






 , Jakub Vejskal 1, Ingrid Menkyova 2,3,4, Marek Peterka 1,5, Marta Vachova 2,6, Dominika Stastna 2,4
, Jakub Vejskal 1, Ingrid Menkyova 2,3,4, Marek Peterka 1,5, Marta Vachova 2,6, Dominika Stastna 2,41 Department of Neurology, Faculty of Medicine in Pilsen and University Hospital Pilsen, Charles University, 323 00 Pilsen, Czech Republic
2 Department of Neurology and Centre of Clinical Neuroscience, First Faculty of Medicine, Charles University in Prague and General University Hospital, 128 08 Prague, Czech Republic
3 Department of Neurology, Faculty of Medicine, Slovak Medical University, 831 01 Bratislava, Slovak Republic
4 ReMuS Registry, ReMuS Endowment Fund, 128 00 Prague, Czech Republic
5 Department of Neurology, Faculty of Medicine and University Hospital Hradec Kralove, Charles University in Prague, 500 05 Hradec Kralove, Czech Republic
6 Department of Neurology, KZ a.s., Hospital Teplice, 415 01 Teplice, Czech Republic
Abstract
Multiple sclerosis is a chronic immune-mediated disease of the central nervous system, marked by demyelination, axonal damage, and progressive neurological decline. T lymphocytes—particularly CD4+, T helper (Th)1 and Th17 cells, as well as cytotoxic CD8+ cells—play a pivotal role in initiating and sustaining central nervous system inflammation. Acute inflammation is driven by peripheral immune activation, while progressive disease reflects compartmentalized, smouldering inflammation within the central nervous system, dominated by CD8+ T cells and microglia. A relative deficiency or dysfunction of regulatory T cells contributes to immune tolerance loss and ongoing neurodegeneration. Although T lymphocytes play a central role, the pathogenesis of multiple sclerosis involves a broader cellular network, including antigen-presenting cells, B lymphocytes, microglia, and astrocytes. While recent therapeutic strategies have increasingly focused on B lymphocytes, most disease-modifying therapies—and many emerging ones—exert at least partial effects by modulating T cell–mediated mechanisms. These insights underpin current T cell-targeted therapies and highlight unmet needs in multiple sclerosis.
Keywords
- multiple sclerosis
- T cells
- Th17
- CD8 T cells
- B cells
- cytokines
- molecular mechanisms
- pathophysiology
- smouldering inflammation
- disease-modifying therapy
Multiple sclerosis (MS) is a chronic demyelinating disease of the central nervous system (CNS) in whose pathogenesis, in addition to cell-mediated inflammatory activity, neurodegeneration also plays a role from the beginning. This disease most commonly affects young adults of productive age, with the average age at diagnosis around 30 years [1]. However, recent studies show a shift toward older age at diagnosis, with approximately 5% of patients being diagnosed in their sixth decade of life [2, 3]. The prevalence in Europe and North America is estimated at around 111–300 cases per 100,000 inhabitants [4, 5]. The aetiology of the disease remains unclear, but available evidence suggests a combination of genetic, epigenetic, and environmental factors (including Epstein-Barr virus (EBV) infection, vitamin D deficiency, smoking, and lifestyle factors) [6, 7, 8, 9, 10]. The influence of the gut microbiome and hormonal factors has also been described, with MS incidence being up to three times higher in women, while male sex is considered a negative prognostic factor [1, 11].
Although the classification of MS based on clinical phenotypes is increasingly viewed as outdated from a pathogenetic perspective, it is still used in clinical practice, primarily for practical reasons. The most common disease course is relapsing-remitting MS (RRMS), characterized by alternating periods of flare-ups and relative stability. During a relapse, (multi)focal neurological dysfunction develops, which may even resolve completely. As the disease progresses, relapse frequency decreases, while neurological impairment, dependent on and independent of relapses (progression independent of relapse activity, PIRA), increases (Fig. 1). This is associated with advanced demyelination, accumulation of axonal and neuronal damage, and reduced neuronal connectivity [12]. Over time, the disease often transitions into a secondary progressive form. “Smouldering” inflammation behind an already closed blood-brain barrier (BBB) typically localizes at the edges of existing lesions and is mainly mediated by activated microglia. Magnetic resonance imaging (MRI) reveals slowly expanding lesions (SELs) and progressive brain atrophy. While the primary target of inflammation is the myelin sheath, the ultimate cause of permanent damage and PIRA is axonal loss. This is attributed to inflammation, accumulation of lesions with subsequent retrograde and anterograde degeneration, mitochondrial dysfunction and oxidative stress, iron accumulation in myelin and oligodendrocytes, ectopic meningeal lymphoid follicles, age-related neurodegeneration, and loss of functional reserves [13]. The degree of disability is also influenced by the development of neural network dysfunction and inadequate repair mechanisms [14]. A rarer form of MS (10–15% of cases) is primary progressive MS (PPMS), which is characterized by the gradual progression of symptoms from the onset, without relapses [4].
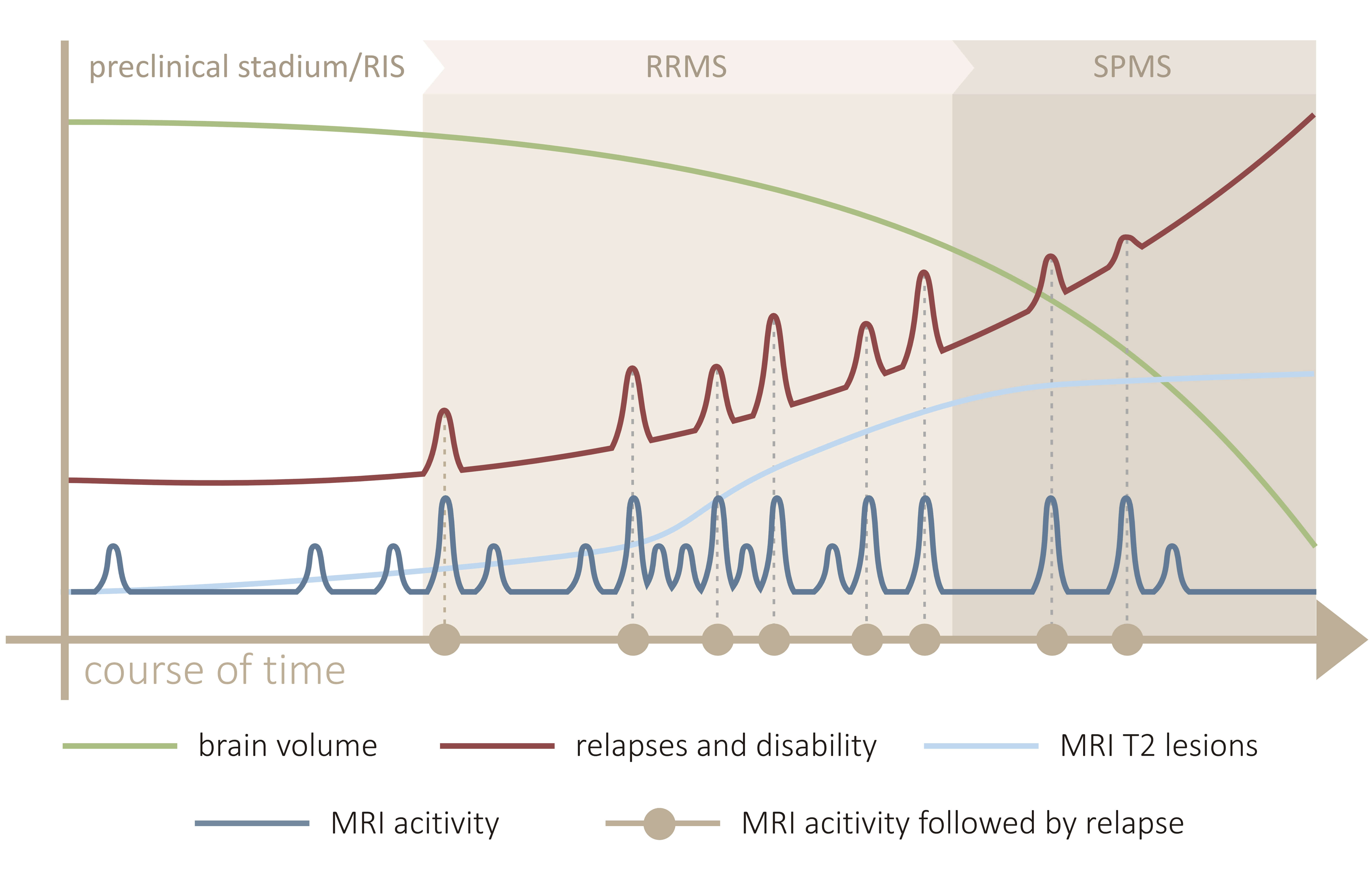 Fig. 1.
Fig. 1.
Course of relapsing-remitting multiple sclerosis. Although we divide MS into three stages - RIS, RRMS and SPMS, the biological course of the disease is continual. Inflammatory activity in the CNS leads to accumulation of T2-weighted lesions and later to axonal loss manifested by progressive brain atrophy. Only some inflammatory flare-ups are accompanied by a clinical relapse, while others are subclinical, but gradually contribute to slowly progressing disability. RIS - A subclinical stage of MS until the first relapse, but already with evident biological and radiological activity. RRMS - A clinical stage of MS associated with relapses, accumulation of T2-weighted lesions, brain atrophy and clinical disability. SPMS - A clinical stage of MS with minimum relapses, but dominant progressive axonal loss, brain atrophy and clinical disability independent of relapses. The line between RR and SP grade is not sharp. MS, multiple sclerosis; RIS, radiologically isolated syndrome; RRMS, relapsing-remitting multiple sclerosis; SPMS, secondary progressive multiple sclerosis; MRI, magnetic resonance imaging; CNS, central nervous system.
According to the currently most widely accepted model (“outside-in”), MS is
considered a disease that begins with peripheral activation of an autoimmune
process, which subsequently extends into the CNS. The interaction between
infiltrating immune cells (T and B lymphocytes) and resident CNS cells
(particularly microglia and astrocytes) plays a key role [15]. Primary activation
of T lymphocytes occurs in the periphery via antigen-presenting cells (APCs).
Activated CD4+ T lymphocytes (especially T helper (Th)1 and Th17 subsets) express
adhesion molecules and chemokine receptors (e.g., very late antigen-4 [VLA-4],
lymphocyte function-associated antigen 1 [LFA-1], CC chemokine receptor 6 [CCR6]) that allow them to interact with BBB endothelial cells [16, 17]. This
interaction leads to endothelial activation, resulting in increased BBB
permeability. Cytokines modulate blood–brain barrier properties in a
context-dependent manner. Interleukin (IL)-17 and IL-22 can promote leukocyte
trafficking by upregulating endothelial adhesion and altering the function of
tight junctions. In contrast, cytokine interferon-gamma (IFN-
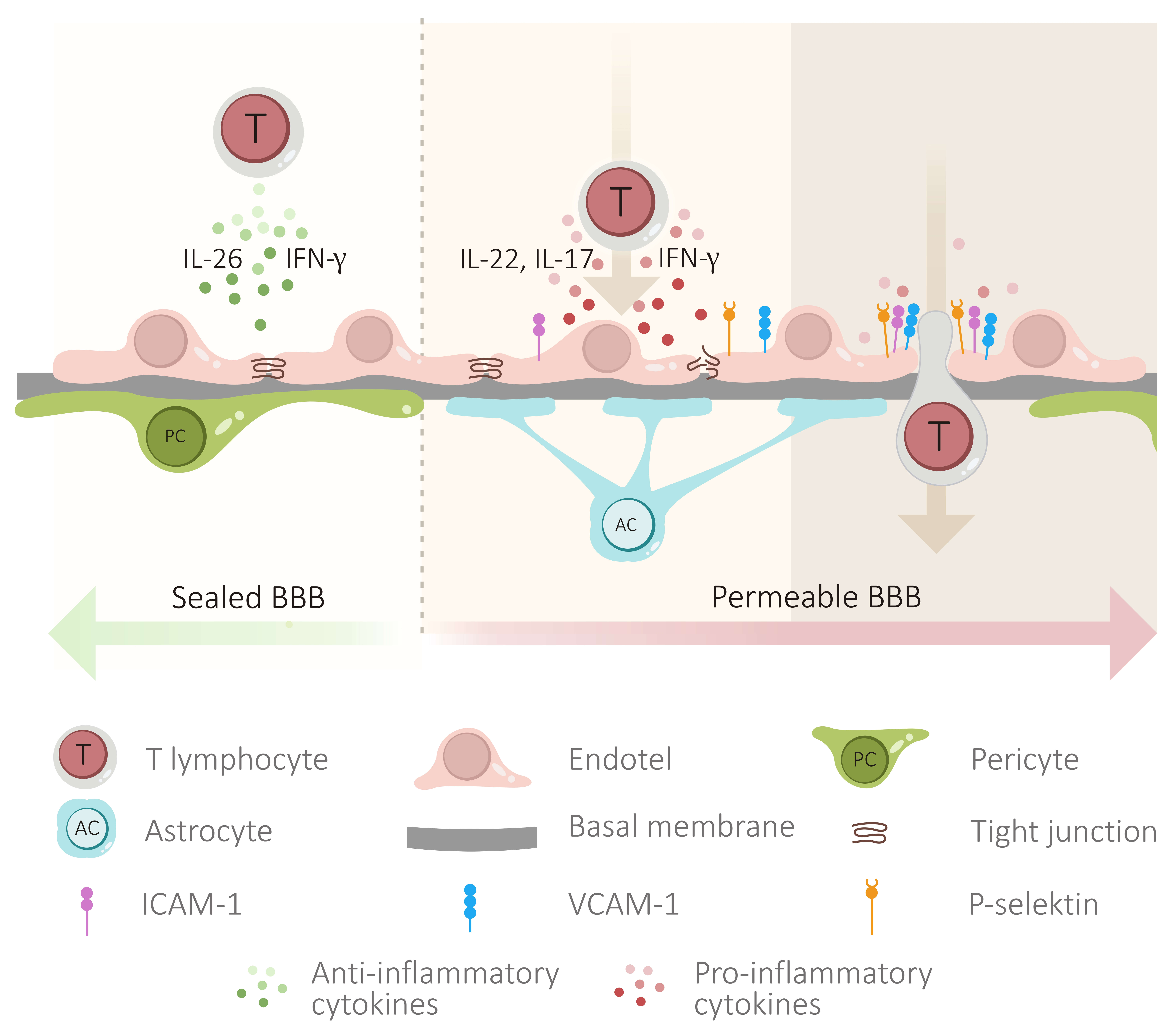 Fig. 2.
Fig. 2.
Transfer of inflammation across the BBB. An autoimmune
inflammation activated in the periphery leads to increased BBB permeability.
Activated CD4+ T lymphocytes produce pro-inflammatory cytokines (IL-22, IL-17
and IFN-
T lymphocytes are pivotal drivers of relapse activity—particularly in RRMS. In contrast, disability accrual and progression reflect an integrated network involving B cells, innate immunity, and CNS-resident glia within compartmentalized inflammation [22]. The predominant lymphocyte type at all stages of MS is the cytotoxic CD8+ T cell, which closely correlates with axonal damage [23]. Regulatory T lymphocytes (Tregs), which protect the brain from further immune-mediated injury, are less abundant [24]. This article aims to provide an up-to-date overview of the influence of T lymphocytes on MS pathophysiology and to highlight the mechanisms of treatment targeting T-cell modulation.
Genome-wide association studies (GWAS) have brought about a fundamental shift in our understanding of genetic and epigenetic factors in the etiopathogenesis of MS. These studies have identified more than 200 single-nucleotide polymorphisms associated with genetic predisposition to MS, most of which are located on chromosome 6 in a region containing genes that regulate the immune response. However, the odds ratio (OR) associated with most of these variants is low (1.1–1.2) [25]. The largest group of genes identified in GWAS is involved in either the signalling, development, and differentiation of T lymphocytes or in the presentation of antigens to these cells [26]. The human leukocyte antigen DR15 (HLA-DR15) haplotype plays a crucial role in promoting the development of an autoreactive CD4+ T cell repertoire in conjunction with environmental factors [27]. Self-reactivity, defined as the “autoproliferation” of peripheral Th1 cells, is increased in individuals carrying the HLA-DRB1*15:01 allele [28]. This autoproliferation is mediated by memory B cells in an HLA-DR-dependent manner, with its intensity decreasing both during in vitro depletion of B cells and during in vivo anti-CD20 therapy [29].
In addition to genetic predisposition, epigenetic mechanisms that influence the expression of genes associated with the inflammatory response also contribute to the pathogenesis of MS. Active and inactive MS lesions exhibit a different spectrum of expressed small non-coding RNAs (microRNAs, miR) miR-155, miR-34A, and miR-326 predominate in active lesions, influencing the expression of pathological cell genes, including by regulating the transcription of integrin-associated protein CD47, which is involved in macrophage activity [30]. Furthermore, miR-155 expression contributes to the differentiation of Th1 and Th17 lymphocytes and worsens the course of experimental autoimmune encephalomyelitis (EAE) and MS [31]. The important proinflammatory cytokine IL-17, produced by Th17 lymphocytes, is elevated in the cerebrospinal fluid of patients with RRMS, and its blockade reduces inflammatory activity in the experimental autoimmune encephalomyelitis model [32]. In addition, hypomethylation of the promoter region of the IL-17A gene has been demonstrated in patients with MS, which is associated with increased expression of this cytokine.
In addition to genetic predisposition and epigenetic factors, external environmental factors also play a significant role in the aetiology and pathogenesis of MS. The importance of environmental factors is underscored by numerous migration studies. Adult migrants from countries with a lower risk of developing MS do not experience an increase in risk after moving to high-risk regions, but an increase is already apparent in migrant children born in high-risk countries [33]. Likely, we are already exposed to risk factors for the development of MS in utero. Early-life influences are suggested by the modest and geographically variable month-of-birth effect, as well as familial concordance patterns. Concordance is highest in monozygotic twins, lower in dizygotic twins, and slightly lower still in non-twin siblings, indicating contributions from both genetics and shared early environmental factors [34].
Among modifiable environmental risk factors, smoking is one of the most significant. Some studies report that it increases the risk of developing MS by up to 50% [35]. A major Swedish study involving more than 9000 patients with MS and an equal number of age-matched controls determined the population attributable fraction to be 13%, meaning that eliminating smoking would potentially prevent the development of MS in this proportion of patients [9]. Smoking is thus one of the few modifiable risk factors. Its influence is well documented not only epidemiologically but also at the level of molecular mechanisms. Cigarette smoke induces oxidative stress and activates various intracellular signalling pathways, including Toll-like receptors (TLRs), via pathogen-associated molecular patterns (PAMPs), which can lead to epigenetic changes in the regulation of immune genes. In addition, changes in T-cell differentiation occur in the lung microenvironment; in an experimental model of EAE, it has been shown that autoreactive T cells acquire the ability to penetrate the CNS after transient exposure in lung tissue. Smoke-induced signals can transform potentially autoreactive T cells into a pathogenic phenotype. A key molecular mechanism involves the aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor expressed in T cells. Polycyclic aromatic hydrocarbons present in cigarette smoke activate AHR, which promotes Th17 cell differentiation and IL-17A production while modulating the Th17/Treg balance [36, 37, 38]. This AHR-mediated pathway provides a mechanistic link between smoking and the Th17-driven neuroinflammatory environment characteristic of MS.
Another important environmental factor is vitamin D deficiency. Its effect on the immune system is mediated, among other things, by epigenetic mechanisms. Vitamin D plays a key anti-inflammatory role in modulating the immune response, and its deficiency has been repeatedly associated with an increased risk of developing MS [8]. The active form of vitamin D acts through the vitamin D receptor, which binds to specific sequences in the genome and regulates the transcription of hundreds of target genes, including those with immunological functions. This mechanism interferes with both innate immunity (e.g., monocytes, dendritic cells) and adaptive immunity, particularly by modulating T and B lymphocytes. Vitamin D inhibits the polarization of pathogenic Th1 and Th17 cells, whose activity plays a key role in the pathogenesis of MS. At the same time, it promotes the formation of regulatory T cells, preferentially stimulating the Tr1 (Type 1 regulatory T cells) subpopulation, which produces the anti-inflammatory cytokine IL-10 [39, 40]. The result is a shift in the immune response towards a tolerogenic profile and a reduction in autoreactivity. The epigenetic effects of vitamin D include histone acetylation and the modulation of microRNAs that regulate gene expression, which contributes to maintaining the balance between effector and regulatory immune responses [41, 42, 43].
Among environmental risks, EBV exposure stands out as one
of the most significant. A history of infectious mononucleosis approximately
doubles the risk of MS [44, 45]. A large cohort study using data from more than 10
million US military recruits found up to a 32-fold higher risk of developing MS
in individuals with serologically proven contact with EBV, opening up potential
opportunities for MS prevention [46]. In patients with MS, an increased and
expanded repertoire of antibody and T-cell immune responses to EBV-encoded
antigens, particularly against the dominant CD4+ T-cell EBV nuclear antigen
1 (EBNA1), has been observed [47]. Two main hypotheses have been proposed: (1)
molecular mimicry, whereby EBV antigens cross-react with CNS components, and (2)
repeated EBV reactivation during the alternation between latent and lytic phases
of infection, leading to chronic stimulation of the immune system. Consistent
with the first, cross-reactivity between EBV and CNS antigens has been documented
in CD4+ T cells, while CD8+ T cells display increased frequency but reduced
functional capacity [48]. Reactivity to antigens of both phases of
infection—latent and lytic—further suggests that cyclic reactivation of EBV
promotes diversity and long-term survival of memory T cells in patients with MS
[7]. Nevertheless, the near-universal prevalence of EBV (up to 99%) suggests
that contact with the virus alone is not sufficient for the development of MS.
The development of the disease probably requires a combination of other genetic
predispositions and environmental factors [25, 33]. Recent exosome profiling has
revealed that EBV-encoded miRNAs (BART9-3p and BART15) are significantly elevated
in the cerebrospinal fluid of RRMS patients, accompanied by upregulation of
proinflammatory cytokines including IFN-
Obesity, particularly during adolescence, is another important modifiable risk
factor. Elevated body mass index has been repeatedly linked to increased MS risk,
with the pathophysiological basis of this relationship lying in impaired
immunological and metabolic regulation. Adipose tissue in obesity exhibits an
overproduction of proinflammatory adipokines (e.g., leptin) and reduced levels of
anti-inflammatory adipokines (e.g., adiponectin), leading to chronic subclinical
inflammation that may facilitate the development of the autoimmune process. Obese
individuals also show elevated circulating proinflammatory cytokines, including
IL-6 and TNF-
Various organs, particularly the intestine and possibly the lungs, perceive environmental signals and shape immune responses via lymphoid tissue. Physiological colonization of the gut is essential for maintaining the balance of mucosal and systemic immunity. Disruption of the normal intestinal microbiome (dysbiosis) predisposes individuals to immunopathological conditions and has been repeatedly linked to the onset and progression of MS [11, 52, 53]. The microbiome produces a number of biologically active substances, including short-chain fatty acids (SCFAs) such as propionate and butyrate. These metabolites induce histone deacetylation, thereby epigenetically regulating gene expression and also promoting the maturation of protective Tregs [54]. Their action leads to a reduction in the activity of Th1 and Th17 effector cells. Functional microbiome analysis has shown that oral administration of propionate leads to increased expression of Treg-inducing genes in the intestine and normalization of mitochondrial function and morphology of these cells in patients with MS [55]. SCFAs also promote the production of the anti-inflammatory cytokine IL-10, thereby further contributing to immunological tolerance [56].
MS patients consistently exhibit reduced gut microbiome diversity compared to healthy controls, with notable depletion of SCFA-producing species, especially certain Clostridia, which support not only remyelination but also the development of Tregs. The result is an imbalance in the T-cell response–a reduced occurrence of Tregs in the lamina propria mucosae, Peyer’s patches, and mesenteric lymph nodes, and at the same time, an increased prevalence of proinflammatory Th1 and Th17 cells [57]. This immunological imbalance can lead to a lower threshold for the development or exacerbation of MS. Other mechanisms are also likely to be involved in the link between the gut microbiome and MS. These include molecular mimicry (so far mainly documented in mouse models), increased binding of intestinal immunoglobulin A (IgA) to specific types of bacteria, and the effects of oxidative stress [58].
Increasing attention is also being paid to the link between the microbiome and the progression of MS. In 2020, a study using metagenomic sequencing of the gut microbiome was published, revealing differences between patients with RRMS and secondary progressive MS (SPMS). These observations support the hypothesis that changes in the microbiome may contribute to the transition to the progressive phase of the disease, among other things by influencing T-lymphocyte balance [59, 60]. Changes in the gut microbiome thus significantly modulate the balance between regulatory and effector T lymphocytes and influence the immunological microenvironment in the CNS. However, microbiome findings show considerable variability between studies, with conflicting results for the same bacterial species across different cohorts. This heterogeneity, likely reflecting geographic variation, dietary differences, and disease modifying therapies (DMT) effects, complicates the translation of these findings into therapeutic interventions [11, 52, 53, 59, 60]. Despite these challenges, the microbiome remains a promising modulator of the autoimmune response and a potential therapeutic target in MS.
When nervous tissue is damaged, for example, by a viral agent or toxic substance, antigenic peptides are presented by APCs via major histocompatibility complex (MHC) molecules to T cells. Among APCs, dendritic cells—a functionally heterogeneous population—are the most effective, as they are capable of processing complex antigen structures. These cells possess pattern-recognition receptors (PRRs), enabling them to identify external threats in the form of PAMPs and internal danger signals known as damage-associated molecular patterns (DAMPs). Recognition of DAMPs or PAMPs via PRRs leads to dendritic cell activation and their targeted migration into secondary lymphoid tissues. There, dendritic cells functionally differentiate based on the specific PRR stimulus and begin to present processed antigenic peptides in the context of MHC molecules to T cells. This interaction triggers the activation of specific T cells, followed by their clonal expansion and functional polarization. Polarization is mediated by co-stimulatory signals from dendritic cells and by the cytokine milieu, largely shaped by these same dendritic cells. The initiation of damaging inflammation takes place in secondary lymphoid organs [61].
T cell development primarily occurs in the thymus, where two major populations emerge: precursors of cytotoxic T cells (CD8+) and helper T cells (CD4+, Th). Upon encountering an antigen, these cells differentiate into effector T cells [62]. For T cells to participate in damaging inflammation, they must exit the secondary lymphoid organs and migrate to target sites. Chemokines regulate this migration but also depend on the sphingosine-1-phosphate (S1P) gradient formed through the metabolism of ceramides. Lymphocytes sense this gradient via S1P receptors, primarily S1P1, which signal through intracellular GTPases. The binding of S1P to the S1P1 receptor induces controlled egress of lymphocytes from secondary lymphoid tissues and supports their subsequent chemokine-guided migration [63]. Specifically, this involves interaction with the chemokine CCL21 bound to intercellular matrix components, mediated through CCR7 receptors on the surface of central memory T cells [64].
Under physiological conditions, the entry and exit of immune cells and macromolecules from the brain are tightly regulated by the BBB, which protects the CNS from circulating immune cells and potentially harmful molecules [65]. In MS, however, the BBB is compromised, leading to increased permeability. T cells adhere to the endothelium during CNS infiltration via adhesion molecules such as LFA-1/Intercellular Adhesion Molecule 1/CD54 (ICAM-1) and VLA-4/Vascular cell adhesion molecule 1/CD106 (VCAM-1) [17]. B-cell trafficking involves classical adhesion pathways (e.g., VLA-4/VCAM-1 and LFA-1/ICAM-1), with contributions varying by activation state and tissue context [66]. Within the CNS, T cells are reactivated through renewed antigen presentation, which initiates a cascade of inflammation and recruits additional immune cells. Autoreactive T cells recognize immunodominant epitopes of neural membrane components, particularly myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), myelin-associated glycoprotein (MAG), proteolipid protein (PLP), and cyclic nucleotide phosphodiesterase. This cascade results in focal oedema, demyelination, oligodendrocyte death, and damage to axons and neurons [4, 67]. Regarding cellular immunity, macrophages, microglial cells, cytotoxic CD8+ T cells, and natural killer (NK) cells are activated.
The role of perivascular B cells associated with active white matter lesions is likely to be to reactivate proinflammatory CD4+ and CD8+ T cells. In later stages of the disease, B cells are, at least in part, gradually transformed into plasma cells [68]. In addition to producing antibodies, B cells contribute to inflammation through cytokine secretion and their role as APCs activating T cells [69, 70]. Humoral immunity further participates via antibody-mediated recognition of myelin surface structures, leading to complement activation. Conversely, regulatory B cells (Bregs) have immunosuppressive properties—they modulate macrophage and dendritic cell function, inhibit CD4+ T-cell proliferation, and enhance the expansion of Tregs [71, 72].
The pathological landscape of MS is highly complex. Mounting evidence suggests the coexistence of two forms of inflammation—one driving multifocal active lesions and another associated with smouldering, slowly evolving lesions (Fig. 3) [73]. Data from MRI and histopathological studies show that progressive neuroaxonal loss, which drives long-term disability, begins early in the disease, indicating a continuum between the relapsing and progressive phases of MS [13].
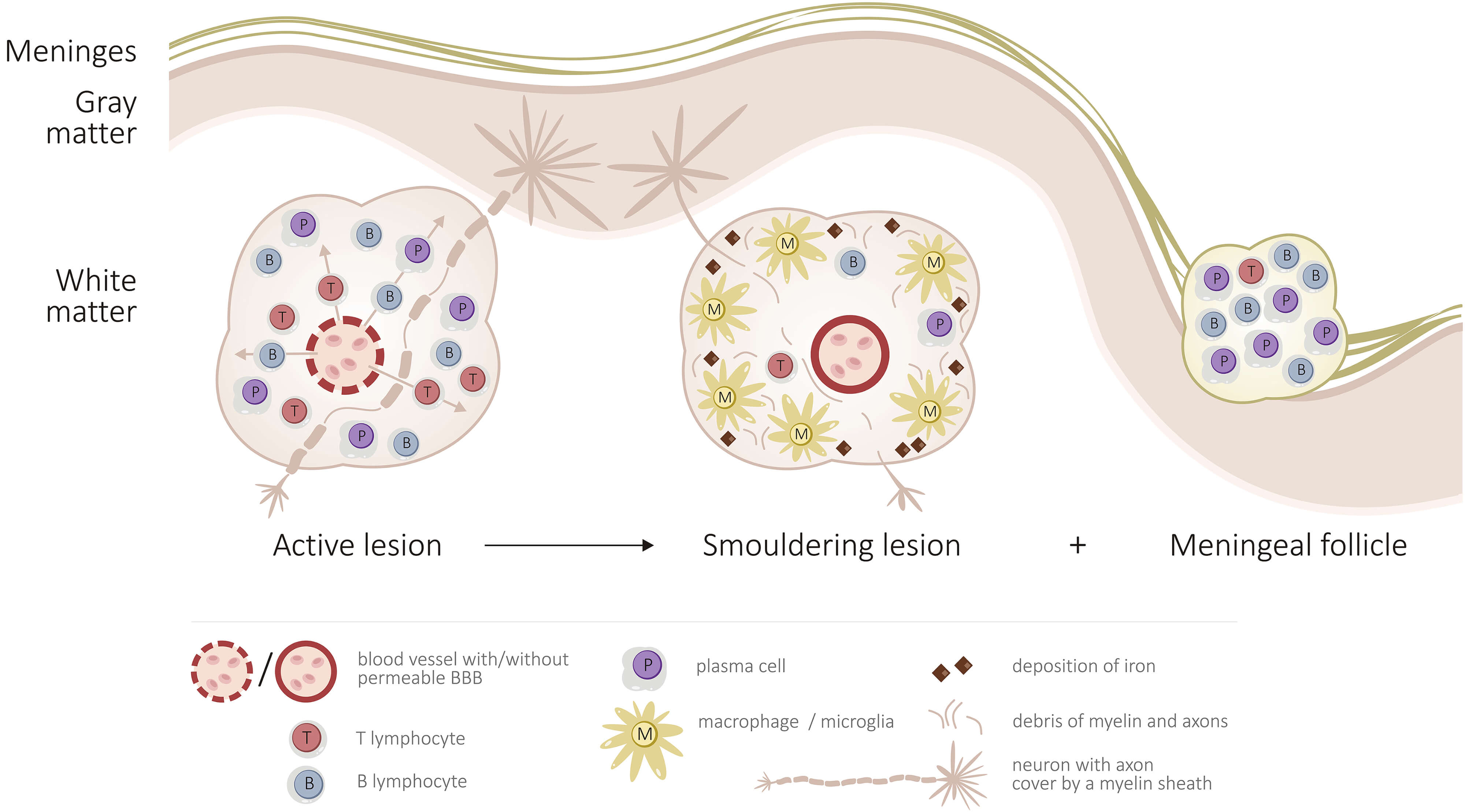 Fig. 3.
Fig. 3.
Development of inflammation and neurodegeneration in multiple sclerosis. Active lesion – The pathogenesis of multiple sclerosis begins with active lesions, where inflammation originates in the peripheral immune system. A proinflammatory cytokine milieu leads to BBB disruption, enabling the infiltration of autoreactive T and B lymphocytes into the CNS. Cellular immunity (CD4+ and CD8+ T cells) and humoral immunity (B cells and plasma cells) contribute to demyelination through oligodendrocyte damage. Although axonal loss is less prominent in this phase, it is already underway. After crossing the BBB, immune cells localize in perivascular areas, which become the initial sites of inflammation. Smouldering lesion – In smouldering lesions, the BBB reseals, leading to compartmentalization of inflammation within the CNS. Innate immune cells (macrophages, activated microglia) and cytotoxic CD8+ T cells dominate the chronic inflammatory response. These cells accumulate at the lesion rim, where they interact with myelin and neuronal degradation products and contribute to iron deposition. This phase marks the transition toward a primarily neurodegenerative process characterized by progressive axonal loss. Tertiary meningeal follicle – Tertiary meningeal follicles represent another manifestation of compartmentalized inflammation beyond the BBB. These ectopic lymphoid structures form in the meninges and are rich in B cells, which undergo maturation into plasma cells, contributing to chronic immune activation in progressive MS.
In acute inflammation, peripheral immune responses dominate. Autoreactive T cells generated in secondary lymphoid tissues produce proinflammatory cytokines that disrupt the BBB and enable immune cell entry into the CNS. The predominant inflammatory cells in early active lesions are CD8+ T cells, which proliferate and contribute to lesion formation [68]. These lesions also include significant numbers of CD4+ T cells, activated macrophages, microglial cells, B cells, antibodies, and complement components [74]. Lymphocyte distribution within lesions appears stratified: CD8+ T cells are often located at lesion borders, while CD4+ T cells are found deeper within [75]. These classic active white matter lesions are common in RRMS but become infrequent as patients transition to the progressive stage [76].
During the acute phase, CNS inflammatory infiltrates may resolve without causing axonal damage or gliosis, enabling recovery and remyelination. However, some infiltrates persist as chronic aggregates resembling tertiary lymphoid structures composed of CD4+ and CD8+ T cells, B cells, and plasma cells, leading to compartmentalized inflammation [77]. In the progressive phase—characterized by chronic or smouldering inflammation—the inflammatory process persists behind a re-sealed BBB. This compartmentalized inflammation drives neurodegeneration and disease progression. It is diffusely present in the meninges and perivascular Virchow–Robin spaces. It is associated with slow expansion of prior white matter lesions, widespread damage to normal-appearing white and grey matter, and subpial cortical demyelination in the brain and spinal cord. These features are hallmarks of progressive MS and are often accompanied by active demyelination and neurodegeneration [73]. Significant infiltration by CD8+ T cells and activated microglia in these regions likely contribute to smouldering inflammation, ongoing cognitive decline, and disability progression [78, 79].
Beyond their co-localization, these cell populations engage in bidirectional
signalling that amplifies neuroinflammation. Microglia express MHC class I and II
molecules, enabling them to present myelin antigens to CD8+ and CD4+ T cells,
respectively, thereby reactivating infiltrated lymphocytes within the CNS
parenchyma [80]. In turn, T cell-derived cytokines, particularly IFN-
While this review focuses on T cells, emerging evidence challenges the traditional T-cell-centric view of MS pathogenesis. Despite CD8+ T cells being the predominant infiltrating population in MS lesions, B-cell depletion therapies have demonstrated superior clinical efficacy compared to many T-cell-targeted approaches. Recent findings reveal that B cells can control responses of myeloid cells through oxidative phosphorylation-regulated cytokine release, revealing their previously underappreciated regulatory role rather than secondary players [83]. This paradox, coupled with the failure of atacicept (which blocks the B-cell activating factor and a proliferation-inducing ligand), which actually worsens disease activity, demonstrates that B cells have complex pathogenic and protective functions that may be equally central to MS pathogenesis [84].
The understanding of the role of B cells in MS has evolved substantially in
recent years. Intrathecal antibody production is a hallmark of multiple
sclerosis, and humoral immunity plays an important role in the inflammatory
response and development of demyelinated lesions. Interplay between B and T
lymphocytes is a central feature of disease pathogenesis [85]. B cells are
well-known efficient APCs, characterized by the expression of MHC class II, and
specialized in capturing soluble and membrane-tethered antigens, with a high
efficiency in presenting antigens and activating T cells. B cells produce
pro-inflammatory cytokines IL-6, GM-CSF, TNF-
In early and active lesions, CD20+ B cells dominate and may have pro-inflammatory functions, while at later stages, plasma cells with possible anti-inflammatory functions increase in number [68]. B cells are mainly located in the meninges and in the large perivascular spaces around the cerebral ventricles. Especially within deep cortical sulci, prominent B cell-rich inflammatory aggregates of ectopic meningeal lymphoid follicles are found. These tertiary lymphoid follicles rich in B and T cells likely mediate the progressive loss of neurological function in MS [77].
MS brain-infiltrating lymphocytes also express and respond to IL-21, the
cytokine that drives follicular T- and B-cell responses [87]. Additionally,
IFN-
Collectively, these findings reveal that the B-T cell axis in MS is more complex than previously appreciated. B cells function not merely as antibody producers but as critical orchestrators of T-cell responses through antigen presentation, cytokine production, and formation of ectopic lymphoid structures. The bidirectional nature of B-T cell interactions—where each population can activate and sustain the other—creates self-perpetuating inflammatory loops that current therapies only partially address. Understanding these intricate cellular interactions will be essential for developing next-generation therapies that can effectively target compartmentalized inflammation and halt disease progression.
Although multiple immune cell types contribute to MS pathophysiology, T lymphocytes have emerged as central regulators of both peripheral and CNS immune responses. Their functional diversity—ranging from highly proinflammatory to regulatory phenotypes—helps explain the immunological heterogeneity observed in MS lesions. A closer look at individual T-cell subsets reveals how specific phenotypes contribute to demyelination, neurodegeneration, or, conversely, immune regulation and provides insight into the cellular targets of current and emerging therapies.
CD8+ cytotoxic T cells protect the CNS from infectious agents and eliminate damaged or malignant cells. These cells express CD8 on their surface, which enables them to recognize antigens presented on MHC class I molecules. Upon recognition, CD8+ cells induce apoptosis in the target cell. They also play a central role in the pathogenesis of MS. During acute demyelination, axons become vulnerable and temporarily express MHC class I molecules, making them targets for CD8+ T cells and macrophages [74]. CD8+ cells are the predominant lymphocyte subtype in all stages of MS and are closely associated with axonal damage [20, 93].
Helper T lymphocytes (Th cells) express CD4 on their surface and are activated
upon recognising antigens presented on MHC class II molecules. Naive CD4+ T cells
can differentiate into various effector subsets (Th1, Th2, Th9, Th17) or Tregs
(Fig. 4). Th1 and Th17 cells mediate responses against intra- and extracellular
pathogens but can contribute to autoimmune pathology if dysregulated. In
contrast, Tregs suppress excessive immune activation and help maintain immune
tolerance [94]. The subsets are classified based on their cytokine profiles and
immunologic functions. Th1 cells mainly produce IFN-
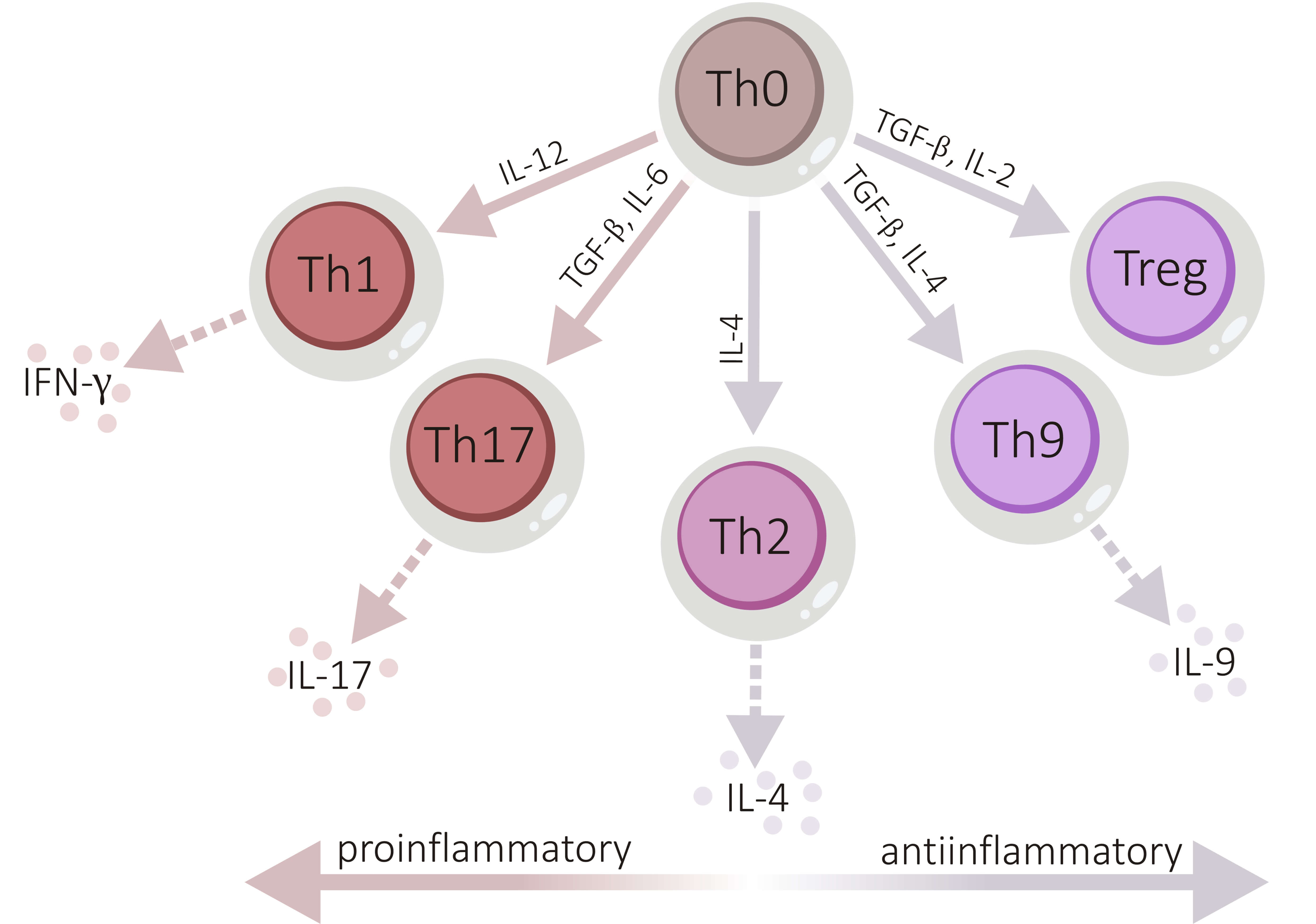 Fig. 4.
Fig. 4.
Differentiation of CD4+ T cell subsets based on cytokine milieu. Naïve Th0 lymphocytes differentiate into various CD4+ T cell subtypes
depending on the surrounding cytokines. Th1 and Th17 cells promote inflammation
via the secretion of IFN-
Th1 cells exert their effects by activating CD8+ cytotoxic T cells, NK cells,
and macrophages by producing IFN-
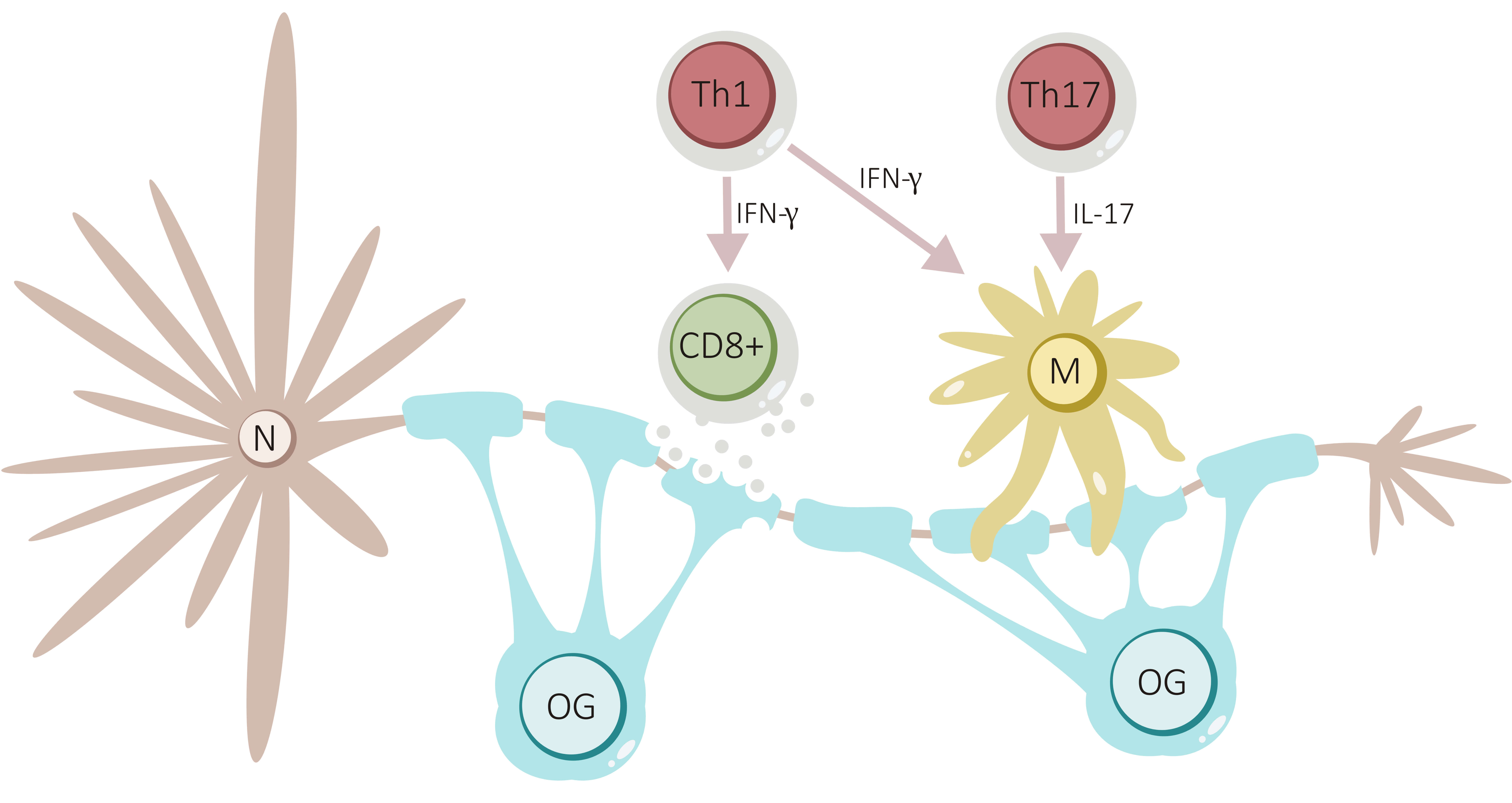 Fig. 5.
Fig. 5.
Role of cellular immunity to demyelination and axonal loss. Th1
cells, through IFN-
Th2 cells are primarily involved in immune responses against extracellular parasites and allergens. They produce IL-4, IL-5, IL-6, IL-9, and IL-10. Their differentiation is promoted by the presence of IL-4 during antigen presentation on MHC class II molecules [99]. In tertiary lymphoid follicles of the CNS, Th2 cells assist in fully activating antigen-stimulated B cells [100]. Nevertheless, Th2 cells generally exert anti-inflammatory effects and can counterbalance Th1-mediated inflammation.
Th9 cells participate in allergic inflammation and immunity against intestinal
parasites. They produce IL-9 and, to a lesser extent, IL-10 and IL-21. IL-9
regulates the balance between Th17 and Tregs. Th9 cell differentiation requires a
specific cytokine environment, particularly the presence of both TGF-
Th17 cells are defined by their secretion of IL-17A, IL-17F, IL-21, IL-22,
TNF-
Th17 cells are highly proinflammatory. They promote the recruitment of
neutrophils and macrophages, drive the expansion of dendritic and T cells, and
amplify the inflammatory response through positive feedback loops. Th17 cells
also induce the production of cytokines, chemokines, antimicrobial peptides, and
matrix metalloproteinases [106]. They facilitate CD4+ T-cell migration across the
BBB by disrupting tight junctions via IL-17 and synergise with IFN-
Tregs play a crucial role in maintaining immune homeostasis by suppressing other
immune cells’ activation, proliferation, and effector functions. They prevent the
emergence of autoreactive T and B cells. Tregs are classified into thymus-derived
(tTregs) and peripherally induced (pTregs) subtypes. tTregs arise in the thymus
upon recognizing self-antigens presented via MHC class II, requiring CD28
co-stimulation. pTregs develop in peripheral tissues from naive CD4+ T-cells
under the influence of IL-2 and TGF-
These insights into T-cell biology not only deepen our understanding of MS immunopathogenesis but also clarify the rationale behind many current and emerging treatment approaches.
T lymphocytes represent critical therapeutic targets in MS for several fundamental reasons. First, they initiate and orchestrate CNS inflammation—autoreactive T cells must be activated in the periphery and migrate across the BBB before any significant demyelination occurs, providing an upstream intervention point [128]. Second, unlike microglia or other CNS-resident cells, circulating T cells are readily accessible to therapeutic agents, eliminating the need for BBB penetration. Third, T cell responses demonstrate remarkable plasticity—pathogenic Th1/Th17 cells can be shifted toward regulatory phenotypes, potentially restoring immune tolerance without permanent immunosuppression [127]. Fourth, the diversity of T-cell trafficking mechanisms offers multiple therapeutic intervention points: preventing activation (teriflunomide), blocking adhesion molecules (natalizumab), sequestering in lymph nodes (S1P modulators), or depleting specific subsets (alemtuzumab), each with distinct safety-efficacy profiles that enable personalised treatment selection [116].
Although not all DMTs for MS treatment were specifically designed to target T lymphocytes, many exert substantial effects on T-cell–mediated immune mechanisms (Table 1, (Ref. [4, 29, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138]) and Table 2, (Ref. [139, 140, 141, 142, 143])). These diverse approaches—from cytokine modulation to lymphocyte depletion—ultimately converge on disrupting autoreactive T-cell function, though with varying efficacy depending on disease stage and individual patient characteristics. Importantly, the clinical efficacy of these T cell-targeting mechanisms varies dramatically depending on disease stage. The therapeutic landscape reveals a critical disconnect between efficacy in relapsing versus progressive disease phases [144]. While therapies highly effective in RRMS often show minimal benefit in progressive MS, this reflects fundamental differences in how T cells contribute to pathology at different stages [145]. In RRMS, where peripheral T-cell infiltration drives acute inflammation, treatments blocking CNS entry (natalizumab, S1P modulators) or depleting circulating lymphocytes (alemtuzumab, anti-CD20 antibodies) demonstrate robust efficacy. However, once compartmentalized inflammation establishes behind a closed blood-brain barrier in progressive MS, these same mechanisms fail to address ongoing neurodegeneration [146]. Only therapies with direct CNS penetration or effects on resident immune cells show promise in progressive disease—explaining why siponimod (which crosses the BBB and affects CNS S1P receptors) and potentially brain-penetrant Bruton’s tyrosine kinase (BTK) inhibitors demonstrate efficacy where purely peripheral immunomodulation fails [147, 148].
| Disease modifying treatmens (DMTs) | Route of administration | Mechanism of action |
| Glatiramer acetate [4] | subcutaneous | shift the immune response from a proinflammatory Th1 phenotype to an anti-inflammatory Th2 profile, reduce production of cytokines IL-2 and IL-12, increase release of IL-4, IL-10, and IL-1, bind to T-cell receptors (TCRs) on autoreactive T-cells without co-stimulatory signals, impede lymphocyte migration across the BBB and increase the number and function of Tregs |
| Interferon-beta 1a and 1b [129] | subcutaneous, intramuscular | enhance the production of anti-inflammatory cytokines (IL-10), suppress proinflammatory cytokines (IL-17), reduce antigen presentation and inhibit T-cell proliferation, strengthen antiviral defence mechanisms, impede lymphocyte migration across the BBB, increase the number and function of Tregs |
| Fumarates (dimethyl fumarate, diroximel fumarate) [130, 131] | oral | activation of the transcription Nrf2 - induce the transcription of antioxidant response genes - antioxidative, anti-inflammatory, cytoprotective, and neuroprotective effects - limit neuroinflammation and microglial activation, immunomodulatory effects on T lymphocytes (reduce the frequency and function of proinflammatory Th1 and Th17 cells and promote a shift toward Tregs), downregulate the production of proinflammatory cytokines (IL-17 and IFN- |
| Teriflunomide [132] | oral | reversibly inhibit dihydroorotate dehydrogenase, reduce proliferation of activated T and B cells impact the CD8+ T-cell compartment, suppress the production of proinflammatory cytokines (TNF- |
| S1P modulators (fingolimod, ozanimod, ponesimod, siponimod) [134] | oral | bind to S1P receptors on lymphocytes - leading to their internalization and functional inactivation, prevent lymphocyte egress from lymph nodes, decrease peripheral lymphocytes, involve in reparative processes in CNS |
| Natalizumab [133] | subcutaneous, intravenous | a monoclonal antibody targeting VLA-4, prevent interaction with VCAM-1 on endothelial cells of the BBB, block the migration of T-cells across the BBB almost entirely |
| Cladribine [138] | oral | a purine nucleoside analogue - increase deoxycytidine kinase expression in lymphocytes and lead to lymphocyte apoptosis |
| Alemtuzumab [137] | intravenous | a monoclonal antibody targeting CD52 (a surface protein expressed on T and B cells), induce profound lymphocyte depletion, subsequent repopulation favour regulatory and tolerogenic immune subsets, promoting a shift toward immune tolerance |
| anti-CD20 monoclonal antibodies (rituximab, ocrelizumab, ofatumumab, ublituximab) [29, 135, 136] | subcutaneous, intravenous | a monoclonal antibody targeting CD20 (a surface protein expressed on pre-B and mature B cells), induce complement-mediated lysis of B cells, indirectly reduce T-cell autoreactivity and proliferation, dampen the proinflammatory cytokine milieu, increase in the proportion of naive CD4+ and CD8+ T cells, decrease in the proportion of T cells producing IFN- |
S1P, sphingosine-1-phosphate; Nrf2, nuclear factor erythroid 2–related factor 2; TNF-
| Emerging therapies | Route of administration | Mechanism of action |
| BTK inhibitors (fenebrutinib, tolebrutinib, remibrutinib, orelabrutinib, evobrutinib) [139] | oral | limit B-cell and microglial activation, supress proinflammatory cytokines (IFN- |
| anti-CD40 ligand (frexalimab) [140, 141] | subcutaneous, intravenous | a monoclonal antibody targeting CD40 ligand (expressed predominantly on antigen-activated T-cells and, to a lesser extent, on B cells, mast cells, eosinophils, NK cells, and other immune cells) - modulate both adaptive and innate immune responses |
| CAR T-cell therapy [142] | intravenous | ex vivo genetic modification of T cells to target CD19+ B cells - eliminate autoreactive B-cell clones |
| anti-CD3 monoclonal antibody (foralumab) [143] | intranasal | promoting regulatory T cell (Treg) expansion and suppressing effector T cell activation without causing lymphocyte depletion |
BTK, Bruton’s tyrosine kinase; CAR, chimeric antigen receptor.
First-generation DMTs primarily function by shifting T cell responses from a
proinflammatory to a regulatory phenotype. Interferon-beta (IFN-
Teriflunomide selectively and reversibly inhibits dihydroorotate dehydrogenase,
a key mitochondrial enzyme essential for de novo pyrimidine synthesis, leading to
reduced proliferation of activated T and B cells. Teriflunomide specifically
impacts the CD8+ T-cell compartment by reducing homeostatic proliferation and
suppressing production of proinflammatory cytokines such as TNF-
A third therapeutic strategy involves preventing T-cell entry into the CNS.
Natalizumab, a monoclonal antibody targeting VLA-4, blocks the migration of T
cells across the BBB almost entirely. VLA-4 is highly expressed in central memory
and effector memory T cells involved in MS pathogenesis. By binding to VLA-4,
natalizumab prevents its interaction with VCAM-1 on endothelial cells of the BBB
[133]. This leads to profound suppression of CNS inflammation but carries a risk
of reactivation of latent infections, most notably John Cunningham virus (JCV)
and the development of progressive multifocal leukoencephalopathy (PML) [149].
PML risk is stratified by three major factors: anti-JCV antibody serostatus and
index level, prior immunosuppressant use, and treatment duration beyond 24 months
[150, 151]. In high-risk patients (JCV-seropositive with an index
Several DMTs work by selectively or broadly depleting lymphocytes, followed by
immune reconstitution. Anti-CD20 monoclonal antibodies (rituximab, ocrelizumab,
ofatumumab, ublituximab) primarily target B cells but have significant indirect
effects on T-cell function. By depleting B cells, these therapies reduce antigen
presentation to T cells and alter the cytokine milieu, indirectly reducing T-cell
autoreactivity and proliferation [135]. In vitro studies confirm that
rituximab suppresses T-cell proliferation and dampens the proinflammatory
cytokine environment [29]. Ocrelizumab, in a study of patients with PPMS, reduced
the number of naive and memory B cells while increasing the relative proportion
of plasmablasts. There was also an increase in the proportion of naive CD4+ and
CD8+ T cells and a decrease in the proportion of T cells producing IFN-
Current therapies for MS reduce both relapses and relapse-associated worsening of disability, which is thought to be mainly related to the transient infiltration of peripheral immune cells into the CNS. However, approved therapies are less effective in slowing the accumulation of disability in MS patients, in part due to their lack of relevant effects on inflammation in CNS compartments, where B cells and microglia are considered key and which is thought to be the cause of disability [156]. Phase-3 trials have demonstrated efficacy in progressive disease for ocrelizumab (primary progressive MS) and siponimod (secondary progressive MS); beyond these settings, effects on progression remain limited or uncertain [147, 157].
BTK inhibitors aim to modulate both B-cell receptor signalling and myeloid/microglial activation, to address peripheral as well as compartmentalized inflammation. CNS exposure varies by molecule, and agents with demonstrable brain penetration are theoretically better positioned to influence microglia-associated pathology [139]. Phase 3 results have been mixed: in non-relapsing SPMS (HERCULES), investigators reported a reduction in 6-month confirmed disability progression with tolebrutinib, whereas in relapsing MS (GEMINI-1/-2), the primary endpoint on annualised relapse rate versus teriflunomide was not met, despite reported signals on disability-worsening metrics [158, 159]. Conversely, evobrutinib did not show superiority on relapse outcomes in phase 3 despite earlier biomarker signals [160]. Safety profiles differ across the class; early transaminase elevations have been observed with some BTK inhibitors, so early liver-function monitoring is advisable, while longer-term risks (e.g., infection, malignancy) require further clarification [139]. Interpretation of these findings is limited by heterogeneous trial designs, relatively short follow-up periods, and the need for longer-term safety surveillance, particularly regarding hepatotoxicity and potential infectious complications. Overall, BTK inhibition remains promising but unfortunately still unproven.
The CD40-CD40L pathway represents an attractive target given its central role in T-B cell crosstalk. However, first-generation anti-CD40L antibodies were abandoned due to thromboembolic complications arising from platelet activation [161]. Frexalimab, engineered without Fc effector function, appears to have overcome this safety hurdle—no thromboembolic events were reported in phase 2 trials [140]. Nevertheless, completely blocking this fundamental co-stimulatory pathway raises theoretical concerns about the potential for opportunistic infections and impaired vaccine responses [162]. In the phase 2 trial, frexalimab demonstrated robust efficacy with an 89% reduction in new gadolinium-enhancing T1 lesions at week 12 and sustained disease control through 48 weeks, with 96% of high-dose recipients remaining free of new active lesions [140]. The drug progressively reduced plasma neurofilament light chain levels, reaching 41% reduction by week 48, suggesting potential neuroprotective effects beyond the suppression of acute inflammation [140]. The ongoing phase 3, which includes trials in both relapsing MS (NCT06141473) and non-relapsing secondary progressive MS (NCT06141486), will be critical in determining whether CD40L blockade can address compartmentalized inflammation and impact long-term disability progression where other therapies have failed [140, 141].
Chimeric antigen receptor (CAR)-T cell therapy represents the most aggressive immune reconstitution strategy currently under investigation, involving ex vivo genetic modification of T cells to target CD19+ B cells. While this approach has revolutionised the treatment of hematologic malignancies, its application in MS remains experimental. Fischbach et al. [142] reported initial cases in progressive MS demonstrating CSF penetration and intrathecal antibody reduction, notably, in these initial cases, without the immune effector cell-associated neurotoxicity syndrome (ICANS) that has complicated oncologic applications. Recent data presented at American Academy of Neurology (AAN) annual meeting 2025 from a small cohort of four patients with treatment-refractory secondary progressive MS showed robust CAR-T expansion in both peripheral blood and CSF, successful elimination of CD19+ B-lymphocytes, and reduction in oligoclonal bands [163, 164]. Although preliminary data suggest a more favourable safety profile than anticipated—with no ICANS observed despite CSF penetration and only mild cytokine release syndrome in one patient—expected cytopenias, including lymphopenia, neutropenia, and hypogammaglobulinemia, occurred [165]. However, mainly due to the limited number of cohort studies so far, these findings are preliminary, require confirmation in larger trials, and the long-term safety and efficacy of this approach remain to be established. Also, significant barriers to wider implementation remain, including the complex manufacturing process that requires apheresis and ex vivo cell modification, the need for lymphodepleting chemotherapy, the unknown long-term consequences of complete B-cell elimination, and questions about efficacy in progressive MS, where CNS-compartmentalized inflammation may persist despite peripheral B-cell depletion. Current phase 1 trials (NCT06451159, NCT06138132) are appropriately restricted to patients with highly active, treatment-refractory disease where conventional therapies, including anti-CD20 antibodies, have failed, and the risk-benefit ratio may justify this experimental approach.
Beyond aggressive B-cell depletion strategies, novel approaches targeting T-cell modulation are emerging. Foralumab, an intranasal anti-CD3 monoclonal antibody, represents a fundamentally different strategy—promoting regulatory T cell (Treg) expansion and suppressing effector T cell activation without causing lymphocyte depletion. By targeting nasal mucosal lymphoid tissue, this approach avoids systemic immunosuppression while potentially modulating CNS inflammation. A phase 2a trial (NCT06292923) in non-active secondary progressive MS is evaluating changes in microglial activity via the translocator protein-positron emission tomography (TSPO-PET) imaging, with preliminary data suggesting reduced fatigue and decreased microglial inflammation without neurological worsening [143]. This non-depleting approach may offer a safer alternative for patients where aggressive immunosuppression poses unacceptable risks.
Despite decades of research, fundamental questions about the pathogenesis of MS remain unanswered. Perhaps the most striking paradox lies in the cellular hierarchy of the disease. B-cell depletion therapies demonstrate superior clinical efficacy, despite CD8+ T cells being the predominant infiltrating population in MS lesions, which challenges our basic understanding of which cells truly drive the disease. Recent evidence suggests B cells may control myeloid cell responses through oxidative phosphorylation-regulated mechanisms, positioning them as potential master regulators rather than secondary players [83]. Yet anti-CD20 therapies, while effectively reducing relapses, fail to resolve chronic active lesions with paramagnetic rim lesions even after two years of treatment, highlighting a disconnect between clinical benefit and persistent tissue pathology [91]. Elucidating the mechanistic basis of this therapeutic paradox—why B-cell depletion confers clinical benefit despite CD8+ T cell predominance in lesions—represents a key research priority [112, 113]. Future studies employing spatial transcriptomics and longitudinal sampling may clarify whether B cells serve as upstream orchestrators of T-cell responses or operate through parallel pathogenic pathways.
The epidemiology of MS presents equally puzzling questions. Recent genetic studies revealed that MS risk alleles emerged in steppe pastoralist populations and spread with their migrations, yet the disease shows dramatic geographic variation that genetics alone cannot explain [166]. The role of EBV remains particularly enigmatic—while nearly universal in MS patients, the mechanistic link between infection and disease onset remains elusive. Recent serological studies have identified specific antibody signatures against the EBV peptidome that precede MS onset by years [46], and T-cell responses to EBV antigens show heightened cross-reactivity with CNS proteins in MS patients [167, 168]. The discovery that up to 47% of expanded CSF T-cell clones specifically target EBV-infected B cells suggests direct pathogenic mechanisms beyond traditional molecular mimicry [50], yet this cannot explain why most EBV-infected individuals never develop MS. Prospective cohorts tracking EBV-specific T-cell responses before MS onset, combined with deep immunophenotyping, may identify the additional host or environmental factors required for disease initiation [46, 167].
Despite recognising MS as a disease continuum with overlapping inflammatory and neurodegenerative processes from onset, predicting individual disease trajectories remains impossible. The concept of PIRA, now recognised to occur even in early “stable” disease, reveals that neurodegeneration proceeds despite apparent clinical stability [169]. Furthermore, the plasticity of T-cell phenotypes within the CNS compartment during disease progression remains poorly understood - particularly how effector T cells may transition between Th1/Th17/Treg phenotypes, acquire tissue-resident memory characteristics, or shift functional programmes in response to the changing CNS microenvironment [170]. Current therapies effectively suppress acute inflammation but fail to address this insidious progression, with no biomarkers able to predict which patients will experience rapid disability accumulation versus those who remain stable for decades [171]. These observations raise the hypothesis that MS may comprise pathogenically distinct subtypes requiring tailored therapeutic approaches. Single-nucleus RNA sequencing has already demonstrated that progressive MS patients can be stratified into distinct groups based on white matter glial responses, with potential implications for treatment selection [172]. Testing this hypothesis will require integrating clinical, imaging, and molecular biomarkers, including neurofilament light chain trajectories, paramagnetic rim lesion burden, and CSF immune profiling, to stratify patients and predict treatment response [173]. Such precision medicine strategies, if validated in prospective trials, could transform MS management from empirical escalation to mechanism-based therapy selection.
Multiple sclerosis is a complex immune-mediated disease of the central nervous system, characterized by both inflammatory and neurodegenerative components. Among immune players, T lymphocytes—particularly CD4+ Th1 and Th17, as well as cytotoxic CD8+ subsets—are key drivers of demyelination, axonal damage, and BBB disruption. Conversely, Tregs play a protective role, and their dysfunction or insufficiency may exacerbate disease progression. Accumulating evidence highlights that genetic and epigenetic factors, along with environmental triggers such as EBV, smoking, vitamin D deficiency, obesity, and gut dysbiosis, modulate T-cell responses and influence MS susceptibility and course. These findings have expanded our understanding of disease mechanisms and opened avenues for targeted therapeutic interventions. DMTs modulate T-cell function through diverse mechanisms, from altering cytokine profiles (e.g., interferons, glatiramer acetate), inhibiting lymphocyte trafficking (e.g., natalizumab, S1P modulators), to inducing immune reconstitution (e.g., cladribine, alemtuzumab) or targeting B cell-T cell interactions (e.g., anti-CD20 antibodies). Recent agents such as fumarates act both peripherally and within the CNS, modulating T-cell subsets and promoting regulatory phenotypes via oxidative stress pathways. Novel targets are under active investigation: BTK inhibitors have shown mixed phase III results, CD40L blockade is in phase III development, and CAR T-cell therapy remains in early-phase trials. Whether these approaches can durably restore immune tolerance and impact long-term outcomes (e.g., sustained neurofilament light chain reduction, disability progression, chronic active lesion resolution) requires further validation. A deeper understanding of T-cell subpopulations and their molecular regulation can guide the development of personalised treatment strategies in MS.
AAN, American Academy of Neurology; APCs, antigen-presenting cells; AHR, aryl hydrocarbon receptor; BBB, blood-brain barrier; Bregs, regulatory B cells; BTK, Bruton’s tyrosine kinase; CCR6, CC chemokine receptor 6; CRISPR-TET1, clustered regularly interspaced short palindromic repeats - ten-eleven translocation 1; CNS, central nervous system; CSF, cerebrospinal fluid; DAMP, damage associated molecular pattern; DMTs, disease modifying therapies; EAE, experimental autoimmune encephalomyelitis; EBNA1, EBV nuclear antigen 1; EBV, Epstein-Barr virus; Foxp3, Forkhead box protein 3; GM-CSF, granulocyte-macrophage colony – stimulating factor; GWAS, genome-wide association studies; HLA, human leukocyte antigen; ICANS, immune effector cell-associated neurotoxicity syndrome; Ig, immunoglobulin; IL, interleukin; IFN-
PP performed the literature search, wrote the manuscript, and approved the final version. JV, MP prepared the figures, revised the manuscript, and approved the final version. DS, IM and MV performed the literature search, contributed to conception and manuscript editing, scientific and English revision, supervision of the work, and approved the final version. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by a Czech Ministry of Health project - Conceptual Development of Research Organization - [University Hospital in Pilsen - FNPl, 00669806], by the National Health and Medical Research Council grant NU23-05-00462, by the General University Hospital in Prague project [grant number MH CZ-DRO-VFN64165] and the Charles University: Cooperatio Program in Neuroscience.
PP received compensations for travel, speaker honoraria and consultant fees from Biogen Idec, Novartis, Merck Serono, Roche, Sanofi Genzyme. MP received compensations for travel, and/or speaker honoraria, and/or consultant fees from Biogen, Novartis, Merck Serono, Sanofi, Roche, Janssen and Teva. MV received compensation for travel and/or speaker honoraria and/or consultant fees from Biogen, Merck Serono, Novartis, Roche, Sanofi, Teva, and Janssen-Cilag. DS has received financial support for conference travel and/or speaker honoraria from Novartis, Biogen, Merck, Teva, Janssen-Cilag, and Roche. None of the other authors has any conflict of interest to disclose.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.