome,
and Rev-erb
It has been demonstrated that the knockdown of Bmal1 specifically within the SCN leads to reduced circadian rhythmicity, desynchronized single-cell rhythms, and exhibit an increase in behaviors indicative of despair, anxiety, and helplessness [135, 136]. In contrast, the functional disruption of the Bmal1 molecular clock in NAc astrocytes is associated with improvements in reward- and emotion-related behaviors, such as increased time spent in the open arms of the EPM and a higher number of center entries in the open field test (OFT) during the day, but not at night [34]. However, another study reported that the deletion of Bmal1 in NAc astrocytes does not influence emotional behaviors such as despair or anxiety [35]. Notably, the effects of disrupting the Bmal1 molecular clock in SCN versus NAc astrocytes on mood regulation appear to be diametrically opposed. The attenuation of activity within the SCN, which serves as the central circadian pacemaker, results in the disruption of rhythmic patterns in downstream brain regions that are directly implicated in emotion regulation [136, 167]. This suggests that disturbances in the SCN’s central rhythmic pacemaker may directly induce alterations in brain regions responsible for emotion regulation, thereby impacting emotional behavior. In contrast, the rhythmic regulation within the NAc is generated locally within this downstream brain region and does not influence the central rhythmic control exerted by the SCN. Similar to the SCN, the NAc is primarily composed of GABAergic neurons, and astrocytes within the NAc play a pivotal role in maintaining glutamate homeostasis [144, 145]. The regulation of glutamate by astrocytes in the NAc is believed to contribute to reward modulation and susceptibility to drug addiction [130, 168, 169]. Moreover, the functional disruption of the Bmal1 molecular clock in NAc astrocytes has been shown to significantly upregulate the expression of genes related to glutamate [34]. Moreover, the astrocyte-specific deletion of Bmal1 across the brain results in astrocyte activation and the expression of inflammatory genes, thereby promoting neuronal death in vitro [36]. The neuroinflammation resulting from Bmal1 deletion may be linked to the onset of mood disorders. Thus, while astrocytic Bmal1 in both the NAc and SCN plays a role in emotion regulation, the underlying pathways differ, with the regulation of glutamate transport likely serving as a key mechanism for astrocytic Bmal1 in the NAc (Fig. 2).
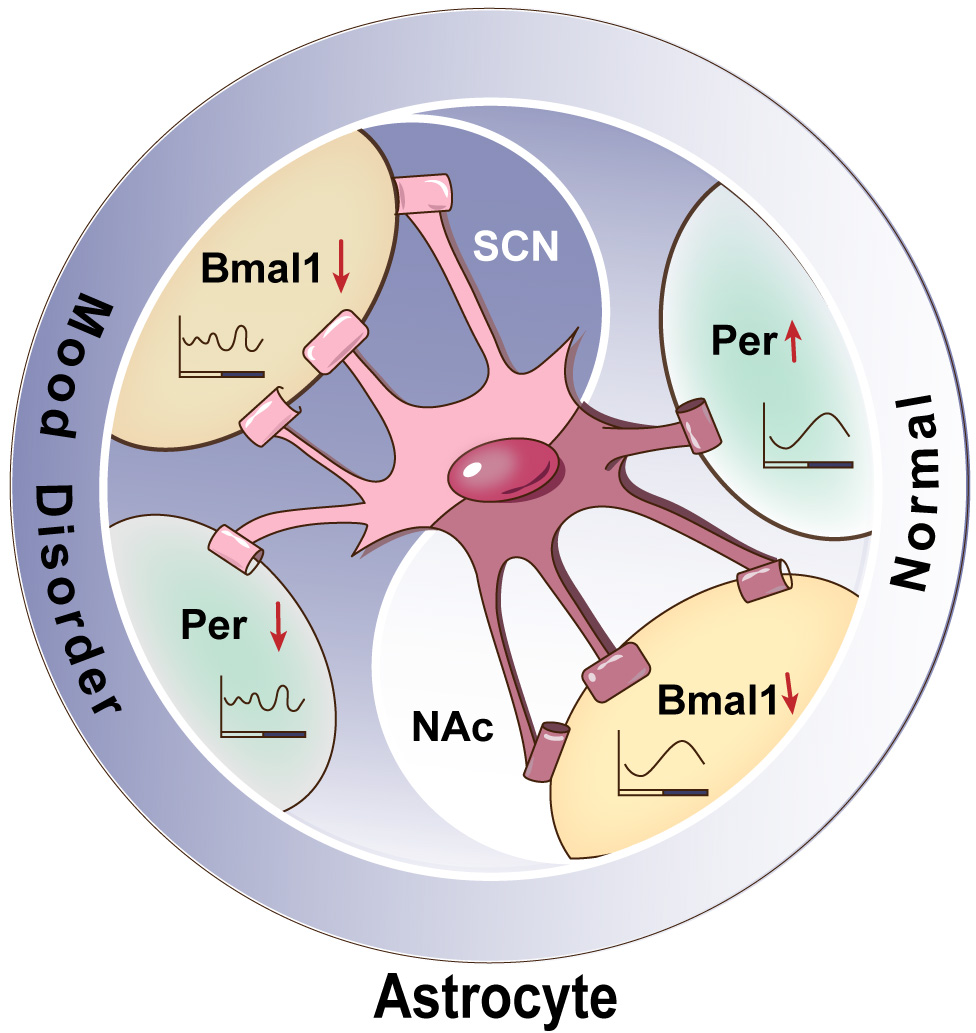 Fig. 2.
Fig. 2.
Illustrates the regulation of astrocytic clock genes in the SCN
and the NAc in the context of mood disorders. In the SCN, the astrocyte-specific
deletion of Bmal1 or Per disrupts circadian rhythms and is associated
with the manifestation of mood disorder phenotypes. In the NAc, the
astrocyte-specific deletion of Bmal1 alleviates depressive-like
behaviors without affecting circadian rhythmicity, while the deletion of Per
results in mood disorder phenotypes while maintaining rhythmicity. SCN,
suprachiasmatic nucleus; NAc, nucleus accumbens;
The ventral tegmental area (VTA), a critical region involved in the regulation
of reward and emotion, demonstrates a distinct rhythmic organization, including
Clock gene rhythms [122, 170]. Diurnal transcriptional regulation of key enzymes
responsible for dopamine (DA) synthesis and degradation within the VTA has been
documented [13, 128], alongside circadian variations in the activity of VTA DA
neurons [171]. Research indicates that Clock-
The evidence for direct neuronal projections from the SCN to downstream regions involved in emotion regulation, such as the NAc and VTA, remains limited. Consequently, it is not possible to definitively ascertain whether the observed astrocytic rhythmic changes in the NAc and VTA are directly regulated by the SCN or if they arise from indirect effects of mutations affecting various unrelated processes in other brain regions. Current evidence supports the presence of a neural connection from the SCN to the striatum, which influences anxiety- and depression-like behaviors in mice. Additionally, SCN lesions or Bmal1 knockout result in altered amplitudes of circadian gene oscillations and total mRNA levels in the striatum, alongside activation of the brain-derived neurotrophic factor (BDNF)-tropomyosin receptor kinase B (TrkB) pathway [172]. Nevertheless, the specific regulatory mechanisms within the NAc and VTA require further investigation.
Astrocytes, a predominant type of glial cell, play a crucial role in modulating glutamate levels through mechanisms of uptake, release, and maintaining neuro-metabolic homeostasis [173]. Investigations utilizing real-time imaging of extracellular glutamate concentrations within the SCN have demonstrated distinct circadian oscillations. Notably, the extracellular glutamate rhythm aligns with the [Ca2+]i rhythm observed in astrocytes [29]. Prolongation of the Ca2+ rhythm period correspondingly extends the period of the glutamate rhythm [31]. In the context of arrhythmic Cry-null SCN, astrocyte-mediated glutamate regulation has been shown to influence neuronal rhythmic recovery, thereby affecting the overall circadian rhythm of the SCN [31]. As the central circadian pacemaker of the brain, the SCN displays disrupted Clock gene expression rhythms in response to various light intervention stimuli [85, 174], which are associated with behavioral changes such as heightened depression-like behaviors and diminished anxiety-like responses [175, 176]. Furthermore, dysregulation of glutamatergic signaling has been observed in preclinical models of depression and in patients with MDD [177, 178]. Neuroimaging and postmortem analyses have consistently shown alterations in glutamatergic signaling in individuals with BD. The NMDAR antagonist ketamine has been validated as an effective antidepressant and anti-suicidal intervention for BD [179, 180, 181]. Therefore, The circadian regulation of extracellular glutamate concentration serves as a crucial mechanism through which astrocytes contribute to mood regulation. This process involves the transmission of temporal information, generated by astrocytes’ cell-autonomous clocks, to the neurons of the SCN, thereby influencing affective states [25, 31, 104]. The question arises as to how the rhythmic fluctuations in astrocytic glutamate are communicated to SCN neurons.
Primarily, astrocytes mediate the removal and recycling of glutamate from the synaptic cleft. Cx43 is the predominant gap junction protein in astrocytes [182], playing a significant role in astrocyte-neuron interactions and capable of modulating circadian clock gene expression in SCN neurons [31]. At night, astrocytes release glutamate through Cx43 hemichannels, which activates NR2C-containing N-methyl-D-aspartate receptors (NR2C-NMDARs) on neuronal presynaptic membranes. This activation results in an increased [Ca2+]i concentration, promoting the release of gamma-aminobutyric acid (GABA), which subsequently inhibits postsynaptic neuronal electrical activity. Conversely, during the day, the release of glutamate from astrocytes diminishes. Concurrently, the efficiency of excitatory amino acid transporters (EAATs) in clearing glutamate is enhanced, leading to a decrease in extracellular glutamate concentration and a reduction in the inhibition of postsynaptic neurons, thereby facilitating peak neuronal electrical activity [29, 33]. Furthermore, rhythmic changes in glutamate levels have also been observed in the hippocampus, a critical region for emotion regulation. Research indicates that the magnitude of long-term potentiation (LTP) in the hippocampus exhibits diurnal variation [183]. Specifically, during the dark phase, astrocytes retract their processes and reduce glutamate uptake, accompanied by a decrease in the rhythmic amplitude of NMDA EPSCs [184]. Circadian regulation influences the rhythmic capacity of astrocytes for glutamate release and recycling. Aberrant expression of astrocyte glutamate-associated proteins may result in an excitation-inhibition imbalance, potentially precipitating mood disorders such as anxiety and depression. Clinical studies have reported diminished expression and content of the glia-specific enzyme glutamine synthetase (GS) and the glutamate transporter 1 (GLT-1) in individuals with depression [118], along with the downregulation of SLC1A2 and SLC1A3 [118], and reduced expression of connexins Cx30 and Cx43 [119]. Furthermore, microinjection of a GLT-1 inhibitor into the rat PFC might induces depression-like behavior [147], suggesting compromised glutamate clearance and metabolism in specific brain regions.
The epithalamic habenula (Hb), a critical component of the brain’s reward circuitry, contains an autonomous circadian clock that is functionally coupled to the SCN [185]. Moreover, the region-specific inhibition or deletion of GLT-1 in the lateral habenula (LHb) induces depression-like phenotypes, as evidenced by the tail suspension test (TST) and the novelty-suppressed feeding test (NSFT). The frequency of neuronal spikes in the LHb increases following pharmacological inhibition or virus-mediated deletion of GLT-1, correlating with depressive symptoms [148]. During daylight hours, the uptake capacity of GLT-1 is enhanced, facilitating the recycling of excess synaptic glutamate and maintaining glutamate homeostasis [29, 30]. Inhibition of GLT-1 can result in glutamate accumulation and subsequent neurotoxicity [186], aligning with the observed effects of region-specific GLT-1 inhibition on emotional behavior. Therefore, it is plausible that GLT-1 inhibition exerts its effects predominantly during the daytime. Conversely, the targeted knockdown of EAAT2 within BLA astrocytes contribute to ameliorated anxiety-like behavior in stressed mice, whereas overexpression of EAAT2 in the BLA might induced anxiety-like behavior [149]. These findings suggest that the regulation of glutamate uptake and emotional behavior is specific to particular brain regions. In conclusion, rhythmic alterations in astrocytic glutamate levels may disrupt the balance between excitatory and inhibitory neurotransmission, potentially contributing to mood disorders. Nevertheless, the pathways vary across different brain regions, and glutamate homeostasis may offer temporal insights into the mechanisms underlying mood disorders.
In addition to inadequate glial glutamate reuptake, mood disorders are also
linked to glutamate receptors, such as NMDA receptors and
In models of depression, enhanced burst activity of Hb neurons has been observed. Activation of Hb neurons induces burst activity via NMDA receptors, which exerts an inhibitory effect on the brain’s reward circuitry, thereby contributing to symptoms of despair, anhedonia, and anxiety [188]. The mechanism of action for rapid-acting antidepressants, such as ketamine, involves modulation of this process. Ketamine primarily targets NMDA receptors, reducing the burst activity of Hb neuron-dependent NMDA receptors, thereby alleviating the inhibition of the reward circuit and rapidly ameliorating mood [189]. Clinical investigations have demonstrated that treatment with the NMDA receptor antagonist ketamine ameliorates depressive symptoms in patients with MDD and enhances the glutamine-glutamate complex to water ratio (Glx/W) as well as the gamma-aminobutyric acid to water ratio (GABA/W) in the mPFC [190, 191]. Corresponding findings have been observed in animal models, where chronic mild stress (CMS) induces depressive-like behaviors and elevates NMDAR expression levels [192]. In contrast, ketamine administration reduces NMDAR expression and significantly increases extracellular glutamate concentrations [193]. Notably, selective inhibition of the GluN2A subunit by the NMDAR antagonist PEAQX markedly attenuates ketamine’s antidepressant effects, whereas activation of the GluN2A subunit does not influence ketamine’s efficacy [189]. These findings suggest that NMDAR inhibition constitutes a potential mechanism for alleviating mood disorders, although the roles of different NMDAR subunits may be divergent. For example, CMS has been shown to upregulate GluN1 receptor levels in the hippocampal synaptic membrane fraction without affecting GluN2A, while significantly increasing GluN2B levels [192]. However, recent study indicates that extensive activation of the GluN2A subunit might elicit antidepressant effects [189]. The effects of GluN2A intervention may vary depending on the specific brain region. Ionotropic NMDARs are emerging as a potential target for the regulation of astrocyte-mediated glutamate rhythms in mood disorders. While ketamine research has predominantly focused on neuronal mechanisms, especially glutamatergic signaling [191, 194], astrocytes also play a crucial role in modulating NMDAR activity by regulating extracellular glutamate rhythms [29, 30, 184], thereby influencing mood disorders. Investigating astrocyte regulation of specific NMDAR subtypes could represent a promising new research avenue.
Neurons within the SCN are primarily GABAergic, with nearly all SCN neurons expressing GABA [45]. Studies suggest that GABA-mediated neurotransmission can be either excitatory or inhibitory, depending on the circadian phase or subregion within the SCN [195, 196], and has the capacity to synchronize or desynchronize the circadian rhythms of SCN cells [197]. Astrocytes are involved in the synthesis, transport, and release of GABA. During synthesis, GABA can be absorbed by astrocytes, converted into glutamine (Gln) via the tricarboxylic acid (TCA) cycle, and subsequently transported back to GABAergic neurons for GABA resynthesis [198]. GABA serves as a critical molecule in astrocyte-mediated circadian regulation. Astrocytes in the SCN facilitate coherent spatiotemporal circadian patterns of neuronal activity by rhythmically regulating glutamate uptake and release, as well as modulating GABAergic tone and receptor signal transduction [199, 200]. Extracellular GABA levels within the SCN display pronounced rhythmicity, oscillating in phase with glutamate, with both reaching their peak during the night. These rhythms are antiphase to neuronal activity. In genetically arrhythmic SCN, GABA rhythms are absent but can be reinstated by activating the TTFL in astrocytes [32]. Furthermore, inhibiting synaptic GABA release disrupts neuronal circadian rhythms in the SCN, and suppressing astrocytic GABA synthesis eliminates the extracellular GABA rhythm and desynchronizes neuronal circadian activity. This indicates that astrocytic GABA synthesis is a vital pathway for intrinsic circadian regulation within the SCN circuitry [201]. Research indicates that a 3-hour exposure to light at the onset of the subjective night suppresses changes in glutamate and GABAergic activity, specifically in the frequency of miniature excitatory postsynaptic currents (mEPSC) and miniature inhibitory postsynaptic currents (mIPSC). In contrast, exposure to a skeleton photoperiod does not influence mEPSC or mIPSC frequency between the subjective day and night [202]. Additionally, the suppression of rhythms in SCN GABA neurons has been shown to increase anxiety-like behavior in mice [150]. These findings suggest that circadian disruptions may predispose individuals to mood and anxiety disorders, with an imbalance between excitation and inhibition potentially affecting emotional states. While there is growing evidence of reduced GABA levels in the PFC of patients with (MDD [203], the mechanisms by which astrocytic GABA release is altered in mood disorders remain poorly understood.
The astrocytic clock can modulate emotional behavior and regulate changes in GABA expression, although this occurs with clock specificity. For example, the targeted deletion of Per2 in astrocytes contribute to alleviate anxiety, increase the mRNA expression of the GABA transporter 2 (GAT-2), and reduce glutamate levels in the NAc. In contrast, the knockout of Per2 in neurons might decreases despair behavior without affecting anxiety, underscoring the specific role of astrocytes [35]. Additionally, research indicates that the deletion of Bmal1 does not influence anxiety-like behavior or alter GAT-2/Slc6a13 expression in the NAc, but it does result in the downregulation of Drd3 expression. These findings suggest that Per2 in astrocytes is the primary gene involved in regulating anxiety within the NAc [35]. Regarding GABA transport, GAT-1 and GAT-3 are expressed in the SCN, with GAT-3 predominantly localized to astrocytes. GATs facilitate the uptake of GABA into astrocytes to maintain normal circadian rhythms [201]. Study demonstrates that astrocytes encode the extracellular GABA rhythm by enhancing GAT-3-mediated GABA uptake during the day to clear synaptically released GABA, with the gene encoding GAT-3, Solute Carrier Family 6 Member 11 (Slc6a11), reaching peak expression in SCN astrocytes at circadian time 6 (CT6) [32]. During nighttime, astrocytes enhance the synthesis of GABA, thereby replenishing extracellular GABA levels and exerting inhibitory effects on neurons within the SCN. Disruption in GABA production or uptake results in the dysregulation of extracellular GABA rhythms, effectively abolishing its circadian cycling [201]. Research indicates that reactive astrocytes influence GABA homeostasis by downregulating GAT-3 expression, which leads to reduced GABA uptake, neuronal disinhibition, and the emergence of anxiety-like behaviors. Conversely, upregulation of GAT-3 has been shown to mitigate these anxiety-like behaviors [151]. Clinical study further suggests that individuals with an evening chronotype demonstrate increased sensitivity to negative emotions and a higher susceptibility to mood disorders [65]. Consequently, the regulation of GABA homeostasis by astrocytic GAT-3 may constitute a pathway contributing to the development of mood disorders during nighttime (Fig. 3).
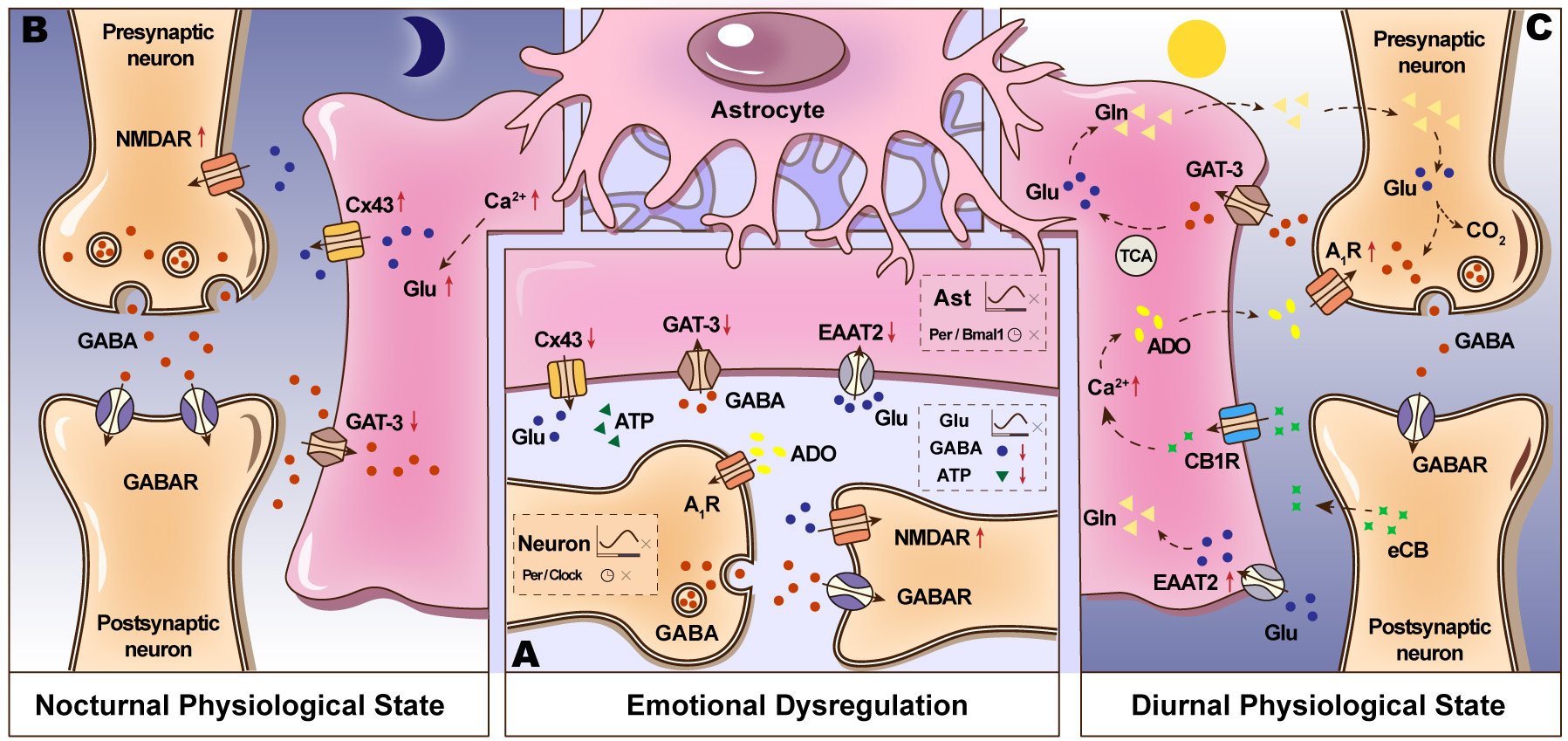 Fig. 3.
Fig. 3.
The communication between astrocytes and neurons in both normal
and mood disorder states in SCN. (A) In models of mood disorders, astrocytes
exhibit a diminished expression of the glutamate transporter EAAT2, Cx43, and
GAT-3. Conversely, neurons display an increased expression of NMDA receptor
subunits GluN1 and GluN2B. Mood disturbances lead to the desynchronization of
neuronal Per and Clock gene rhythms, while also disrupting the
rhythms of glutamate in astrocytes. The specific ablation of core clock genes,
such as Bmal1 or Per, in astrocytes results in circadian fragmentation
and exacerbates anxiety- and depressive-like behaviors. The disruptions in the
rhythms of GABAergic interneurons can also lead to mood disorder. (B) During
nocturnal periods (the active phase of an organism), astrocytes release glutamate
via Cx43 hemichannels, which subsequently activates NR2C subunit-containing
NMDA-type glutamate receptors (NR2C-NMDARs) in presynaptic neurons. This
activation facilitates the release of GABA, thereby inhibiting the electrical
activity of postsynaptic neurons. Astrocytic GAT-3 exhibits a diminished capacity
for GABA uptake. (C) During diurnal periods, neurons in the SCN are active. GABA
uptake by astrocytes via GAT-3 can lead to its conversion into glutamine through
the TCA cycle, followed by its transport back into GABAergic neurons for GABA
re-synthesis. Endocannabinoids released from postsynaptic neurons can interact
with CB1 receptors on astrocytes, activating [Ca2+]i signaling
pathways. This activation may result in the release of adenosine, which acts on
A1Rs on neurons, thereby reducing GABA release. Furthermore, astrocytic
EAAT2 proteins enhance the capacity for glutamate uptake. EAAT2, excitatory amino
acid transporter 2; Cx43, connexin 43; GAT-3, GABA transporter 3; TCA,
tricarboxylic acid; A1R, adenosine A1 receptor; GluN1, Glutamate
Ionotropic Receptor NMDA Type Subunit 1; GluN2B, Glutamate Ionotropic Receptor
NMDA Type Subunit 2B;
Additionally, ATP functions as a signaling molecule, facilitating astrocyte-neuron communication and purinergic signaling [204], and plays a role in regulating circadian variations [205]. Astrocytes within the SCN have the capacity to influence rhythmic oscillations via the release of ATP. In the SCN of rats, ATP concentrations exhibit rhythmic fluctuations, reaching their zenith during the dark phase and their nadir during the light phase [206]. Additionally, ATP demonstrates rhythmic oscillations in both cortical astrocytes and astrocyte cultures, which are synchronized with the rhythmic fluctuations of mitochondrial calcium ion concentration [Ca2+]i within astrocytes [207]. Among various neurotransmitters, extracellular ATP has been extensively investigated in relation to depression-like behaviors [208, 209]. For example, mice that are susceptible to chronic stress display reduced levels of extracellular ATP [152], and diminished ATP levels in the hippocampus and mPFC are correlated with depression-like behaviors [152, 210]. The astrocyte-specific expression of a dominant-negative soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE), which inhibits vesicular ATP release from astrocytes [211], help to increased immobility time in male mice during the FST [152]. In contrast, the deletion of glucocorticoid receptors (GR) in the mPFC decreases astrocytic calcium signaling and ATP release, thereby inducing depression-like behavior [39]. The administration of ATP has been shown to mitigate depression-like behaviors [152, 210, 212, 213]. This evidence implies a potential association between low extracellular ATP levels and depression-like behaviors, which may also impact the rhythmic oscillations of the SCN [214]. However, research indicates that disrupting the vesicular release mechanism does not affect the circadian rhythm of ATP release in cultured cortical astrocytes [206, 215], highlighting an apparent contradiction. Given that ATP release occurs through various pathways, including lysosomal release, connexin/pannexin hemichannels, volume-regulated anion channels, and calcium homeostasis modulators [216, 217, 218], and considering the rapid degradation of extracellular ATP by ectonucleotidases such as cluster of differentiation 39 (CD39), it may be necessary to analyze these alternative pathways [219].
Recent study has demonstrated that reduced expression of Cx43 in mPFC astrocytes results in decreased ATP release, which in turn induces depression- and anxiety-like behaviors, whereas overexpression of Cx43 reverses these phenotypes [40]. Furthermore, ATP release from SCN astrocytes can also be modulated through P2X7 and P2Y receptors [220]. Research has demonstrated that P2 purinergic receptors display diurnal variations in response to ATP stimulation, reflecting the rhythmicity of ATP. Notably, P2X receptors are more prevalent during the circadian dark phase [204, 221]. In parallel, mice vulnerable to chronic stress exhibit reduced extracellular ATP levels, and the activation of P2X2 receptors in GABAergic interneurons of the mPFC modulates depression-like behaviors in male mice [152, 222]. Additionally, extracellular ATP can mitigate depression-like phenotypes through P2Y1 receptors in both neurons and glial cells [223, 224]. Electroacupuncture has been shown to enhance the expression of P2Y1 receptors in the PFC, significantly ameliorating social avoidance and anxiety-like behaviors in socially isolated male mice [224]. Intriguingly, P2Y receptors exhibit higher expression during the circadian light phase, which is antiphase to ATP rhythmicity [221]. Thus, while P2 receptor activation enhances mood and increases ATP release, ATP rhythmic oscillation reaches its nadir during the light phase. It is established that P2X receptors are more abundant during the dark phase, aligning with the peak levels of their primary agonist, ATP. In contrast, P2Y receptors demonstrate increased abundance during the light phase, positioning them in antiphase relative to the ATP peak [214]. Consequently, further research incorporating additional intervention time points is warranted to elucidate the rhythmic mechanisms through which P2 receptors modulate ATP release in models of mood disorders.
Astrocytes undergo dynamic changes throughout the sleep-wake cycle, releasing sleep-inducing molecules that regulate brain activity and sleep architecture [225]. Adenosine is a pivotal sleep-regulating factor. Recent study indicates that, in addition to the conventional sleep homeostasis pathways in the basal forebrain (BF) and cortex, activation of astrocytes in the parafacial zone (PZ) specifically leads to increased extracellular adenosine via the ATP hydrolysis pathway, which subsequently activates A1Rs to promote wakefulness [226]. Beyond its role in sleep regulation, adenosine and its metabolic enzymes, such as cytosolic 5′-nucleotidase (cN-II) and adenosine kinase (ADK), exhibit circadian oscillations [227]. Research indicates that adenosine mediates sustained synaptic inhibition by attenuating neuronal activity through A1Rs [211] and influences the transmission of circadian photic signals [228]. The SCN neurons receive input from the RHT, thereby synchronizing cellular activity and the TTFL with external environmental rhythms [78, 79]. Activation of adenosine A1Rs located on RHT axon terminals has been demonstrated to inhibit the release of glutamate and mitigate light-induced phase shifts within the SCN [229]. Additionally, the activity of adenosine and Adenosine A2A receptor (A2ARs) is regulated by the retinal clock, with peak levels occurring during nighttime. This clock-controlled activation of endogenous retinal cone A2ARs enhances nocturnal cone gap junction coupling and increases rod input to cones and horizontal cells (cHCs) [230]. Exposure to light during nighttime disrupts circadian rhythms and is associated with negative health outcomes [231]. The study using animal models suggests the existence of an SCN-independent pathway, mediated by ipRGCs that provide excitatory synaptic input to neurons in the posterior habenula (PHb) of the dorsal thalamus, which plays a role in mood regulation [82]. Conversely, although astrocytes do not directly receive input from the RHT, the reciprocal interaction between astrocytes and neurons can induce phase advances through neuron-astrocyte activation of cannabinoid receptors (CBRs), which subsequently feedback to activate neuronal adenosine receptors [153]. Thus, A1Rs may play a crucial role in mood regulation through the transmission of photic signals within the central pacemaker.
Research indicates that the expression of adenosine A1Rs and A2ARs in the SCN follows a rhythmic pattern, with elevated levels observed at circadian time 5 (CT5) and diminished levels at CT17. Activation of adenosine receptors by agonists stimulates the classical cAMP-CREB signaling pathway, resulting in increased expression of the clock genes Per1 and Per2 and an extension of the rhythm period. This pathway is similarly activated in SCN neurons in response to light stimuli [154, 232]. In vitro study has further demonstrated that activation of A2ARs significantly enhances the expression of the clock genes Clock and Bmal1 [155]. Additionally, targeted knockdown of Bmal1 within the SCN contribute to diminished circadian rhythmicity, desynchronized single-cell rhythms, and heightened behavioral despair, anxiety-like behaviors, and helplessness [135, 136]. Consequently, adenosine receptors in the SCN may modulate mood disorders through their regulatory effects on circadian clock mechanisms. Beyond the SCN, distinct subregions of the amygdala exhibit daily variations in Clock gene expression [233]. For example, Per2 mRNA levels in the BLA exhibit significant circadian fluctuations over a 24-hour period and are associated with affective changes [234]. The targeted activation of astrocytes within the centromedial amygdala (CeM) leads to a reduction in the expression of learned fear responses. Synaptic modulation of CeL-evoked IPSCs and BLA-evoked EPSCs is inhibited by antagonists of A2ARs and A1Rs, respectively [235]. Additionally, research conducted in the lateral septum (LS) demonstrates that the application of an adenosine A1Rs antagonist can reverse the reduction in sEPSC frequency in LS neurons, whereas activation of A1Rs further diminishes sEPSC frequency. Conversely, blocking adenosine A2ARs reverses the increase in sIPSC frequency in LS inhibitory (LSi) neurons, while activation of these receptors further elevates sIPSC frequency. This modulation of LS neural circuitry plays a role in regulating social avoidance and anxiety-like behaviors in mice [236]. Collectively, these studies suggest that ATP/adenosine functions as a critical gliotransmitter in the regulation of emotions. By modulating presynaptic mechanisms, astrocytes can selectively regulate synaptic activity and exert inhibitory effects on neurons, thereby influencing emotional behavior. The mechanisms underlying the action of A1R may vary between the CeM and the LS.
Furthermore, the expression of the hippocampal clock demonstrates circadian rhythmicity, and the factors that regulate this clock in the hippocampus have a significant impact on behaviors related to depression. For instance, the phosphorylation of the Clock protein in the hippocampus is involved in the regulation of the circadian clock [237]. Elevated levels of extracellular ATP and adenosine in the ventral hippocampal CA1 (vCA1) subregion predominantly originate from activated astrocytes. Inhibition of astrocyte function or blockade of the gap junction protein Cx43, which is widely distributed on astrocytes, substantially reduces extracellular ATP and adenosine levels in vCA1 and effectively mitigates anxiety and depression-like behaviors. Similarly, local administration of an A2AR antagonist in vCA1 produces comparable effects [238]. Recent study proofed that VTA astrocytes activated by stressors reduced the inhibition of DA neurons via presynaptic A1Rs on GABAergic interneurons, which helped alleviate anxiety- and depression-like behaviors [41].
In conclusion, there exists a complex interplay between astrocytic ATP/adenosine rhythms and mood states within the SCN and other emotion-related brain regions. Despite this, circadian research in this specific domain remains sparse. Further investigation into these mechanisms could yield valuable insights into the connection between circadian rhythms and mood regulation.
A bidirectional regulatory relationship is evident between disruptions in
circadian rhythms and psychoaffective disorders, with circadian phenotypes
exhibiting variability across different mood disorder types, wherein astrocytes
play a pivotal role. Bipolar disorder is a prevalent and severe mood disorder,
characterized by recurrent major depressive episodes interspersed with hypomanic
or manic episodes [239]. BD is also linked to significant disruptions in
metabolic processes [240] and circadian rhythms [241]. Clinical investigations
have found that individuals with BD experience elevated daytime melatonin levels
and a delayed nocturnal melatonin peak during manic episodes [242]. Furthermore,
melatonin secretion is delayed in bipolar depression compared to unipolar
depression [242]. Genetic research on circadian clock genes indicates that
PER3 and Rev-erb
In patients with MDD, abnormalities in mood and circadian parameters are
evident. Notably, depressive symptoms tend to be more pronounced in the morning
[265, 266], and there is a marked advancement in the rhythm of melatonin secretion
[267]. Additionally, there is a disruption in the expression of clock genes such
as BMAL1, PER1-3, and Rev-erb
Emotional disorders are frequently associated with comorbid disruptions in
circadian rhythms, and the restoration of these rhythms can significantly
influence the progression of such disorders. Astrocytes play an independent role
in regulating the circadian rhythms of the SCN [25, 27, 28, 29, 30, 31]. For example,
extracellular levels of GABA and glutamate in the SCN exhibit pronounced
rhythmicity, with both reaching their peak at night [32]. Similarly, ATP
concentrations demonstrate rhythmic oscillations, peaking during the dark phase
and reaching their lowest levels during the light phase [206]. In contrast,
the rhythmic oscillations of receptor proteins exhibit an antiphase pattern:
Slc6a11 (encoding GAT3) [32], P2Y receptors [221], and A1/A2A receptors [154, 232]
peak during the daytime. Comparable rhythmic alterations are observed in brain
regions associated with emotion; for instance, the capacity for glutamate uptake
by hippocampal astrocytes decreases during the dark phase [183, 184], and
disruptions in circadian function in NAc astrocytes impair glutamate homeostasis,
subsequently affecting emotional behavior [34]. Furthermore, diminished nocturnal
melatonin levels, phase delays [242], and dysregulated cortisol release [275, 276]
may contribute to emotional dysregulation. Empirical evidence indicates that
activation of the NOD-like receptor family, pyrin domain containing protein 3
(NLRP3) inflammasome is associated with stress and lipopolysaccharide
(LPS)-induced depression in animal models [277]. Exogenous administration of
melatonin has been demonstrated to mitigate LPS-induced depression-like behaviors
by downregulating NLRP3 activation in the brain [278]. The inhibitory effect of
melatonin on the NLRP3 inflammasome is mediated through various proteins and
pathways, including the NF-
Currently, rhythmic genes and the aforementioned targets are under investigation for their potential in therapeutic development for mood disorders. Notable examples include light therapy, which targets the RHT photic input [96], the NMDA receptor antagonist ketamine [289, 290], and AMPA receptor modulators [291]. Research indicates that exposure to one hour of high-intensity light during the active phase leads to the upregulation of Per1, Per2, and Cry1 expression in the SCN, thereby modulating pathophysiological processes [96]. Furthermore, evening administration of melatonin facilitates phase advances in the circadian clock of individuals with delayed sleep-wake phase disorder [99]. The NMDA receptor antagonist ketamine can modulate the upstream CREB signaling pathway of clock genes to improve depressive symptoms [289, 292]. Additionally, ketamine also manifests distinct circadian motor activity patterns and temporal responses, such as the association between a low-amplitude 24-hour activity rhythm and rapid depressive relapse [293]. Notably, the clinical investigation of the timing effects in subanesthetic ketamine has yet to be addressed, but such as are being planned [290]. In individuals with bipolar disorder, skin fibroblasts transfected with Per2 luciferase reporter constructs (Per2::luc) exhibited extended circadian periods compared to healthy controls, and lithium, a common therapeutic agent for bipolar disorder, was found to resynchronize and enhance the amplitude of the attenuated molecular rhythms in these cells [294]. The investigation of astrocyte-associated neurotransmitter rhythms provides a neurobiological basis for chronotherapeutic interventions in mood disorders. Consequently, examining the “astrocyte-circadian rhythm-emotional circuit” axis through the perspective of astrocytic rhythmic proteins holds significant promise for the advancement of astrocyte-targeted chronotherapeutics with precise timing. This research provides compelling evidence for expanding time-of-day therapies and exploring chronopharmacological treatments for mood disorders.
A1R, adenosine A1 receptor; A2AR, Adenosine A2A receptor; ACC, anterior cingulate cortex; ADK, adenosine kinase; Aldh1l1, Aldehyde dehydrogenase 1 family member L1; AQP4, aquaporin-4; ATP, Adenosine triphosphate; AVP, arginine vasopressin; BD, bipolar disorder; BDNF, brain-derived neurotrophic factor; BF, basal forebrain; Bhlhe40, Basic helix-loop-helix family member e40; BLA, basolateral amygdala; BMAL1, brain and muscle ARNT-like 1; CA1, Cornu Ammonis 1; CBR, cannabinoid receptors; CD39, cluster of differentiation 39; CeM, centromedial amygdala; cHCs, cones and horizontal cells; ChR2, Channelrhodopsin-2; CLOCK, circadian locomotor output cycles kaput; cN-II, cytosolic 5′-nucleotidase; CREB, cAMP response element-binding protein; CRY1, Cryptochrome 1; Cx30, Connexin 30; Cx43, connexin 43; DA, dopamine; dACC, dorsal anterior cingulate cortex; dlPFC, dorsolateral prefrontal cortex; Drd3, dopamine receptor D3; DRN, dorsal raphe nucleus; EAATs, excitatory amino acid transporters; EPM, elevated plus maze; FST, forced swim test; GABA,
Conceptualization, LK, PL and YH; literature search and organization, HL and YZ; writing—original draft preparation, PL and SS; writing—review and editing, HL; visualization, SS; supervision, LK and YH; funding acquisition, YH. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We gratefully acknowledge the support by the Key Laboratory of Sports Medicine of Sichuan Province.
This work was supported by the National Natural Science Foundation of China (81704190, 82074576), Sports Medicine Key Laboratory of Sichuan Province & Key Laboratory of Sports Medicine, General Administration of Sport of China, Excellence Research Program (2025-A032), Expert Centre Foundation of Sichuan Province (A2020-SCZJJCB02), Sichuan Provincial Joint Fund Project for Science and Technology Education (2025NSFSC2165), Sichuan Provincial Administration of Traditional Chinese Medicine Special Scientific Research Project (25MSZX524).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.