






 , Xiaoxuan Li 1,2,†, Yuanyuan Chen 1,2, Minghao Xu 1,2, Shilin Liu 1,2, Xinyu Xu 1,2, Jiuyu Gao 1,2, Chuandong Cheng 3,4,*
, Xiaoxuan Li 1,2,†, Yuanyuan Chen 1,2, Minghao Xu 1,2, Shilin Liu 1,2, Xinyu Xu 1,2, Jiuyu Gao 1,2, Chuandong Cheng 3,4,* , Tao Jiang 1,2,*
, Tao Jiang 1,2,*1 Department of Neurosurgery, The First Affiliated Hospital of Anhui Medical University, 230001 Hefei, Anhui, China
2 Department of Neurosurgery, Anhui Public Health Clinical Center, 230001 Hefei, Anhui, China
3 Department of Neurosurgery, The First Affiliated Hospital of University of Science and Technology of China, 230001 Hefei, Anhui, China
4 Division of Life Sciences and Medicine, University of Science and Technology of China, 230001 Hefei, Anhui, China
†These authors contributed equally.
Abstract
Secondary cerebral oedema following traumatic brain injury (TBI) is a major cause of poor prognosis, primarily driven by neuroinflammation. High-mobility group box 1 (HMGB1) is a key damage-associated molecular pattern that initiates a potent inflammatory cascade, yet targeted pharmacological interventions face clinical translation challenges. Non-invasive transcutaneous auricular vagus nerve stimulation (taVNS) has shown anti-inflammatory potential, but its efficacy and specific mechanisms in treating traumatic cerebral oedema remain unclear.
A controlled cortical impact (CCI) model was established in male C57BL/6 mice. The animals were randomly divided into five groups: sham, TBI, TBI + taVNS, TBI + HMGB1 agonist (high glucose), and TBI + HMGB1 antagonist (glycyrrhizic acid). taVNS was administered daily for 7 days. Cerebral oedema volume was quantified via magnetic resonance imaging (MRI) on days 3 and 10 post-injury. Neurological function was assessed using the open field test and modified neurological sign score (mNSS). Molecular mechanisms were investigated through transcriptomic sequencing, enzyme-linked immunosorbent assay (ELISA), western blotting, and immunofluorescence to analyze HMGB1 and downstream inflammatory factors (interleukin-1β (IL-1β), interleukin-6 (IL-6)).
Transcriptomic analysis revealed that taVNS reversed TBI-induced dysregulation of genes enriched in HMGB1-related pathways (e.g., Ras-associated protein-1 (Rap1), mitogen-activated protein kinase (MAPK)). Compared with the TBI group, taVNS significantly accelerated the resolution of cerebral oedema (reduction rate: 74.7 ± 12.1% vs 53.5 ± 16.2%, p < 0.05) and improved neurological function. Mechanistically, taVNS markedly suppressed the upregulation of HMGB1, IL-1β, and IL-6 in both serum and brain tissue. Crucially, the therapeutic effects of taVNS were abolished by HMGB1 agonism (high glucose), while HMGB1 antagonism (glycyrrhizic acid) alone mimicked the benefits of taVNS.
This study demonstrates that taVNS effectively promotes the resolution of post-traumatic cerebral oedema and facilitates neurological recovery by specifically inhibiting the HMGB1-mediated inflammatory pathway. These findings position taVNS as a promising, non-invasive therapeutic strategy for the early management of secondary brain injury.
Keywords
- traumatic brain injury
- cerebral edema
- vagus nerve stimulation
- HMGB1 protein
- neuroinflammatory diseases
- transcutaneous electric nerve stimulation
Traumatic brain injury (TBI) represents a significant global public health
concern, characterized by high incidence, disability rates, and mortality [1].
The pathology of TBI encompasses two phases: primary and secondary brain injury.
Primary brain injury results directly from the impact and is therefore largely
untreatable. Within hours to days after the initial mechanical insult, secondary
damage evolves into cerebral edema, intracranial hemorrhage, brain swelling,
cerebral ischemia, and elevated intracranial pressure [2]. The initial physical
impact initiates complex molecular and cellular cascades of secondary injury
mechanisms, with cerebral edema being a key factor. Post-traumatic edema is a
strong predictor of poor prognosis [3]. Although decompressive craniectomy and
osmotic diuretics (e.g., mannitol and hypertonic saline) may temporarily
alleviate intracranial hypertension, approximately 35% of patients still
experience delayed exacerbation within 72 hours. This fundamentally stems from
neuroinflammation-driven blood-brain barrier disruption and glial cell-mediated
toxicity [4]. Recent studies indicate that high-mobility group box 1 (HMGB1),
released during early neuronal necrosis (2–6 hours post-injury), activates the
Toll-like receptor 4 (TLR4)/nuclear factor
HMGB1 is typically expressed in the cell nucleus but may be passively released
by necrotic cells or actively secreted by inflammatory cells in response to
injury. HMGB1 may also be released from peripheral tissues as part of the
systemic inflammatory response, underscoring its broad role in inflammatory
processes. Once released, extracellular HMGB1 functions as a damage-associated
molecular pattern (DAMP) [6], participating in numerous inflammatory diseases by
directly activating receptors, forming complexes with other cytokines, and
influencing clearance from the extracellular space [7]. Interventions targeting
HMGB1 (e.g., monoclonal antibodies and glycyrrhizic acid) have demonstrated
anti-inflammatory potential in animal models. However, clinical translation
remains challenging due to low blood-brain barrier penetration (
The vagus nerve, as the tenth cranial nerve, serves as a primary conduit for transmitting biofeedback to the brain [8]. Extensive research indicates that invasive cervical VNS suppresses inflammatory responses by activating the cholinergic anti-inflammatory pathway (CAP). Due to its relative minimal invasiveness and broad applicability, VNS has become one of the most frequently employed neuromodulation techniques [9]. However, the requirement for surgical electrode implantation limits its application in acute cerebral injury. Transcutaneous auricular vagus nerve stimulation (taVNS) emerges as a non-invasive alternative. By stimulating the auricular branch of the vagus nerve in the concha region, it indirectly mimics the therapeutic effects of VNS, offering a novel strategy for modulating inflammation. Presently, the clinical indications for taVNS resemble those of VNS, as supported by previous studies [10, 11]. Nevertheless, such research has predominantly focused on epilepsy and depression, leaving the temporal control mechanisms and optimal parameter protocols for traumatic brain injury largely unexplored. Therefore, this study aims to evaluate the therapeutic potential of taVNS in TBI. We sought to determine the therapeutic efficacy of taVNS for traumatic cerebral edema, and to elucidate the pathways and molecular mechanisms through which taVNS modulates brain injury, using HMGB1 agonists and inhibitors for mechanistic validation.
This study employed male SPF (Specific Pathogen Free) C57BL/6 mice (Hefei Jisai
Biotechnology Co., Ltd., Heifei, Anhui, China) aged 8–12 weeks, each housed
individually for one week prior to experimentation. Mice were maintained under a
12-hour light-dark cycle at 25–26 °C and 50
Using a desktop animal anaesthesia ventilator system (Ruiwode Life Sciences Co., Ltd, Hefei, Anhui, China), set the isoflurane concentration to 4%, with a N2O to O2 ratio of 2:1 with an oxygen flow rate of 0.5–1 L/min. Place the mouse in the induction box for anaesthetic induction. Observe the mouse until loss of righting reflex is confirmed, then position it on the CCI impactor workbench (YHKJCI990313, Wuhan YiHong Sci.&Tech. Co., Ltd., Wuhan, Hubei, China) and secure it. Connect the inhalation anaesthesia mask (set to 2.5% isoflurane concentration) and maintain body temperature using a heated pad. The scalp hair was removed from the mouse’s cranial region. Following alcohol disinfection, the scalp was incised to expose the skull. A square bone window measuring 3–4 mm in diameter was created in the right cerebral hemisphere (2 mm posterior to the anterior fontanelle, 2 mm right of the sagittal line) using a cranial drill, exposing the intact dura mater. Subsequently, employ a 3mm impactor to strike the brain tissue according to pre-set parameters (velocity: 4.5 m/s, depth: 2 mm, dwell time: 200 ms), inducing CCI. Following modelling completion, suture the wound site. After disinfection with povidone-iodine, administer antibiotics via intraperitoneal injection. Upon anaesthetic recovery, transfer the animal to a mouse cage.
Following one week of adaptive feeding, mice were randomly assigned to five groups (n = 15 per group): (1) Sham group (cranial window opened without brain tissue impact), (2) TBI group, (3) TBI + taVNS group, (4) TBI + taVNS + Agonist group (receiving intraperitoneal injection of 50% glucose solution at 6 g/kg/day for 7 consecutive days starting 3 days post-surgery), (5) TBI + Inhibitor group (intraperitoneal injection of glycyrrhizic acid dissolved in 5% DMSO + physiological saline at 50 mg/kg/day for 7 consecutive days starting 3 days post-surgery). For the sake of brevity, the ‘TBI + taVNS + Agonist’ group is hereafter referred to as the ‘Agonist’ group, and the ‘TBI + Inhibitor’ group as the ‘Inhibitor’ group in the text and figures. The experimental timeline is illustrated in Fig. 1.
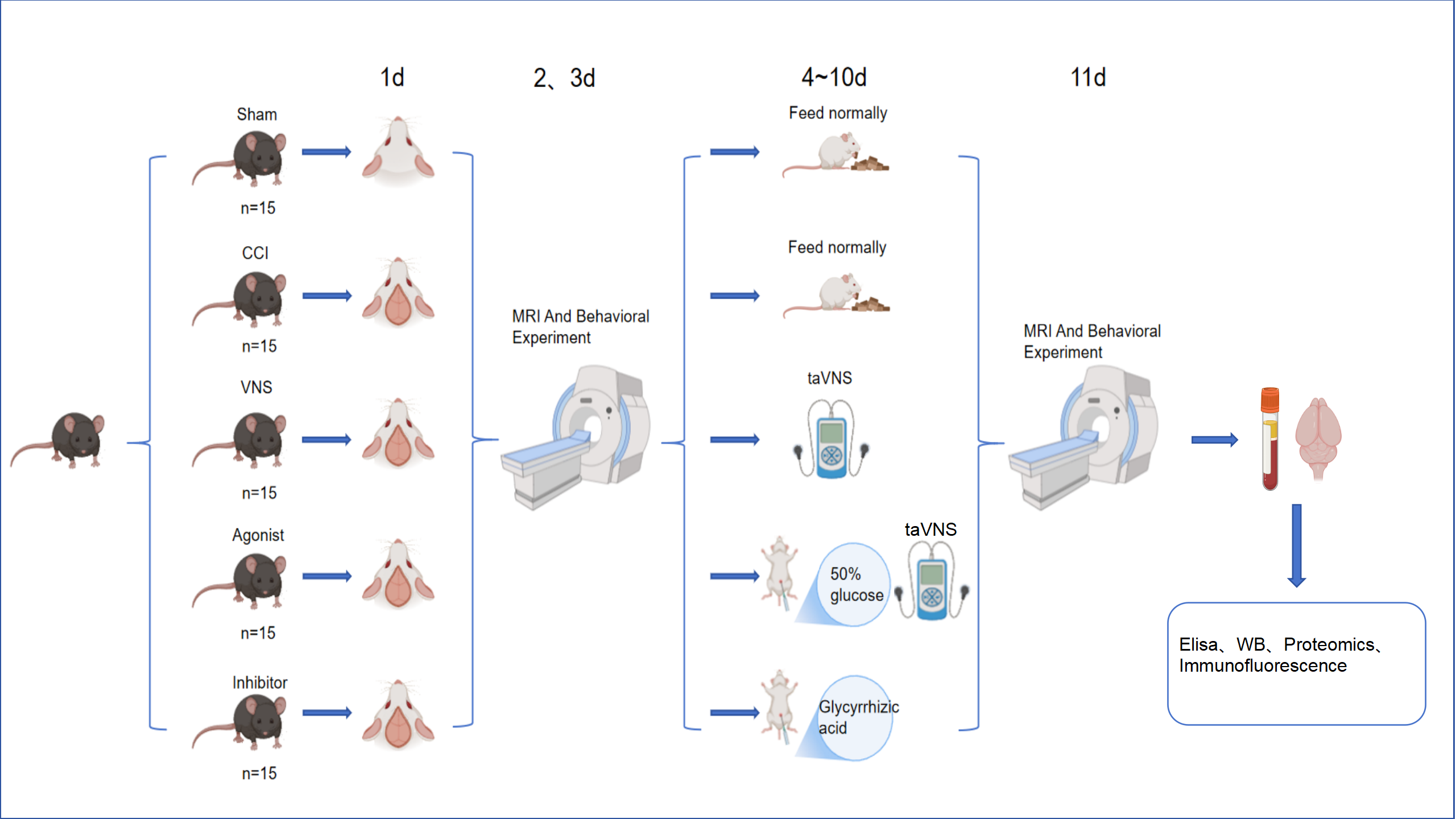 Fig. 1.
Fig. 1.
Experimental timeline and group design. Mice (n = 15/group)
were divided into five groups: Sham, CCI, VNS, Agonist + VNS, and Inhibitor. Except
for the Sham group, all animals were subjected to CCI. The experimental timeline
was as follows: CCI modelling
The taVNS stimulation parameters for this experiment were established based on prior research. The apparatus employed a vagus nerve electrical stimulator (YJT1-240710002) manufactured by Hangzhou Yijian Technology (Hangzhou, Zhejiang, China). To ensure mice could tolerate prolonged vagus nerve electrical stimulation, a bench-top animal anaesthesia ventilator (Ruiwode Life Sciences Co., Ltd.) was employed. Mice were placed in an induction chamber and anaesthetised with 2% isoflurane (0.5%–1% oxygen) to achieve and maintain anaesthesia. Both auricles were swabbed with alcohol. Electrodes were secured to both auricles, and electrical stimulation commenced. Parameters were set as follows: 0.5 mA, 30 Hz, 30 minutes daily, with 30-second on-intervals followed by 4.5-minute off-intervals. Stimulation was administered daily between 21:00 and 22:00 for a total of 7 days.
The experiment employed a Bruker Biospec 9.4T small animal magnetic resonance
imaging system (Bruker Biospec MRI, Ettlingen, Germany) with a 20 mm
mouse-specific brain coil. Under 4% isoflurane induction anaesthesia (maintained
at 1.5%), axial T2-weighted imaging was performed on days 3 and 10 post-TBI
surgery (Parameters: repetition time/echo time (TR/TE) = 3272/48 ms, rapid
acquisition with relaxation enhancement (RARE) factor = 8, acquisition runs = 3,
matrix = 256
Brain tissue samples were collected from mice in the taVNS group, TBI group, and
control group (n = 3 per group). Sequencing analysis was performed using the
DNBSEQ platform (https://www.mgi-tech.com/DNBSEQ-Technology.html). Key modules
were screened for core pathways via Kyoto Encyclopedia of Genes and Genomes/Gene
Ontology (KEGG/GO) enrichment analysis (ClusterProfiler). Target specificity
validation was conducted using western blot and enzyme-linked immunosorbent assay
(ELISA) cross-platform validation for HMGB1 and its upstream/downstream molecules
(including IL-6 and IL-1
The experiment employed a standardised open field apparatus (40 cm
The experiment employed an ELISA to detect concentrations of HMGB1 and IL-6 in mouse serum. The mouse HMGB-1 ELISA detection kit (Lianke Bio, SEKM-0032, Shenzhen, Guangdong, China; detection range 15.6–1000 pg/mL, sensitivity 4.7 pg/mL) and the mouse IL-6 ELISA Detection Kit (EMC004, NeoBioscience Technology Co., Ltd., Shenzhen, Guangdong, China; detection range 7.8–500 pg/mL, sensitivity 2.3 pg/mL).
At the end of the experiment, all animals were deeply anesthetized with 3%
isoflurane delivered via an inhalation chamber (0.5 L/min oxygen flow) until loss
of the righting and pedal withdrawal reflexes. Once deep anesthesia was achieved,
the animals were humanely euthanized by cervical dislocation to ensure death, in
accordance with the AVMA Guidelines for the Euthanasia of Animals (2020). Whole
blood was collected via ocular puncture. After standing for 30 minutes, serum was
separated by centrifugation at 3000
Experimental samples comprised mouse brain tissue perfused and fixed with 4%
paraformaldehyde (BL539A, Hefei Biosharp Biotechnology Co., Ltd., Hefei, Anhui, China), rapidly frozen in liquid nitrogen and stored at –80
°C for subsequent analysis. Protein samples (approximately 20 µg
per well) were denatured in loading buffer and subjected to electrophoresis on a
precast polyacrylamide gel (concentration gel: 80 V for 20 minutes; separation
gel: 120 V for 60 minutes). Proteins were subsequently transferred to
Polyvinylidene Fluoride (PVDF) membranes (Beyotime, Shanghai, China) using a
semi-dry transfer method (PVDF membranes were activated with methanol prior to
transfer; both gel and membrane were equilibrated in ice-cold transfer buffer).
Transfer conditions were 25 V for 30 min (Buffer: 48 mM Tris, 39 mM glycine,
0.04% SDS, 20% methanol). The membrane was blocked with 5% skimmed milk powder (or bovine serum albumin [BSA] for
phosphorylated proteins) at room temperature for 1 hour or overnight at 4
°C. It was then incubated sequentially with specific primary antibodies
(glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:8000, 60004-1-Ig, Proteintech Group, Inc., Rosemont, IL, USA); HMGB1 (1:1000, ab18256, abcam, Cambridge, UK);
IL-1
Mouse brain tissue was perfused and fixed with 4% paraformaldehyde, followed by gradient dehydration and embedding in paraffin. Coronal sections (5 µm thickness) were prepared using a rotary microtome (Leica RM2016; Leica Microsystems (Shanghai) Trading Co., Ltd., Shanghai, China). Deparaffinization and rehydration were performed by sequential immersion in xylene (10023418, Sinopharm, Beijing, China, three changes, 10 min each) and a graded ethanol series (100%, 100%, 95%; 5 min each), followed by a final rinse in distilled water.
Antigen retrieval was carried out by heating the sections in sodium citrate buffer (C1013, Solarbio, 10 mM sodium citrate, 0.05% Tween 20 (ST825, Beyotime, Shanghai, China), pH 6.0) using a pressurized decloaking chamber for 3 minutes. Sections were allowed to cool naturally to room temperature. After washing with PBS, sections were permeabilized with 0.1% Triton X-100 in PBS for 20 minutes at room temperature and then blocked with 3% BSA for 30 minutes at room temperature to prevent non-specific binding.
Sections were incubated overnight at 4 °C in a humidified chamber with
the following primary antibodies diluted in PBS: rabbit anti-HMGB1 (1:100, 10829-1-AP, Proteintech
Group) and rabbit anti-IL-6 (1:100, 83747-5-RR, Proteintech Group). After thorough washing with PBS (3
Finally, the sections were washed with PBS, briefly air-dried, and mounted with
an anti-fade mounting medium (P0126, Beyotime). Images were captured using a
fluorescence microscope (Nikon Eclipse E100, Shanghai, China) at 400
The modified neurological sign score (mNSS) was assessed blindly by two
researchers, who were completely unaware of the experimental groups and
treatments, on postoperative days 2 and 11, respectively. The scoring system
encompasses four dimensions: motor function (limb symmetry, crawling ability),
sensation (tactile/pain reflexes), balance (balance beam walking, righting
reflex), and abnormal behaviour (tremors, circling). The total score ranges from
0 to 18 points (0 = no deficit, 18 = severe deficit). Prior to assessment, mice
were acclimatised for 30 minutes in a quiet environment (25
Transcriptomic analysis revealed 1170 differentially expressed genes in the CCI group, comprising 511 upregulated and 659 downregulated genes. taVNS, the number of differentially expressed genes decreased to 929, including 455 upregulated and 474 downregulated genes. KEGG enrichment analysis confirmed that HMGB1-related pathway genes, such as Rat sarcoma (Ras) and mitogen-activated protein kinase (MAPK), were significantly enriched among the upregulated genes in the CCI group. Moreover, VNS, HMGB1-related pathway genes, including Ras-related protein 1 (Rap1), Ras, and MAPK, were markedly enriched among the downregulated genes in the VNS group compared to the CCI group (Fig. 2). Preliminary transcriptomic findings suggest that taVNS may regulate HMGB1 levels and inflammation by modulating HMGB1-associated pathways.
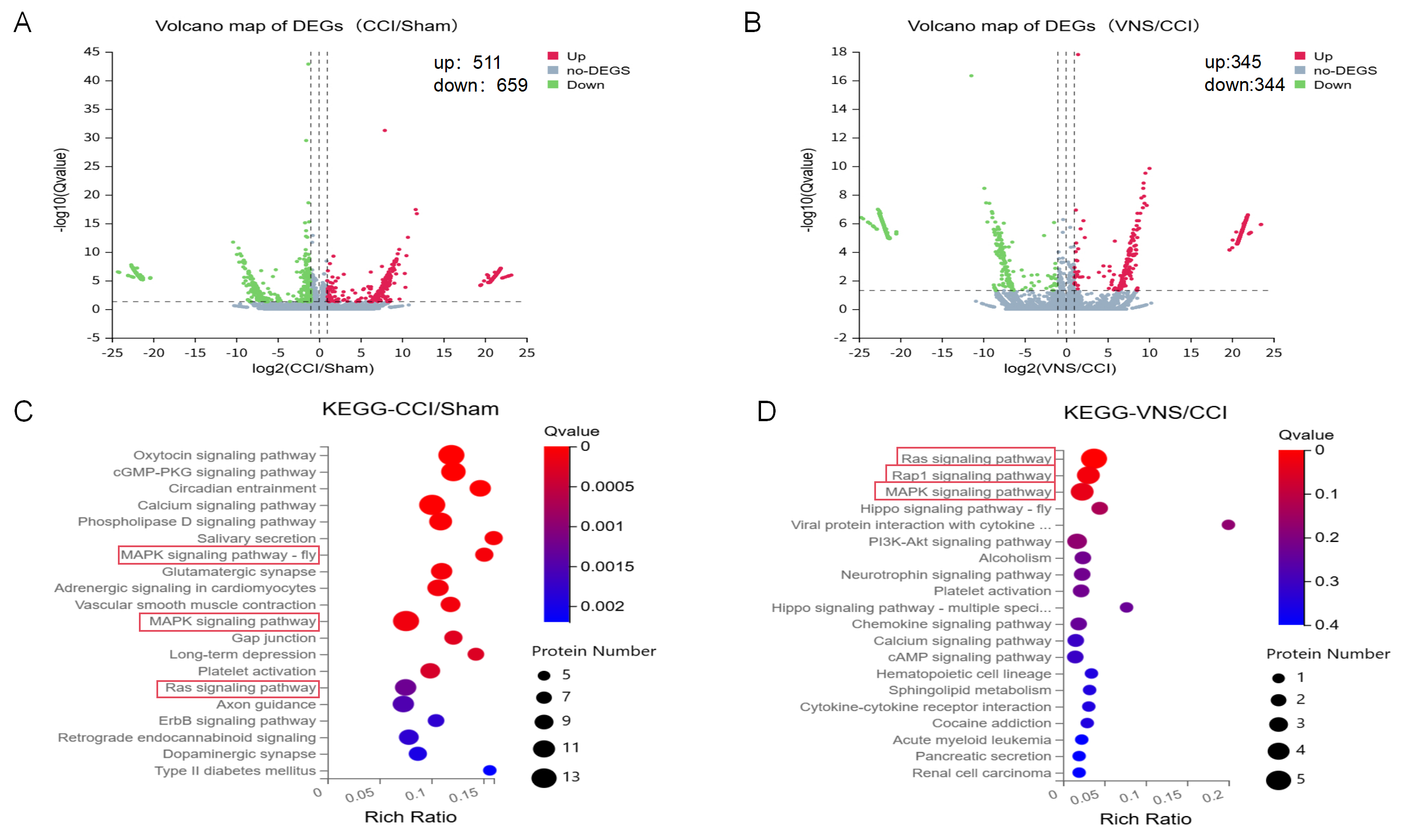 Fig. 2.
Fig. 2.
Transcriptome analysis image. (A) DEGs in CCI versus Sham (volcano plot). (B) DEGs in VNS versus CCI (volcano plot). (C) Pathway enrichment of CCI-upregulated genes identifies HMGB1 signaling as a key upregulated node. (D) Pathway enrichment of VNS-downregulated genes confirms HMGB1 signaling as a primary target for reversion. DEGs, differentially expressed genes; KEGG, Kyoto Encyclopedia of Genes and Genomes; cGMP, cyclic guanosine monophosphate; PKG, protein kinase G; PI3K-Akt, phosphoinositide 3-kinase–protein kinase B signaling pathway; cAMP, cyclic adenosine monophosphate; MAPK, mitogen-activated protein kinase; HMGB1, high-mobility group box 1.
Magnetic resonance imaging revealed marked cerebral edema in the group compared to the
control group at days (Fig. 3A). Combined with analysis, this indicated
significant neurological deficits in the group at day 3 distance travelled in the
central zone of the open field test decreased by 60% compared to controls
(915.20
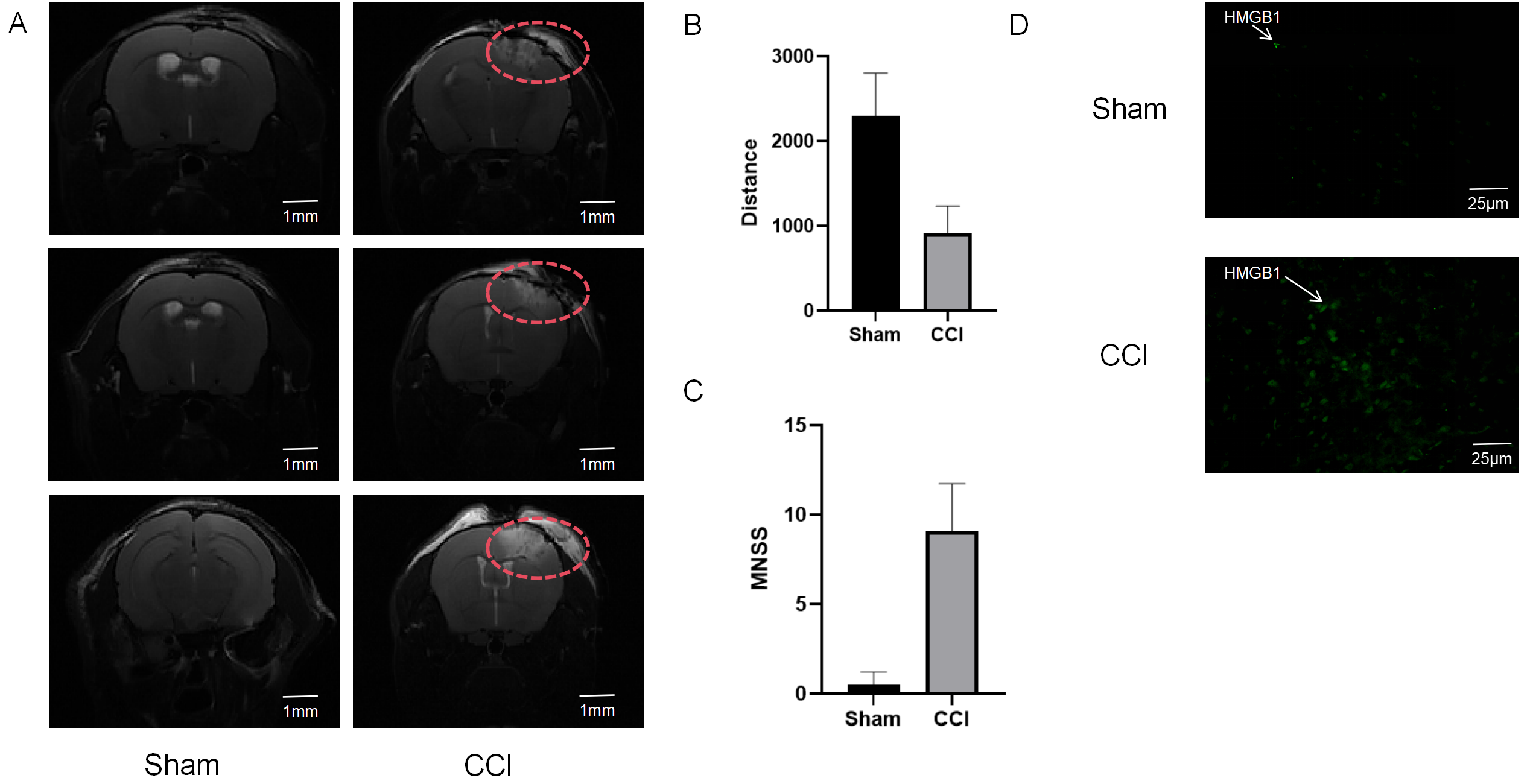 Fig. 3.
Fig. 3.
Successful establishment of the CCI model in mice. (A) Representative MRI images of the brain from the Sham and CCI groups at 3 days post-surgery, showing the distinct lesion area (The area outlined in red dashed lines). Scale bar = 1 mm. (B) Quantitative analysis of the distance traveled in the center of the open field, demonstrating reduced exploratory behavior in the CCI group compared to the Sham group. (C) mNSS scores assessed at 3 days post-surgery, indicating significant neurological deficits in the CCI group. (D) Representative immunofluorescence images showing enhanced expression of HMGB1 (green) in the CCI group compared to the Sham group. Cell nuclei were counterstained with DAPI. Scale bar = 25 µm. mNSS, modified neurological sign score; DAPI, 4′,6-diamidino-2-phenylindole.
ITK-SNAP three-dimensional reconstruction revealed that the taVNS group
exhibited a significant reduction in cerebral volume of 74.7
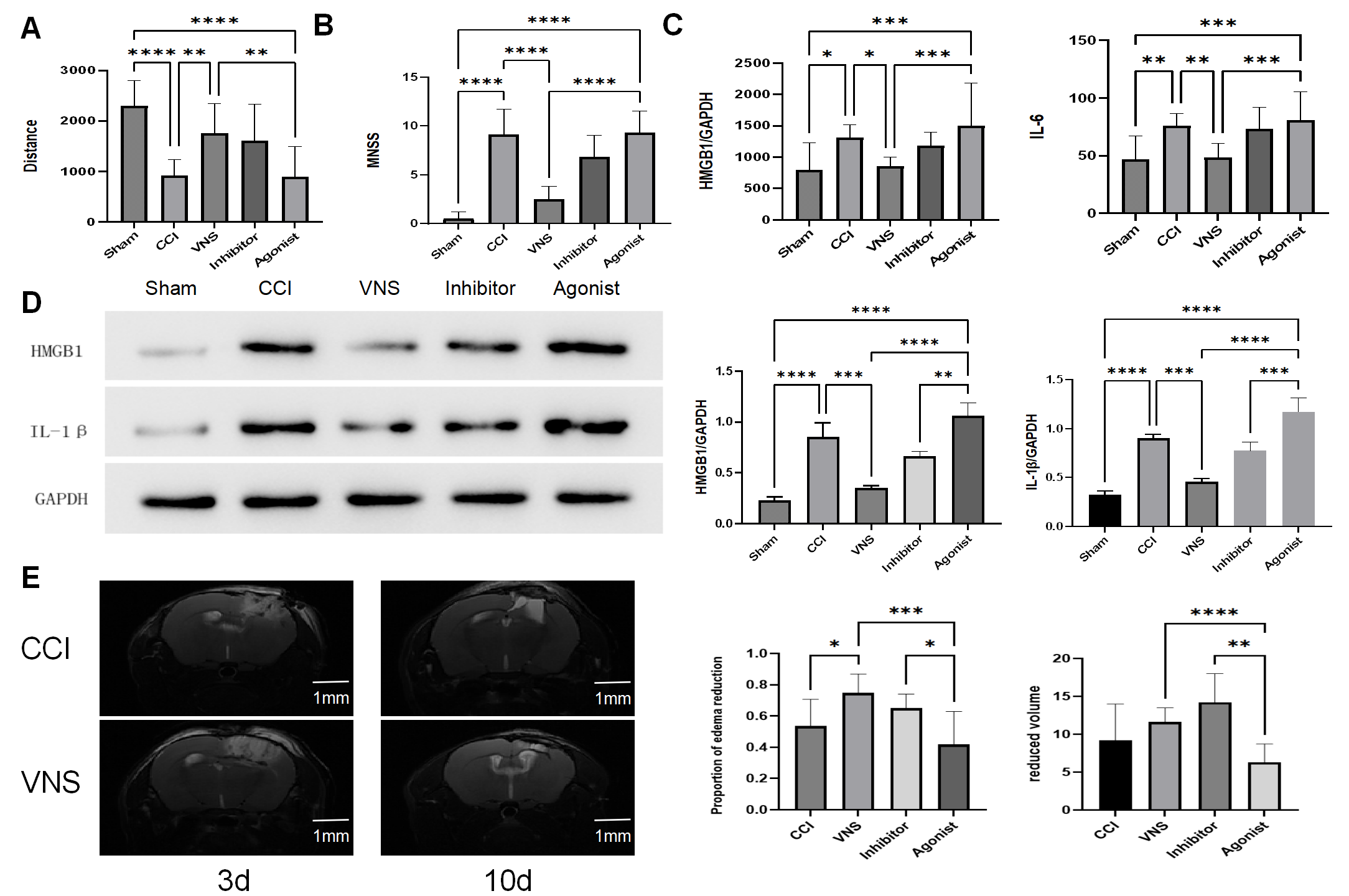 Fig. 4.
Fig. 4.
taVNS improves functional recovery, suppresses
neuroinflammation, and promotes edema resolution post-TBI. (A) Analysis of the
distance traveled in the center of the open field for all five experimental
groups. (B) mNSS neurological scores across all five groups. (C) Serum levels of
HMGB1 and IL-6 were quantified by ELISA in the five groups. (D) Protein
expression levels of HMGB1 and IL-1
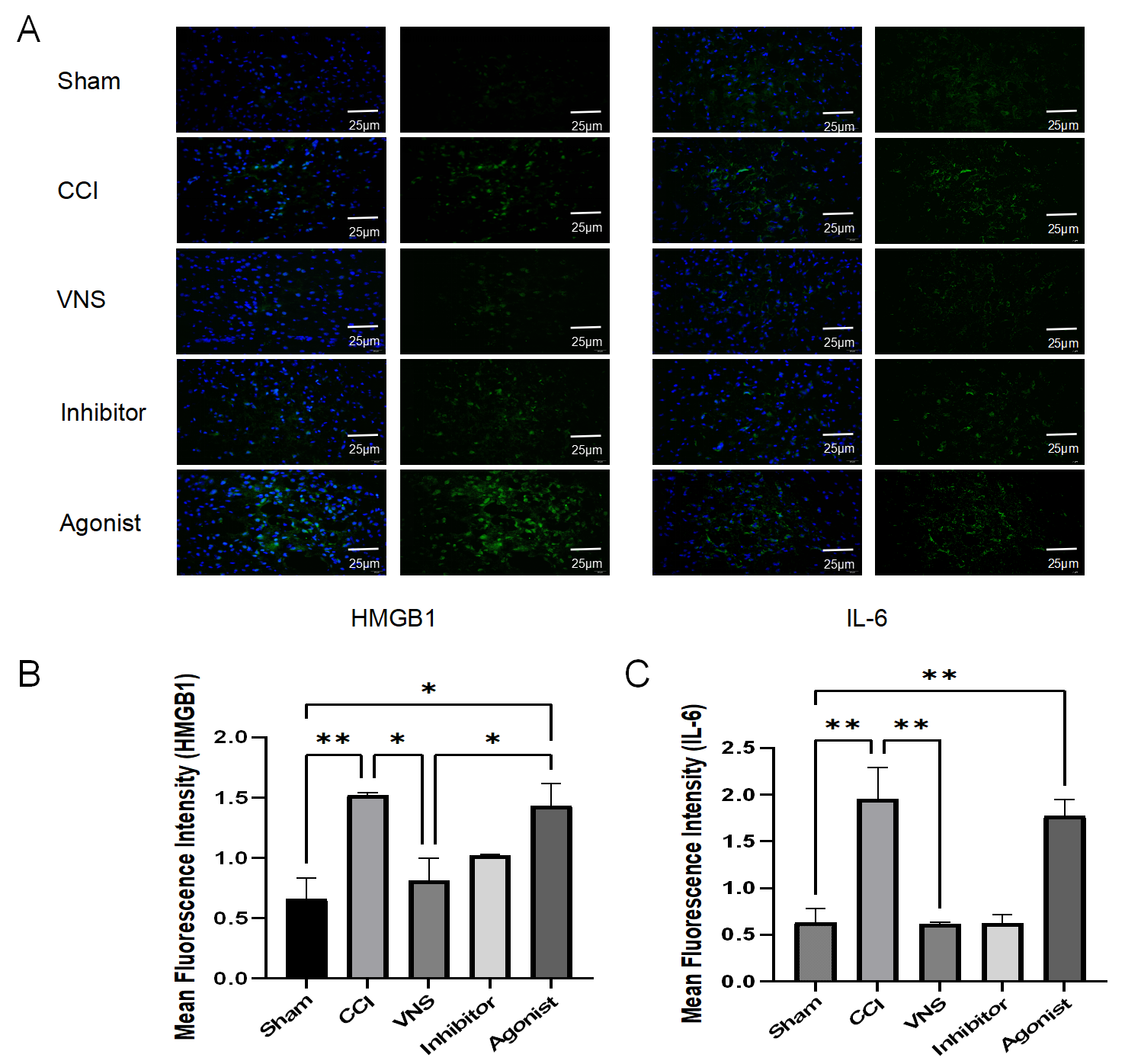 Fig. 5.
Fig. 5.
taVNS suppresses HMGB1 and IL-6 expression in brain tissue
post-TBI. (A) Representative immunofluorescence images from the five
experimental groups. Cell nuclei were counterstained with
4′,6-diamidino-2-phenylindole (DAPI) (blue). Positive signals for HMGB1 and IL-6
are shown in green. Scale bar = 25 μm. (B,C) Quantitative analysis of the mean fluorescence
intensity for (B) HMGB1 and (C) IL-6 in the peri-lesion cortex. Data are
presented as mean
To substantiate that the HMGB1 pathway specifically mediates the anti-edema
effects of taVNS, we specifically examined the phosphorylation levels of its key
downstream effector molecules, NF-
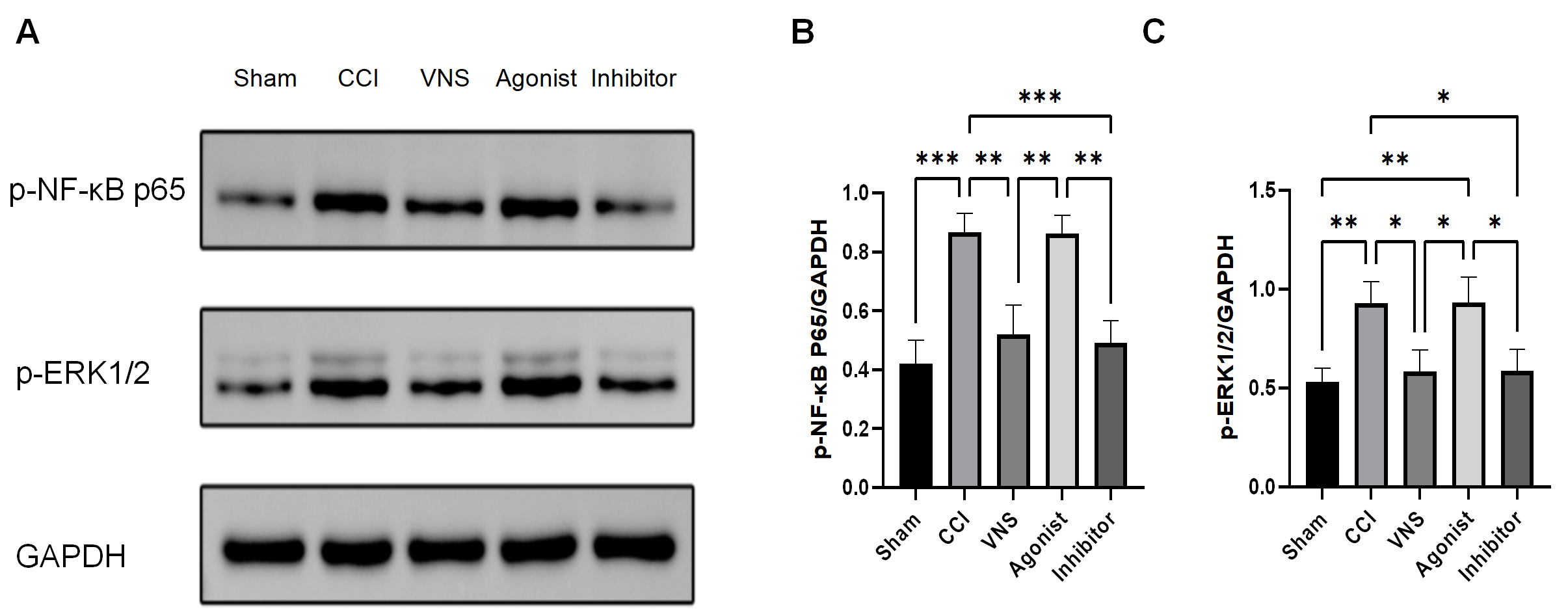 Fig. 6.
Fig. 6.
taVNS suppresses the activation of key downstream effectors in
the HMGB1 signaling pathway. (A) Representative western blot images of
phosphorylated NF-
To complement our western blot findings and spatially resolve the expression of key inflammatory mediators, we performed a quantitative analysis of immunofluorescence (IF) images. Consistent with the molecular data, a significant increase in the fluorescence intensity of both HMGB1 and IL-6 was observed in the peri-lesion cortex of the CCI group compared to the Sham group (HMGB1: p = 0.0084; IL-6: p = 0.0045; Fig. 5B,C). taVNS treatment markedly reduced the signal intensity for both HMGB1 and IL-6 compared to the CCI group (HMGB1: p = 0.0192; IL-6: p = 0.0042). The therapeutic effect of taVNS was abolished by the HMGB1 agonist, as the Agonist group exhibited fluorescence intensity levels comparable to those in the CCI group (HMGB1: p = 0.9579; IL-6: p = 0.8543). Conversely, monotherapy with the HMGB1 inhibitor mimicked the effect of taVNS, leading to a significant suppression of IL-6 (p = 0.0043 vs CCI group) and a strong, albeit non-significant, trend toward reduced HMGB1 (p = 0.0745 vs CCI group). Representative IF images are shown in Fig. 5A. These quantitative IF results reinforce the conclusion that taVNS exerts its anti-inflammatory effects by specifically modulating the HMGB1 pathway at the cellular level.
To determine whether the therapeutic effects of taVNS specifically on the HMGB1
pathway, this study employed a combined intervention with an agonist (high
glucose) and an antagonist (glycyrrhizic acid) at the mechanism level. Behavioral
results demonstrated that, by day 10 post-operation, the central area movement
distance in the inhibitor group was restored to 69.8% of that in the control
group (1648.04
TBI encompasses a spectrum of damage resulting from cranial trauma. Beyond its high mortality rate, severe TBI can lead to varying degrees of cognitive impairment and neurological dysfunction, resulting in lifelong disability and imposing substantial economic and health burdens on families and society [12, 13, 14, 15]. The primary cause of these sequelae is secondary brain injury; however, effective pharmacological interventions remain limited [16, 17]. Following TBI, HMGB1 released from injured cells triggers a cascade of inflammatory mediators, leading to blood-brain barrier (BBB) disruption, neuronal damage, and secondary cerebral edema [18]. This edema is a pivotal pathological mechanism in neurological deterioration. Current clinical management primarily relies on osmotic agents (e.g., mannitol and hypertonic saline) and surgical decompression [19]. However, these measures offer only transient relief from intracranial hypertension without addressing the self-perpetuating cycle of neuroinflammation-driven BBB impairment and glial cell-mediated toxicity.
VNS has attracted interest for its ability to suppress systemic inflammatory
responses via activation of the CAP [20]. Nevertheless, conventional invasive VNS
requires surgical electrode implantation on the cervical vagus nerve, which
limits its clinical applicability due to high costs, impracticality in acute
settings, and potential side effects [21]. In contrast, taVNS has emerged as a
non-invasive neuromodulation approach. By stimulating the auricular concha
region—which corresponds to the “heart” and “brain” acupoints in
Traditional Chinese Medicine and is densely innervated by auricular vagal
branches—taVNS directly activates the solitary nucleus-locus coeruleus pathway.
Animal studies have demonstrated that taVNS exerts anti-inflammatory effects
comparable to invasive VNS [22], while avoiding surgical risks and enabling
timely intervention. This study aimed to evaluate the role of taVNS in early
intervention for post-traumatic cerebral edema and inflammation through targeting
HMGB1. Our results indicate that taVNS initiated within 72 hours after TBI
significantly enhanced edema resolution (volume reduction rate: 74.7
The release of HMGB1 from necrotic neurons into the extracellular microenvironment following TBI is widely recognized as a central element of the DAMP response. In this study, we employed molecular techniques to assess HMGB1 levels in blood and brain tissue, incorporating interventions such as taVNS, HMGB1 agonists, and inhibitors. Our findings establish that the therapeutic benefits of taVNS are fundamentally associated with modulation of the HMGB1 pathway and its upstream and downstream components. Specifically, high-glucose administration abolished the inhibitory effect of taVNS on HMGB1 and impeded edema resolution, whereas glycyrrhizic acid monotherapy replicated the therapeutic benefits of taVNS. The HMGB1 agonist enhanced the expression of HMGB1 and downstream inflammatory factors in both serum and brain tissue, whereas the antagonist facilitated edema resolution to a certain extent.
Therefore, we propose that taVNS, by attenuating HMGB1 release, disrupts the
pro-inflammatory signaling axis, leading to reduced neuroinflammation and
subsequent cerebral edema. This conclusion is strongly supported by our novel
data showing that taVNS effectively inhibits the activation of NF-
This discovery redefines the role of HMGB1 in neuroinflammation: it is not
merely a messenger of injury signals, but rather a ‘modulable hub’ for
therapeutic intervention. taVNS may regulate inflammation by modulating HMGB1
levels rather than directly interfering with downstream pathways such as
TLR4/NF-
In this context, our findings gain significant clinical relevance when contrasted with the current standard of care. The mainstay pharmacological treatment for cerebral edema, glucocorticoids such as dexamethasone, is increasingly recognized for its detrimental immunosuppressive effects, which can antagonize modern immunotherapies and potentially worsen patient outcomes [24]. Our study demonstrates that taVNS achieves comparable anti-edema efficacy without systemic immunosuppression, positioning it as a promising non-invasive and steroid-sparing alternative for managing neuroinflammation post-TBI.
Furthermore, the trajectory of neurological recovery itself could provide a direct and clinically relevant guide for tailoring treatment length. In this study, the taVNS group demonstrated significantly improved mNSS scores and open field performance by day 11 (Fig. 4A,B). We hypothesize that patients whose functional recovery plateaus or lags behind expectations—as measured by serial assessments using standardized scales such as the extended Glasgow Outcome Scale (GOSE) or mNSS—may represent ideal candidates for an extended course of taVNS. This approach would leverage the treatment’s anti-inflammatory and potential pro-repair effects to support ongoing neural repair and plasticity during the critical subacute recovery phase.
Although this study provides valuable insights, several limitations should be acknowledged. First, all experimental subjects were male mice; future studies should include female and aged animals to enhance the generalizability of the findings. Second, although the dry-wet weight method is commonly used to assess cerebral edema, we employed cerebral MRI, which is considered the gold standard for quantitative evaluation. However, this approach is still subject to technical constraints such as partial volume effects at tissue boundaries and significant motion artifacts due to high respiratory rates in rodents (80–120 breaths per minute), which are 3–5 times more pronounced than in humans. Future work could benefit from integrating deep learning-based segmentation and multimodal image registration to improve accuracy. Finally, with regard to stimulation parameters, we used a fixed set of parameters determined during pre-experimental optimization and did not evaluate multiple parameter configurations within the experimental groups. Subsequent investigations should explore parameter optimization and develop adaptive parameter-adjusting systems tailored for multimodal data analysis.
taVNS significantly promotes cerebral edema resolution and neurological recovery by specifically inhibiting the HMGB1-mediated inflammatory pathway. Its noninvasive nature and potential for early intervention offer an innovative pathway to overcome therapeutic limitations in secondary brain injury. Future studies should focus on parameter, multiclinical validation, and the development of intelligent devices to advance taVNS from basic research to clinical application, improving long-term outcomes for patients with.
TBI, traumatic brain injury; CCI, controlled cortical impact; taVNS, transcutaneous auricular vagus nerve stimulation; VNS, vagus nerve stimulation; HMGB1, high mobility group box 1; MAPK, mitogen-activated protein kinase; IL-6, Interleukin-6; IL-1
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
YFF and XXL conceived and designed the study. YFF performed the majority of the experiments and data analysis. XXL supervised the project. TJ and CDC contributed to the experimental design, specifically refining the surgical and stimulation protocols, and were solely responsible for the acquisition, processing, and quantitative analysis of all magnetic resonance imaging (MRI) data. MHX performed the statistical analysis and interpreted the behavioral and molecular data. YYC conducted the immunofluorescence experiments and performed quantitative image analysis. JYG and XYX contributed to the study by establishing and optimizing the post-operative animal care protocol, which was critical for ensuring animal welfare and standardized recovery, thereby directly supporting the integrity of the longitudinal data acquisition. SLL contributed to the manuscript by drafting the initial sections on methodology and results, and by participating in the interpretation of the transcriptomic data. All authors participated in revising the manuscript critically for important intellectual content, read, and approved the final version to be published. All authors agree to be accountable for all aspects of the work.
All experimental procedures were approved by the Ethics Committee of Anhui Medical University (Approval Number: 20252156) and were conducted in accordance with its relevant guidelines and regulations (Guidelines for the Management and Use of Laboratory Animals).
The authors would like to thank all colleagues who provided general support and stimulating discussions during the course of this research.
This research was funded by Anhui University Scientific Research Project, grant number AH2023AH040076.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN47066.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.