



 , Shiqi Zhang 1,2,4,†, Hangxiu Che 1,2,4, Yiwen Wan 1,2,3, Hanyu Gao 1,2,5, Dongyuan Yao 1,2,*
, Shiqi Zhang 1,2,4,†, Hangxiu Che 1,2,4, Yiwen Wan 1,2,3, Hanyu Gao 1,2,5, Dongyuan Yao 1,2,*
1 Neurological Institute of Jiangxi Province, and Department of Neurology, Jiangxi Provincial People’s Hospital and The First Affiliated Hospital of Nanchang Medical College 330006 Nanchang, Jiangxi, China
2 Department of Neurology, Xiangya Hospital of Central South University at Jiangxi, 330006 Nanchang, Jiangxi, China
3 Department of Clinical Medicine, The Second School of Clinical Medicine, Nanchang University, 330038 Nanchang, Jiangxi, China
4 Joint Program of Clinical Medicine/Biomedical Sciences, Queen Mary College, Nanchang University, 330031 Nanchang, Jiangxi, China
5 Department of Clinical Medicine, The First School of Clinical Medicine, Nanchang University, 330209 Nanchang, Jiangxi, China
†These authors contributed equally.
Abstract
Rapid eye movement sleep behavior disorder (RBD) is a parasomnia characterized by the loss of muscle atonia during rapid eye movement sleep, leading individuals to physically act out their dreams, often resulting in injuries. This condition is linked to SubCoeruleus nucleus dysfunction (homologous to the sublaterodorsal tegmental nucleus in rodents) and related neural circuits. Despite its profound impact on patient safety and quality of life, the role of environmental and lifestyle factors in RBD pathogenesis remains underexplored. To bridge this gap, we conducted a review of observational and interventional studies published between 2000 and 2025, using PubMed, Web of Science databases, and ScienceDirect. After systematic screening, 129 studies were selected, covering pathophysiological mechanisms, biomarkers, diagnostic approaches, and therapeutic interventions. Emerging evidence suggests that early detection through biomarkers and neuroimaging, combined with targeted therapeutic interventions, may help delay or prevent progression to more severe neurodegenerative diseases. This review underscores significant advances in identifying RBD biomarkers and targeted interventions, while highlighting the critical need for future research on modifiable environmental and lifestyle risk factors to guide preventive strategies.
Keywords
- REM sleep behavior disorder
- neurodegenerative diseases
- α-synucleinopathies
- biomarkers
- pathogenesis
The human sleep cycle, a fundamental aspect of daily physiological rhythms, is conventionally divided into rapid eye movement (REM) and non-REM sleep. During REM sleep, the brain is remarkably active and often accompanied by vivid dreams, while the body experiences reduced muscle tone. This muscle atonia is crucial for REM sleep, and its disruption can result in dreaming-enacting behaviors such as vocalizations and twitches that mirror dream content, potentially resulting in REM sleep behavior disorder (RBD) [1, 2, 3].
RBD is categorized into idiopathic (iRBD) and secondary RBD (sRBD), with the latter being associated with neurological disorders or medications such as antidepressants. Globally, the prevalence of RBD in the general population ranges from 0.5% to 1.25%, while in older individuals, the rate doubles to approximately 2% [4]. Although the exact pathogenesis of RBD has not been fully elucidated, it is widely recognized that it primarily involves dysfunction in the pontine and midbrain regions of the brainstem. In addition, environmental risk factors (occupational hazards such as fine particulate matter in the air with a diameter of 2.5 µm or less (PM2.5), pesticides, or heavy metals) may also exacerbate neurodegenerative processes through mechanisms such as oxidative stress and neuroinflammation, which may promote the pathogenesis of RBD [5].
This review aims to provide an integrative overview of RBD pathophysiology, explore potential biomarkers for early detection, and discuss the latest therapeutic strategies to mitigate the impact on patients’ quality of life.
For this narrative review, a comprehensive literature search was performed using PubMed (https://pubmed.ncbi.nlm.nih.gov/), Web of Science (https://webofscience.com), and ScienceDirect (https://www.sciencedirect.com/), for articles published between 2000 and 2025. Key search terms included “REM sleep behavior disorder”, “pathogenesis”, “biomarkers”, and “therapeutic strategies”. The selection focused on original research and review articles, emphasizing scholarly relevance. The authors examined the included papers for their relevance to the narrative review, resulting in the inclusion of 129 publications. Although earlier works (pre-2000) provide seminal observations on REM sleep atonia circuitry and parasomnia phenomenology, these foundational findings have since been summarized and expanded in more recent reviews. Consequently, limiting the reference search to 2000–2025 captures the consolidated evidence while avoiding redundancy.
During REM sleep, cholinergic neurons in the pontine reticular formation and
midbrain exhibit heightened activity [6]. Glutamatergic neurons in a small
pontine nucleus located ventral to the laterodorsal tegmental nucleus (LDT) play
a key role in muscle atonia. In rats, this nucleus is termed the sublaterodorsal
tegmental nucleus (SLD), while in humans, it corresponds to the SubCoeruleus
(SubC) nucleus [7]. In rats and mice, the absence of glutamatergic neurons or
impairment of their neurotransmission in SLD leads to the loss of muscle atonia
and the emergence of abnormal RBD-like behaviors during REM sleep [8, 9]. Recent
studies have demonstrated that SLD glutamatergic neurons play a critical role in
maintaining muscle atonia during REM sleep [8, 10]. Beyond atonia, these neurons
are now also recognized as the key trigger of REM sleep themselves. Crucially, a
pontine-medullary loop involving reciprocal connections between
corticotropin-releasing hormone binding protein (CRHBP)-expressing glutamatergic
neurons and Nos1-expressing neurons in the dorsomedial medulla (dorsal
paragigantocellular nucleus/prepositus hypoglossal nucleus, DPGi/Pr) constitutes
a core circuit for REM sleep induction and maintenance. Optogenetic activation of
this loop potently initiates REM sleep, while its inhibition suppresses REM
episodes [11]. In addition to the SLD (a REM-on region), the pontomesencephalic
junction, the ventrolateral periaqueductal gray (vlPAG), the adjoining
lateral pontine tegmentum (LPT), and the lateral hypothalamus (LH) contain
numerous REM-off neurons that inhibit REM sleep [12]. The subset of “REM-off”
The vlPAG/LPT GABAergic neurons also project to multiple other regions, including the LDT and locus coeruleus (LC), both of which are involved in regulating REM sleep. Notably, REM sleep inhibition has been observed following the activation of vlPAG/LPT GABAergic terminals in the LH [12]. Additionally, most orexin (Orx)-producing neurons in the LH inhibit REM sleep by activating vlPAG/LPT neurons, which suppress REM sleep initiation [13]. However, approximately 8% of these neurons may promote or stabilize REM sleep by directly influencing the SLD, thereby highlighting their dual regulatory role [13].
The pathology of RBD is multifactorial, arising from dysregulation of brainstem
nuclei that control REM sleep atonia, frequently associated with
The fundamental mechanism of RBD involves the disruption of brainstem-to-spinal cord pathways that maintain muscle paralysis during REM sleep. Recent studies have shown that projections from glutamatergic neurons in the SubC nucleus (equivalent to the SLD in mice) to the ventromedial medulla (VMM) and spinal cord form the basis of REM sleep atonia in skeletal muscles [13]. Tracing studies have confirmed that glutamatergic neurons in the directly project to GABAergic and glycinergic (Gly) neurons in the VMM, inducing glycine-mediated spinal cord postsynaptic inhibition of motoneurons. Additionally, glutamatergic neurons in the SubC nucleus directly project to layer VII of the spinal cord, which contains GABAergic/glycinergic interneurons [14, 16].
The specific loss or inactivation of GABAergic/Gly neurons in the VMM results in REM sleep without atonia, characterized by exaggerated phasic twitching and involuntary motor activity. Furthermore, experiments in mice have shown that the absence of glutamatergic neurons in the SLD or disrupted glutamatergic neurotransmission from SLD neurons can lead to REM sleep without atonia and dream-enacting behaviors [8, 9, 16]. Therefore, the core pathophysiological mechanism underlying RBD involves the disruption of the descending SLD-VMM-spinal cord pathway, leading to a failure of glycinergic/GABAergic inhibition of motor neurons during REM sleep. Neurodegeneration involving glutamatergic SubC nucleus and/or GABAergic/glycinergic medullary neurons may disrupt these inhibitory systems, ultimately compromising the ability to induce muscle relaxation during REM sleep [17]. Thus, the core pathophysiological mechanism underlying RBD involves the disruption of the descending SLD-VMM-spinal cord pathway, leading to a failure of glycinergic/GABAergic inhibition of motor neurons during REM sleep (Fig. 1).
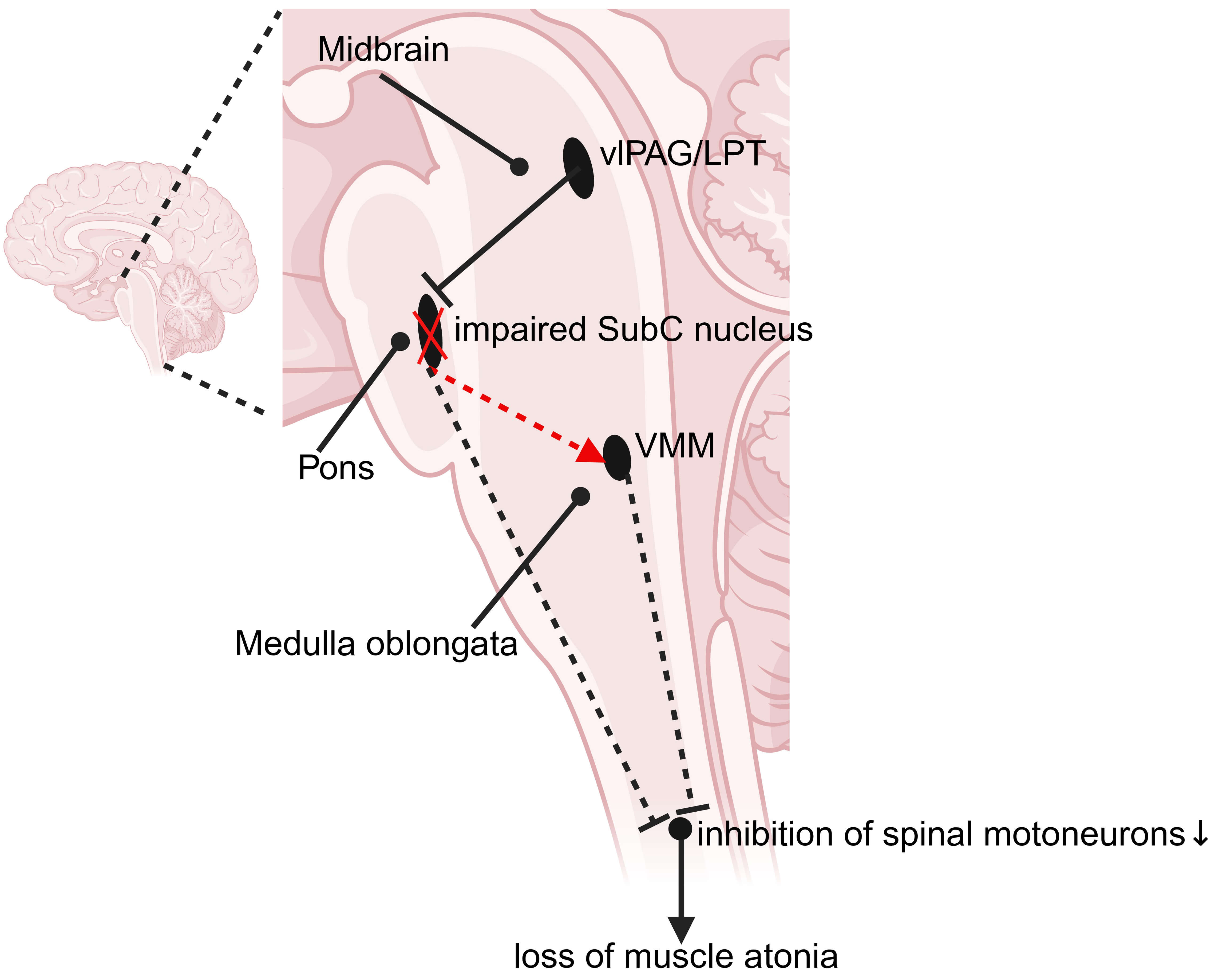 Fig. 1.
Fig. 1.
Circuit dysfunction underlying RBD-like behavior disorder in the human brain. Loss or dysfunction of glutamatergic neurons in the SubC nucleus disrupts glutamatergic neurotransmission, failing to activate GABAergic and glycinergic neurons in the VMM. As a result, the inhibitory control over spinal motor neurons is compromised, leading to their aberrant activation and the subsequent loss of muscle atonia during REM sleep. LPT, lateral pontine tegmentum; RBD, rapid eye movement sleep behavior disorder; REM, rapid eye movement; SubC nucleus, SubCoeruleus nucleus; vlPAG, ventrolateral periaqueductal gray; VMM, ventromedial medulla. The black solid line represents functional inhibitory synapses. Red and black dashed lines indicate excitatory and inhibitory synaptic pathways with impaired or ineffective transmission, respectively. Black solid arrow: spinal motoneurons project to skeletal muscles. Created with biorender (https://www.biorender.com/); Agreement number: KD285OGHOM.
Peripheral
Gastrointestinal biopsies show that abnormal
The precise causal link between
As neuropathological aggregates propagate rostrally through the brainstem, they
ultimately infiltrate the SLD in rats (homologous to the SubCoeruleus, SubC, in
humans), which is a core component of the REM sleep regulatory circuit [26]. This
nucleus contains a population of glutamatergic “REM-on” neurons that, during
normal REM sleep, become active and send excitatory projections to
glycinergic/GABAergic premotor neurons in the ventral medulla and spinal cord.
This excitatory drive is essential for inducing hyperpolarization and atonia in
somatic motoneurons. The accumulation of
In RBD patients,
In addition, several pathways have been identified to facilitate
In summary, when
Non-
In recent years, an increasing number of case reports have demonstrated that acute-onset RBD is closely associated with focal lesions in the pontine tegmentum caused by ischemia, tumors, hemorrhage, demyelination, or inflammation [36]. For example, lesions in the right pontine tegmentum cause RBD, characterized by the loss of muscle atonia during REM sleep and dream-enacting behaviors. In one case, a right pontine infarct led to RBD symptoms that were alleviated by clonazepam [37]. Similarly, ischemic stroke affecting the left rostrodorsal pons can disrupt the LC and LPT regions, interfering with REM sleep pathways and visual processing [38].
Acute encephalitis affecting the brainstem can also lead to inflammatory lesions in the dorsomedial pontine tegmentum characterized by inflammatory infiltration, neuronal loss, and gliosis. Patients with such lesions often develop RBD, supporting the hypothesis that inflammatory damage in this region can induce acute RBD [39]. Furthermore, in patients with multiple system atrophy associated with RBD, degeneration of the pedunculopontine (PPN) and laterodorsal tegmental nuclei results in the depletion of cholinergic neurons, which is considered a key pathological feature of RBD [40]. Similarly, demyelinating lesions in the dorsal pontine region can disrupt pathways that regulate skeletal muscle tone and movement suppression during REM sleep, resulting in the loss of muscle atonia [41].
Although less frequently implicated than in the brainstem, cortical lesions can also affect sleep regulation. Inflammatory demyelinating diseases, such as multiple sclerosis, can lead to lesions in white and grey matter, including regions involved in sleep control [42]. Degenerative changes in subcortical structures, such as the thalamus, putamen, and amygdala, have been observed in patients with RBD. These regions are involved in motor control and emotional regulation during sleep [43]. Neurodegeneration in brainstem nuclei such as the substantia nigra, PPN, and LC may disrupt REM sleep atonia mechanisms, contributing to abnormal motor activation and the hallmark dream-enactment behaviors of RBD [43].
In summary, RBD pathogenesis may result from structural impairments within critical neural networks spanning the brainstem nuclei and cortico-subcortical pathways. These neuroanatomical components, which are essential for mediating muscle atonia during REM sleep, can be compromised by various pathological mechanisms, including ischemic damage, inflammatory processes, neurodegenerative changes, and demyelinating disorders.
Longitudinal cohort studies indicate that a significant proportion (81%–91%)
of patients with iRBD develop definitive neurodegenerative diseases or mild
cognitive impairment within 14 years of follow-up [44]. RBD has been recognized
as an early manifestation of several neurodegenerative diseases, particularly
disorders associated with
During REM sleep, sleep disruption and neural inflammation may lead to an
imbalance between the production and clearance of
Body-first PD (RBD-positive at the onset of motor symptoms) and brain-first PD
(RBD-negative at the onset of motor symptoms) have been distinguished using RBD
as a clinical identifier [49]. In body-first PD, pathological
Patients with body-first PD typically exhibit more prominent autonomic symptoms,
including upright hypotension and constipation, more frequent pathological
aggregation of
In investigating the progression of iRBD to PD, research identified common and
distinct genetic factors for both disorders (Fig. 2). Genome-wide association
studies have identified five genetic loci associated with RBD, primarily
involving the autophagy-lysosome pathway [53]. A high proportion of patients with
iRBD diagnosed via polysomnography carry pathogenic glucocerebrosidase
(GBA) variants, providing strong evidence of a link between GBA genetic variants and RBD [54]. Mutations in the GBA gene impair
lysosomal glucocerebrosidase activity, potentially disrupting lysosomal
degradation and leading to
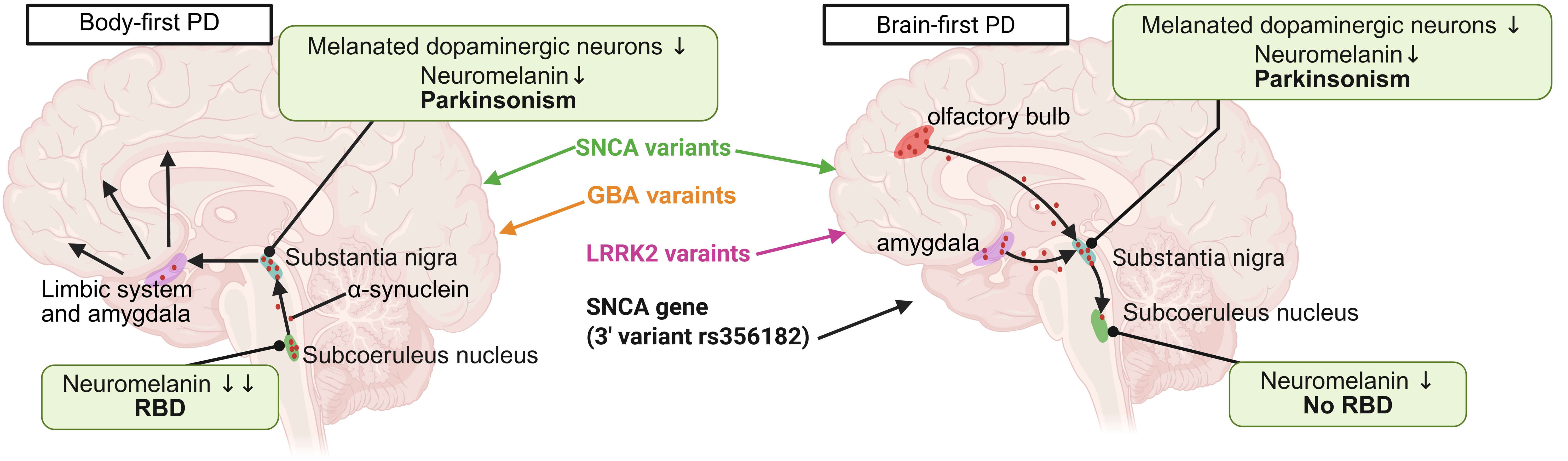 Fig. 2.
Fig. 2.
The relationships between genetic variants and the subtypes of
PD. PD primarily affects dopamine-producing neurons in the substantia nigra, but
the involvement of other brain regions varies among individuals. RBD is linked to
dysfunction in specific brainstem areas controlling muscle atonia during REM
sleep, such as the SubCoeruleus nucleus and pontine tegmentum, and if these areas
remain unaffected, RBD may not develop. GBA variants are more commonly associated
with body-first PD, while LRRK2 variants are predominantly linked to brain-first
PD, though some overlap exists. The SNCA variants are evenly distributed across
the two PD subtypes. Notably, the SNCA gene (3′ variant
rs356182), while linked to an increased PD risk, has been suggested to reduce
the likelihood of RBD. Black arrows indicate the direction of spread of
However, not all patients with PD develop RBD, as disease manifestation varies among individuals. The prevalence of RBD in patients with PD is approximately 40% [56]. Differences in affected brain regions and genetic factors may explain why some patients do not develop RBD [19, 49, 54]. Furthermore, disease subtypes and progression patterns influence RBD occurrence in PD [3, 57]. Clinical heterogeneity suggests that PD comprises multiple subtypes, with some forms predominantly affecting motor pathways while sparing sleep regulation centres [57]. Additionally, symptom timelines vary; RBD may emerge before, during, or after the onset of motor symptoms, meaning some patients develop RBD later in the disease course [3].
iRBD is considered an important pre-clinical indicator of PD and DLB. Long-term follow-up studies indicate that approximately 95% of patients with iRBD will eventually be diagnosed with DLB and PD, with each occurring at roughly equal rates (45%), while an additional 5% develop MSA [58, 59]. DLB and PD are often discussed together because of their overlapping pathological features, both involving dopaminergic innervation loss in the nigrostriatal pathway, as observed on dopamine transporter imaging. However, patients with DLB exhibit a more uniform and symmetrical reduction in dopaminergic activity across the putamen and caudate nuclei. As PD progresses, the dopaminergic deficit becomes increasingly symmetric, making dopamine imaging patterns resemble those seen on DLB [60].
The association between DLB and iRBD primarily highlights marked cognitive
decline, particularly in attention and executive function, as assessed by
specific tests such as the Trail Making Test B and immediate recall in word
lists. In contrast, iRBD-associated PD manifests as early impairments in a
broader range of cognitive domains, including attention, executive function,
memory, and visuospatial abilities [61]. Patients with DLB and RBD may exhibit a
‘bottom-up’ disease progression pattern, in which
MSA is a progressive neurodegenerative disorder characterized by a heterogeneous combination of autonomic failure, cerebellar syndrome, and Parkinsonian features that respond poorly to levodopa and pyramidal signs [66]. Patients presenting primarily with Parkinsonian features are classified as MSA-P, typically characterized by striatal degeneration, whereas individuals with predominant cerebellar ataxia are classified as MSA-C, commonly associated with olivopontocerebellar atrophy [67].
The development of RBD in MSA occurs when pathological changes affect the LDT, PPN, and LC [68]. Study indicates that RBD is highly prevalent in MSA, with up to 90% of patients experiencing RBD at some stage of the disease course. No significant difference in prevalence has been observed between MSA-P and MSA-C subtypes [69]. However, RBD preceding motor symptom onset tends to be more severe in patients with MSA-C than in patients with RBD developing later in the disease course [70].
The pre-RBD patients develop RBD before MSA, and MSA tends to initially present
with autonomic dysfunction (e.g., orthostatic hypotension, urinary issues), rather than typical motor symptoms of Parkinsonism. These patients are more
likely to develop stridor, pyramidal signs, and urinary dysfunction at an earlier
stage, and tend to have a shorter overall disease duration [71]. The post-RBD
patients develop RBD after the onset of MSA, typically following the emergence of
autonomic or motor symptoms [71]. Compared with the post-RBD patients, the
pre-RBD patients have a lower incidence of typical motor symptoms of Parkinsonism
not only at disease onset but also throughout the disease course [71]. The
SNCA gene encodes
Current understanding remains limited by the inability to predict or differentiate specific phenotypic transformations in individuals with iRBD. The duration of RBD varies considerably, with some patients progressing within months while others remain stable for decades [45]. Addressing this challenge requires targeted therapies leveraging biomarkers identified in prior studies. A summary of key candidate biomarkers categorized by their biological or clinical nature is provided in Table 1. (Ref. [64, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86]). The following sections explore the most extensively researched markers in this context.
| Category | Biomarker | Method of detection | Key genetic variant/locus | Phenoconversion endpoint | Sensitivity (%) | Specificity (%) | Ref |
| Molecular | RT-QuIC | / | PD/DLB | 90.4 | 90.0 | [75] | |
| SAA | / | PD/DLB/MSA | 63.6 | 90.3 | [76] | ||
| NfL | Simoa HD-1 | / | PD/DLB/MSA | 75.0 | 83.3 | [73] | |
| Plasma p-Tau181/A |
Elecsys® immunoassays, cobas® e 601 and e 801 analyzers | / | DLB | 89.0 | pTau181: 73.0 A |
[77] | |
| Genetic | SNCA-AS1 | GWAS + eQTLs colocalization | rs3756059 (SNCA 5′ region) | PD/DLB/MSA | / | / | [64] |
| sGBA | Genetic screening/Sequencing | LP444, c.8insG4, IVS2+1G |
Presence of RBD in PD patients | / | / | [74] | |
| MAPT | Genetic Genotyping | rs12185268 | / | / | / | [78] | |
| Imaging | DAT SBR-Putamen | DAT-SPECT Imaging | / | PD/DLB/MSA | 86.0 | 83.0 | [79] |
| VMAT2 binding (SUVR) -Posterior Putamen | 18F-AV-133 VMAT2 PET imaging | / | PD | / | / | [80] | |
| PDRP Expression (Network Activity)- Pons | 18F-FDG PET or ECD SPECT | / | PD/DLB | / | / | [81] | |
| Free-water value- Posterior Substantia Nigra | dMRI; Free-water imaging | / | PD or other synucleinopathy | 61.8 | 75.0 | [82] | |
| Iron overloa- Nigrosome 1 | QSM | / | PD/DLB | / | / | [83] | |
| Electro-physiological | RWA (mentalis) | vPSG + EMG (mentalis) | / | PD/DLB | 68.4 | 80.0 | [84] |
| alpha phase synchronization increased | HD-EEG during resting-state wakefulness | / | PD/DLB | / | / | [85] | |
| Probable napping episodes (during active period) | 7-day actigraphy (Actiwatch Spectrum Plus) | / | PD/DLB/MSA | / | / | [86] |
Notes: CSF, cerebrospinal fluid; DLB,
dementia with Lewy bodies; DAT, Dopamine Transporter; SBR, specific binding ratio; SPECT, Single-Photon
Emission Computed Tomography; dMRI, diffusion magnetic resonance imaging; eQTLs,
expression quantitative trait loci; ECD SPECT, ethylcysteinate dimer
single-photon emission computed tomography; EMG, electromyography;
18F-AV-133 VMAT2 PET, fluorine-18 AV-133 vesicular monoamine transporter 2
positron emission tomography; 18F-FDG PET, fluorine-18 fluorodeoxyglucose
positron emission tomography; GWAS, genome-wide association study; HD-EEG,
high-eensity electroencephalography; LP444, c.8insG4,
IVS2+1G
Alpha-syn plays a central role in RBD pathogenesis, and its aberrant aggregation is a key indicator of disease progression. Current therapeutic approaches increasingly focus on preventing the early stages of protein aggregation, particularly protofibril formation [87].
In vitro studies have demonstrated that 4-arylidene curcumin and
bis-chalcone polyphenols effectively inhibit
Recent investigations into the gut microbiome suggest that oral administration
of resveratrol-selenium peptide nanocomposites can modulate specific bacterial
taxa, such as Desulfovibrio, potentially reducing
The peripheral clearance mechanisms play an important role in
Accurate detection of
These advancements in understanding and detecting
Neurofilament light chain (NfL), a cytoskeletal protein predominantly expressed in large-calibre myelinated axons, has provided valuable insights into neuronal injury mechanisms. Its detection in CSF offers promising avenues for diagnosing and monitoring neurodegenerative disorders [96]. A recent study investigated the potential of plasma NfL levels and cardiac metaiodobenzylguanidine imaging as predictors of phenotypic transformation in patients with iRBD. Baseline plasma NfL levels were significantly elevated in patients with RBD and MSA. The sensitivity and specificity of NfL concentrations exceeding 21.3 pg/mL in predicting the transformation of iRBD to MSA were 100% and 94.3%, respectively. Thus, elevated plasma NfL levels are associated with conversion to MSA, whereas decreased cardiac metaiodobenzylguanidine uptake is indicative of conversion to Lewy body dementia [73, 97].
Moreover, neither olfactory nociceptive sensitization nor a quantitative assessment of striatal dopamine uptake appears to reliably predict phenotypic transition, contradicting previous studies [98, 99]. This challenges the conventional view of these biomarkers and underscores the need to refine and optimize existing biomarker combinations. Based on these findings, controlling plasma NfL levels may be a feasible therapeutic approach for RBD. For instance, nonsteroidal anti-inflammatory drugs have been shown to reduce NfL release following nerve injury. Similarly, neuroprotective agents such as butylphthalide, which mitigate oxidative stress and inflammation, may help lower NfL levels and slow disease progression.
As research advances, NfL has emerged as a key biomarker for predicting and monitoring various neurological disorders. Recent studies have emphasized several detection assays, including traditional enzyme-linked immunosorbent assay (ELISA), and single-molecule array (Simoa) technologies [100, 101]. These techniques have not only improved the early diagnosis and monitoring of neurological disorders but have also enhanced our understanding of the role of NfL in neuropathology. Investigating NfL as a biomarker paves the way for early diagnosis and treatment of RBD while establishing a scientific foundation for understanding the interplay between sleep disorders and cognitive function.
In patients with iRBD, REM sleep without atonia (RWA) has been established as a
reliable electrophysiological biomarker for predicting phenotypic conversion to
synucleinopathy [102]. The current assessment protocol uses video-polysomnography
(vPSG) combined with advanced computer algorithms to accurately calculate the
percentage of submental muscles and bilateral upper limb muscles (flexor
digitorum superficialis) containing excessive electromyography (EMG) activity,
which has been rigorously developed and widely accepted by the international RBD
research group [103]. Compared with single-channel monitoring, multi-channel
monitoring can provide more comprehensive muscle activity information in the
diagnosis of RBD, significantly improve the accuracy of diagnosis, and ensure
accurate identification of RBD patients. Continuous advances in technology have
further optimized the RWA analysis process. The introduction of deep learning
algorithms enables RWA analysis to achieve a high degree of agreement with expert
visual scoring, while significantly reducing the analysis time, which strongly
promotes the widespread application of this technology in clinical practice [104, 105]. In one study, 221 consecutive patients with clinically suspected RBD
underwent DAT single photon emission CT (SPECT) and vPSG. vPSG confirmed 176
patients with RBD, and most RWA parameters were significantly correlated with
Dopamine Transporter Single-Photon Emission Computed Tomography (DAT-SPECT)
ratios. Therefore, the range of RWA values is important to estimate whether
patients are in the early, middle, or late prodromal phase of
In addition to the molecular and electrophysiological biomarkers discussed above, genetic factors play a crucial role in the pathogenesis and progression of RBD. Genome-wide association studies and single-gene mutation analyses have identified several genetic variants associated with synucleinopathies and increased risk of developing RBD. Notably, variants in the SNCA gene, which encodes alpha-synuclein, and the GBA gene, encoding glucocerebrosidase, are among the most well-established genetic markers.
The SNCA Antisense RNA 1 (SNCA-AS1) variant (rs3756059) has been implicated through genome-wide association study (GWAS) and colocalization analyses, highlighting the role of SNCA in RBD and its progression to PD, DLB, or MSA [64]. Similarly, GBA pathogenic variants (such as L444P, c.84insG, and others) are strongly associated with the presence of RBD in Parkinson’s disease patients, further supporting the link between lysosomal dysfunction and alpha-synuclein aggregation [74]. These genetic biomarkers not only enhance our understanding of RBD etiology but also offer potential targets for personalized therapeutic strategies.
Recent research trends show a shift from the traditional detection of RWA to the collaborative application of multi-dimensional indicators to improve the ability of early diagnosis and pathological mechanism analysis. At the REM sleep microarchitecture level, refined electroencephalography (EEG) oscillation analysis revealed the core features of brain stem-thalamo-cortical circuit dysregulation: proportion of phasic REM in iRBD patients is significantly increased, indicating excessive activation of the motor commands system, while the density of sawtooth waves and the abnormal correlation between sawtooth waves and ponto-geniculo-occipital waves in PD-RBD patients reflect the dysfunction of thalamo-cortical information integration. These findings confirm that REM sleep disorders in RBD not only involve muscle tone loss and dream behavior interpretation but also extend to specific electrophysiological characteristics [107]. Such microstructure parameters can be obtained by secondary mining of PSG data, which has high specificity and clinical practicability.
At the level of cortical degeneration, cortical mean diffusivity (cMD) derived
from diffusion MRI, as a new imaging marker, is significantly more sensitive to
neuronal microstructure damage than traditional cortical thickness measurement. A
study has shown that the cMD values of PD-RBD patients are increased in the left
superior temporal gyrus, superior frontal gyrus, precentral gyrus, precuneus, and
the right middle frontal gyrus, postcentral gyrus, and paracentral lobule [108].
In addition, although prolonged REM latency is not a specific marker of
Symptomatic treatment for RBD is crucial for preventing sleep-related falls or injuries; however, only a limited number of patients with RBD seek medical attention. Current treatment guidelines include two main categories: nonpharmacological and pharmacological interventions. Nonpharmacological approaches primarily involve comprehensive environmental modification and safety counselling to reduce injury risk, which are the cornerstone of non-pharmacological management. Recommended measures involve placing mattresses on the floor or lowering the bed height, padding furniture edges, securing windows, and removing potential hazards (such as sharp objects and fragile items) before bedtime [110]. It is also advised to maintain a certain distance from the bed partner or even sleep in separate rooms for safety [111]. Beyond environmental adjustments, establishing good sleep hygiene is important. This includes maintaining a regular sleep schedule (adhering to fixed bedtimes and wake-up times), and avoiding napping, compensating for sleep, or lying in bed for extended periods while awake. Incorporating moderate physical activity, such as daily mindful movement exercises or aerobic exercise for about an hour, may also be beneficial for overall sleep quality [112]. For anxiety and psychological burden associated with RBD, relaxation techniques like progressive muscle relaxation (PMR) and mindfulness practices (e.g., breath watching and body scanning) can help patients relax and reduce excessive focus on symptoms. Although psychotherapy (e.g., cognitive behavioral therapy, CBT) has been explored, current evidence for its efficacy in reducing RBD symptoms remains limited [113].
A summary of pharmacological treatments is shown in Table 2 (Ref. [114, 115, 116, 117, 118, 119]). Pharmacological treatment includes first-line agents such as clonazepam and melatonin, as well as additional agents, including pramipexole, ramelteon, rotigotine [3, 120], and clonazepam, a benzodiazepine, enhances the activity of the inhibitory neurotransmitter GABA, leading to chloride influx, neuronal hyperpolarization, and inhibition of postsynaptic potentials. This process suppresses central nervous system activity, producing anticonvulsant, anxiolytic, and muscle relaxant properties. Clonazepam effectively reduces RBD symptoms by inhibiting electromyographic activity during REM sleep, with reported clinical efficacy rates up to 90% [114]. However, it may cause side effects, including falls and related injuries [121].
| Drug | Dose (mg/day) | Benefits | Side effects | Targeted patients | Ref |
| Clonazepam | 0.25–2.00 | Effective inhibition of neuronal excitability and muscle activities during REM sleep | Daytime excessive sedation, impotence, dyskinesia, confusion, memory loss, etc. | Patients with confirmed RBD, but it should be used with caution in those with concomitant dementia, gait disorders, or OSA | [114, 117] |
| Melatonin | 3.00–12.00 | Improvements of sleep quality and regulation of circadian rhythms with neuroprotective effects | Morning headaches, daytime sleepiness, delusions and hallucinations | RBD patients with DLB, Parkinson’s disease, and MSA | [115] |
| Pramipexole | Reduction of the potential side effects of clonazepam in older adults or patients with RBD with sleep disordered breathing | Patients with DLB may experience hallucinations and delusions | Patients with RBD not diagnosed with neurodegenerative diseases | [116] | |
| Rotigotine | Limited data | Significant improvements in morning motor function, sleep quality, and non-motor symptoms | No information available | PD patients with concomitant RBD | [116] |
| Paroxetine | 10.00–40.00 | Reduction of RBD symptoms by reducing REM sleep | Nausea, dizziness, diarrhea, thirst, etc. | Patients with RBD who have not yet developed neurodegenerative disease | [118] |
| Donepezil | 10.00–15.00 | Improvement of cognitive function | Seizures due to cholinergic effects | RBD patients with mild cognitive impairment, PD, or DLB | [119] |
| Alprazolam | 1.00–3.00 | Anxiolytic and sedative effects may be more effective in RBD patients with concomitant anxiety symptoms | No information available | Patients with RBD who have not yet developed neurodegenerative disease | [117] |
| Temazepam | 10.00 | Sedative and anti-anxiety effects | No information available | RBD patients with symptoms of insomnia | [117] |
| Desipramine | 50.00 | Effective inhibition of REM sleep | No information available | Secondary RBD | [117] |
OSA, obstructive sleep apnea.
Melatonin is an endogenous hormone that regulates the sleep-wake cycle and modulates muscle tone during sleep by enhancing GABAergic inhibitory effects [122]. Experimental studies confirm its efficacy in reducing clinical behavioral manifestations and muscle tone during REM sleep. Moreover, melatonin has a more favorable safety and tolerability profile than does clonazepam [115]. Schaefer et al. [123] found that prolonged-release melatonin improved RBD symptoms coexisting with obstructive sleep apnea syndrome. However, neither clonazepam nor melatonin significantly alters sleep architecture [124].
Recent studies suggest that prazosin, an
Nonetheless, the role of dopamine agonists in ameliorating the symptoms of RBD remains controversial. Short- to medium-term randomized clinical trials involving small cohorts have reported variable efficacy for rivastigmine, memantine, 5-hydroxytryptophan, and the herbal medicine yokukansan. Additionally, the excitatory effects of selective serotonin reuptake inhibitors, such as paroxetine, on serotonin receptors may influence cortical functions related to mood and sleep regulation, although no direct experimental evidence supports their role in RBD treatment. However, previous studies suggest that paroxetine improves sleep quality and depressive symptoms in patients with comorbid sleep disorders [126]. Trazodone, a medication widely used for treating insomnia and other symptoms, has demonstrated sedative effects and potential benefits in improving early symptoms of attention deficit disorders, REM sleep abnormalities, concussion, and olfactory memory deficits [127]. Its role in modulating early tau pathology suggests it may slow disease progression. This highlights the need for further research into the interaction between RBD and tau proteins, particularly in validating the predictive role of tau proteins in phenotypic transitions [128]. Barrow and Vendrame [129] recently reported significant clinical improvement in three iRBD cases following trazodone treatment, reinforcing its potential therapeutic role. Further details on pharmacological treatments are summarized in Table 2.
It is noteworthy that although the aforementioned medications demonstrate efficacy in treating RBD, further research is needed to elucidate their mechanisms of action and optimize treatment regimens. Long-term, multicenter, randomized, placebo-controlled clinical trials are essential for evaluating symptomatic and preventive therapies. Additionally, patient-specific factors, such as comorbid sleep disorders (e.g., obstructive sleep apnea, restless legs syndrome), should be carefully considered to ensure timely intervention. Until RBD symptoms are effectively controlled, cohabitating partners may need to sleep in separate rooms for safety. Future studies should explore novel medications and innovative therapies to provide safer and more effective treatment options for patients with RBD.
We reviewed literature on RBD published between 2000 and 2025, providing an
integrated overview of its pathophysiological mechanisms, biomarkers used for
early detection, and therapeutic strategies. This comprehensive approach might
deepen the understanding of RBD’s intricate relationship with neurodegenerative
disorders and underscores the importance of early intervention strategies.
Special emphasis was placed on key biomarkers such as
RBD is increasingly recognized as a prodromal stage for neurodegenerative
diseases, and (e.g.,
Furthermore, this review emphasizes the need for future research to explore multimodal biomarker combinations—integrating neuroimaging, fluid biomarkers, and electrophysiological signals—to enhance diagnostic accuracy and prognosis. Clinical trials targeting environmental risk reduction, such as air pollution and occupational exposure interventions, should also be prioritized to assess their preventive potential.
Regarding pharmacotherapy, clonazepam and melatonin remain first-line treatments for RBD; however, treatment strategies must be individualized to account for disease complexity, comorbidities, and potential side effects. As research progresses, further exploration of therapeutic targets and biomarker-driven interventions will be crucial in refining management strategies for RBD.
HZ: Conceptualization, Methodology, Investigation, Writing—Original Draft. SZ: Investigation, Data Curation, Validation, Writing—Original Draft. HC: Investigation, Data Curation, Writing—Review & Editing. YW: Visualization, Writing—Review & Editing. HG: Validation, Visualization. DY: Conceptualization, Supervision, Project Administration, Writing—Review & Editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.