



 , Yuanxiang Lin 1,2,3,4, Dezhi Kang 1,2,3,4, Tong Zhao 1,2,3,4,*
, Yuanxiang Lin 1,2,3,4, Dezhi Kang 1,2,3,4, Tong Zhao 1,2,3,4,*
1 Department of Neurosurgery, Neurosurgery Research Institute, The First Affiliated Hospital, Fujian Medical University, 350005 Fuzhou, Fujian, China
2 Fujian Provincial Institutes of Brain Disorders and Brain Sciences, First Affiliated Hospital, Fujian Medical University, 350005 Fuzhou, Fujian, China
3 Department of Neurosurgery, National Regional Medical Center, Binhai Campus of the First Affiliated Hospital, Fujian Medical University, 350212 Fuzhou, Fujian, China
4 Fujian Provincial Clinical Research Center for Neurological Diseases, First Affiliated Hospital, Fujian Medical University, 350005 Fuzhou, Fujian, China
Abstract
Amyotrophic lateral sclerosis (ALS) is a complex neurodegenerative disease. No effective treatments have yet been found for ALS, primarily because the molecular mechanisms that underlie its pathogenesis are unknown. Although animal models are suitable for ALS research, species differences between these models and human spinal cord organs make it difficult to accurately predict the progression of disease in humans. Therefore, the development of more suitable models is urgently needed. Human stem cells have unlimited development potential and can be used to make three-dimensional organoid structures that mimic the architecture and function of actual organs. Organoid models can be used to overcome some of the species differences and accelerate experimental research, leading to the development of practical applications for the treatment of ALS. This article discusses the pathological mechanisms and cell types involved in ALS, as well as the genes associated with this disease. We also discuss the possible applications of spinal cord organoids (SCOs) in ALS research, such as the modeling of disease characteristics, study of pathological mechanisms, and drug screening. Finally, the prospects for SCOs in ALS treatment are highlighted, while acknowledging the need for further development of relevant technologies.
Keywords
- amyotrophic lateral sclerosis
- spinal cord
- organoids
- application
- prospect
- review
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the degeneration of upper and lower motor neurons [1], leading to gradual muscle weakness, atrophy, and ultimately paralysis and respiratory failure [2]. The pathogenesis of ALS is complex and involves a variety of abnormal processes, such as protein misfolding, defects in RNA metabolism, mitochondrial dysfunction, and cytoskeletal abnormalities [3]. Despite significant advances in basic research on ALS, the prognosis for patients is still poor, with an average survival time of 3 to 5 years due to the lack of effective treatments [4, 5].
Traditionally, ALS research has relied primarily on animal models, particularly mutant mouse models [6]. However, due to physiological differences between species, animal models have a limited ability to mimic human ALS pathology, making it difficult to translate findings into potential treatments [7]. With the development of stem cell technology in recent years [8], three-dimensional (3D) organoid models based on induced pluripotent stem cells (iPSCs) have attracted widespread attention [9, 10, 11]. The spinal cord organoid (SCO) can reproduce the cellular structure and function of the spinal cord in vitro. This overcomes the limitation of traditional cell cultures not being able to reproduce the complex phenotype of the natural spinal cord, while also eliminating the problem of interspecies differences between humans and animals [12]. SCOs therefore provide an innovative platform for research into the pathological mechanism of ALS, screening for therapeutic drugs, and exploring personalized medicine [13, 14]. In this article, we review the recent advances and prospects for SCOs in ALS research, as well as discuss the remaining challenges and future directions.
One of the most important findings in ALS pathology is the demonstration that disease onset and progression are influenced by multiple cell types and genes. This has been well-described in much of the literature and is important for understanding ALS disease mechanisms [15].
The pathological processes associated with ALS can be broadly classified into five main categories: abnormal protein aggregation, impaired RNA metabolism, cytoskeletal and transport defects, mitochondrial dysfunction, and endosomal pathway defects [1].
An important feature of ALS is the aggregation of abnormal proteins, such as transactive response (TAR)
DNA-binding protein 43 (TDP-43) and fused in sarcoma (FUS) proteins. Aberrant
aggregation of these proteins leads to an imbalance in protein homeostasis and
autophagy dysfunction, resulting in intracellular accumulation of damaged or
misfolded proteins, thereby increasing oxidative stress and impairing neuronal
survival. The aberrant aggregation of FUS protein affects RNA metabolism, and its
zinc finger domain also aberrantly binds to the Sm site of U1 Small Nuclear
RNA (U1 snRNA), which directly and competitively inhibits the assembly of Sm
proteins. This pathological dual-domain binding pattern disrupts snRNP
biosynthesis, while abnormal FUS aggregation under stress conditions solidifies
snRNP intermediates, forming irreversible pathological aggregates and
accelerating neuronal death [16, 17, 18, 19]. Several ALS-associated genes, such
as chromosome 9 open reading frame 72 (C9orf72), TARDBP and FUS cause disruptions in RNA metabolism and
impair normal cell function. Never in mitosis gene A (NIMA) Related Kinase 1 (NEK1) regulates
In addition to the five primary processes in ALS, several secondary mechanisms also play a role, including inflammatory mechanisms. Activation of immune cells, such as abnormal involvement of natural killer cells and T lymphocytes, can lead to neuroinflammation and neuronal damage [25, 26].
ALS patients often exhibit hypermetabolic features, including increased energy expenditure and weight loss. Metabolomics study has shown that several metabolic pathways, such as lipid and amino acid metabolism, are abnormal in ALS patients and may be associated with disease progression [27]. The relevant mechanisms that are thought to be involved in ALS are depicted in Fig. 1.
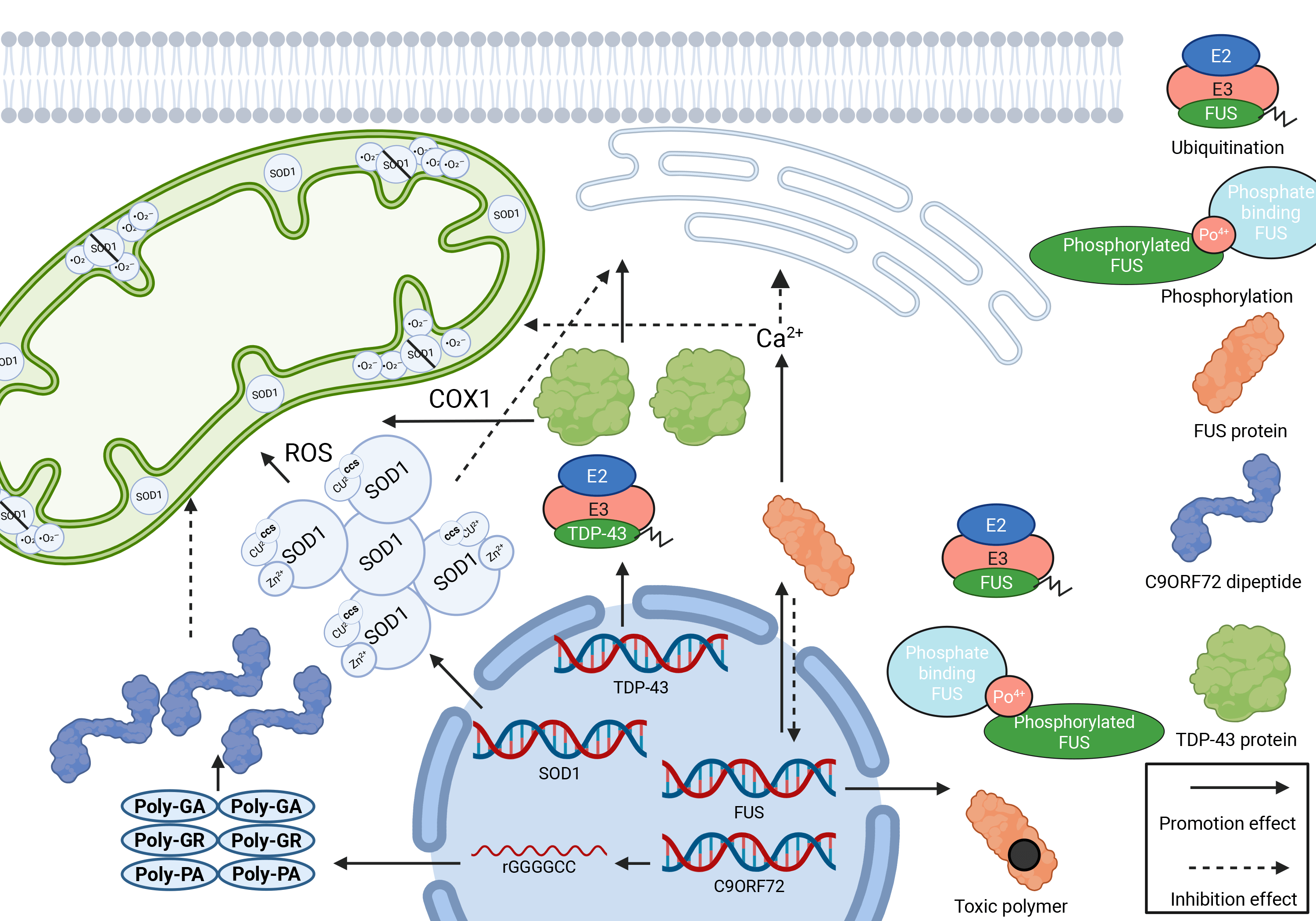 Fig. 1.
Fig. 1.
Molecular pathogenesis of ALS. This image summarizes the core molecular pathological mechanisms of neurodegenerative diseases such as ALS, focusing on three interrelated key processes: mitochondrial oxidative stress, the collapse of protein homeostasis, and failure of the ubiquitin system. ROS, reactive oxygen species; COX-1, cytochrome c oxidase 1; CCS, copper/chaperone for SOD1; GA, glutamine aggregates; PA, proteasomal autophagy; PO, prionic organelles. Created in https://BioRender.com.
The pathogenesis of ALS involves a variety of abnormal cells [28]. The possible roles of such cells in ALS in various mouse models and humans are summarized in Table 1.
| Cell type (Model) | SOD1 mouse | TDP-43 mouse | FUS mouse | C9orf72 mouse | OPTN mouse | MATR3 mouse | TBK1 mouse | Human ALS (C9orf72 mutant) |
| Motor Neurons | Rapid degeneration, mitochondrial dysfunction, elevated NfL | Local protein aggregation, impaired TDP-43 transport | Spinal motor neuron loss, FUS aggregates | RNA toxicity, late motor neuron death | Slow degeneration linked to OPTN mutation | RNA metabolism disruption, nuclear-cytoplasmic barrier breakdown | Autophagy impairment, neuroinflammation | Gradual degeneration, elevated NfL, mitochondrial dysfunction |
| Astrocytes | Rapid activation, glutamate release, increased TNF- |
Impaired support, mild IL-6 increase | Mild inflammation, late-stage effects | Metabolic dysfunction, moderate inflammation | Decreased function, increased inflammatory markers | Reduced support, oxidative stress | Early dysfunction, autophagy impairment | Inflammatory cytokine secretion, exacerbating neuron damage |
| Microglia | Rapid activation, strong inflammatory response (IL-1 |
Slow activation, localized late inflammation | Late activation, FUS involvement in inflammation regulation | Slow activation, involvement in metabolic and inflammatory regulation | Mild inflammation, worsening over time | Strong inflammatory response, exacerbating neurodegeneration | Early activation, high cytokine release | Early activation, inflammation worsens disease |
| Oligodendrocytes | Reduced myelination, early demyelination | Mild dysfunction, slower neural transmission | Intact myelination, mild functional loss | Gradual demyelination, slower signal conduction | Impaired myelination, slower neural signals | Abnormal myelination, reduced support | Inflammation-induced oligodendrocyte loss | Gradual demyelination, severe late-stage myelin loss |
| Muscle Cells | Rapid muscle atrophy following neuron loss, muscle weakness | Localized atrophy, worsening later | Slow muscle atrophy, lower limb weakness | Mid-stage atrophy, reduced muscle tone | Mild atrophy, worsening over time | Atrophy linked to nuclear-cytoplasmic barrier breakdown | Muscle atrophy associated with neuron degeneration | Progressive muscle atrophy leading to paralysis as neurons die |
This table summarizes the development status of motor neurons, astrocytes,
microglia, oligodendrocytes, and muscle cells during ALS in the SOD1, TDP-43,
FUS, C9orf72, OPTN, MATR3, and TBK1 mouse models, as well as human ALS. ALS, amyotrophic lateral sclerosis; SOD1,
superoxide dismutase 1; TAR, transactive response; TDP-43, TAR DNA-binding protein 43; FUS, fused in sarcoma; C9orf72, chromosome 9 open reading frame 72; OPTN, optineurin;
MATR3, matrin-3; TBK1, TANK-binding kinase 1; NfL, neurofilament light
chain; IL-6, interleukin-6; TNF-
A central feature of ALS is motor neuron dysfunction, including corticospinal motor neurons and spinal motor neurons [29]. Dysfunction and loss of corticospinal motor neurons are common in ALS patients, but are poorly understood due to the different patterns of corticospinal connections in humans and mice [30]. Dysfunction of spinal motor neurons is manifested by muscle weakness, muscle atrophy, reduced muscle tone, and fascicular tremor, which are typical symptoms of ALS [31]. Furthermore, in some ALS patients, neurons are damaged in the context of frontotemporal lobe dementia (FLD), leading to cognitive and behavioral changes [29] that are pathologically related to TDP-43 deposition [32]. Astrocytes play an important role in ALS, and reactive astrocytes are one of the hallmarks of ALS and other neurodegenerative diseases [33]. ALS astrocytes have been shown to reduce the glutamate transporter excitatory amino acid transporter 2 (EAAT2) and mediate disease progression [34]. Oligodendrocytes also exhibit pathological changes in ALS, including areas of patchy demyelination, proliferation of oligodendrocyte precursor cells, and loss of monocarboxylate transporter protein 1. This may affect their supporting role for motor neurons, thus contributing to the progression of ALS [35, 36]. Previous studies suggested that microglia are mediators of disease progression after onset [37, 38]. Activation of microglia is associated with reactive microglial proliferation in pathological areas of ALS patients [39]. Interestingly, microglia and astrocytes can form a synergistic deterioration mechanism in ALS through a bidirectional inflammatory factor cascade. Kwon and Koh [40] found that pro-inflammatory microglia activate astrocytes and convert them to the A1 phenotype, causing the release of factors such as chemokine (C-C motif) ligand 2 (CCL2), C-X-C motif chemokine ligand 10 (CXCL10), and granulocyte-macrophage colony-stimulating factor (GM-CSF) that further amplify microglial inflammatory responses, constituting a positive feedback loop. It is worth noting that targeting these synergistic mechanisms requires intervention strategies tailored to the disease stage [40]. In addition, non-neuronal cells such as T lymphocytes, myocytes, Schwann cells, NG2 glial cells, and pericytes may also be involved in the pathogenesis of ALS [28, 41, 42, 43, 44]. In this regard, Månberg et al. [45] found that perivascular fibroblasts (PVFs) play a key role in ALS pathology. PVFs are located in the Virchow-Robin spaces around spinal cord vessels and are activated in the pre-symptomatic phase, leading to expansion of perivascular spaces and separation of the basement membrane through secretion of collagen type VI alpha 1 chain (COL6A1) and secreted phosphoprotein 1 (SPP1). These changes may affect blood-brain barrier function, cerebral blood flow, and metabolism, independent of neuronal damage [45]. However, a limitation of these studies is that most of the relevant conclusions were drawn from experiments on mutant superoxide dismutase 1 (SOD1) (mSOD1) mice, and may therefore be less relevant to humans [46].
To date, researchers have identified more than 40 genes associated with ALS [47]. These vary significantly in mutation frequency, inheritance pattern, and epistasis [48]. Mutations in genes such as C9orf72, TARDBP, SOD1, and FUS are among the more common and highly epistatic conditions [49, 50]. However, it is important to note that the presence of variants in some ALS genes, such as angiogenin (ANG), ataxin-2 (ATXN2), and dynactin subunit 1 (DCTN1), may increase an individual’s risk of developing ALS [51]. Fig. 2 provides a general description of the impact of individual gene mutations on ALS.
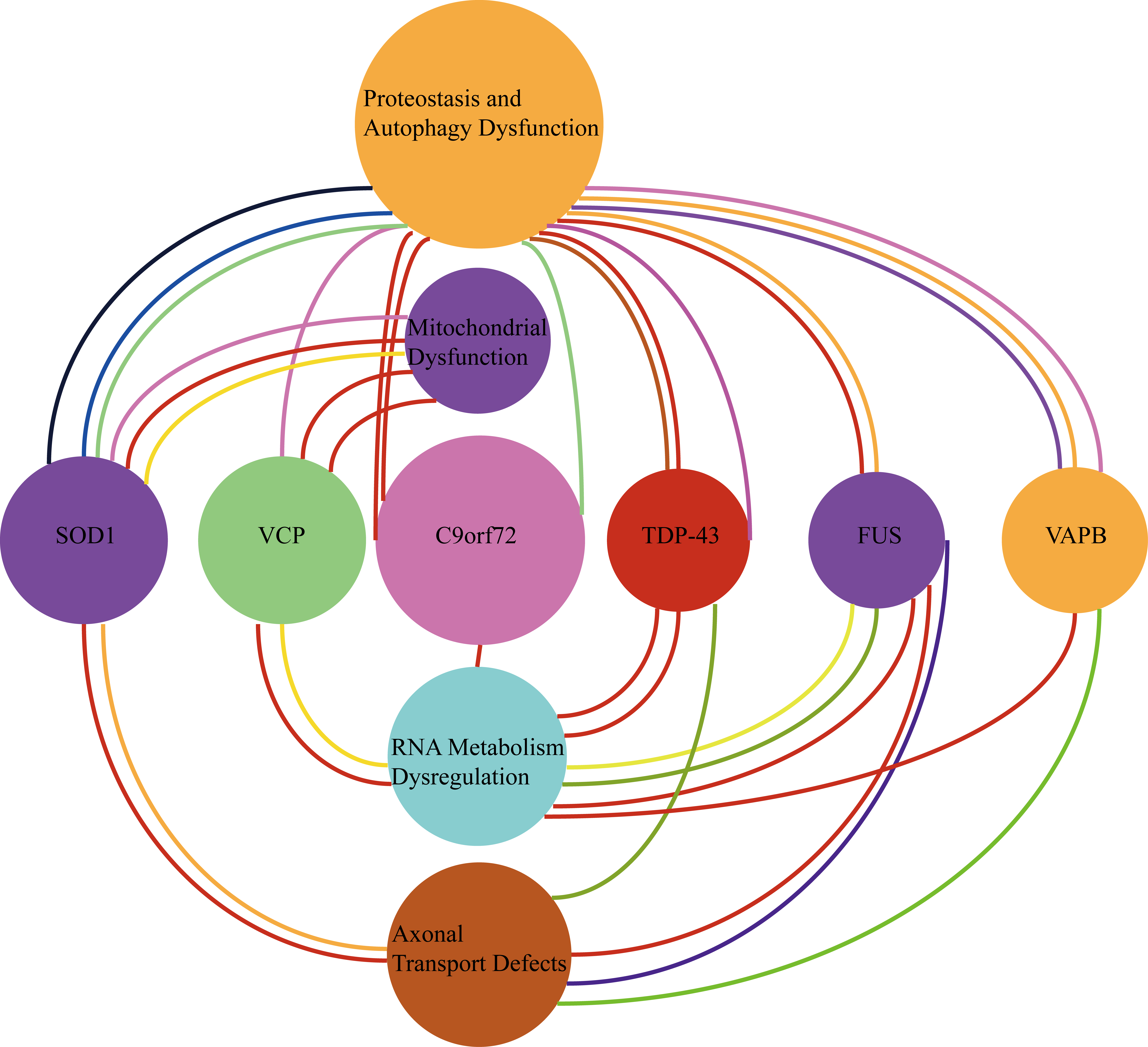 Fig. 2.
Fig. 2.
Gene mutations and their association with ALS. The links between four pathological mechanisms of ALS and specific gene mutations are shown. Most gene mutations are directly linked to protein damage. VCP, valosin-containing protein; VAPB, vesicle-associated membrane protein (VAMP)-associated protein B and C.
In particular, amplification of the C9orf72 gene leads to an increased risk of ALS. Mutations in C9orf72 result in increased toxicity of dipeptide repeat protein (DPR), which may promote heterochromatin abnormalities and TDP-43 aggregation, thereby exacerbating neurodegeneration [52, 53]. C9orf72 mutations have also been associated with medullary morbidity and shortened patient survival [54]. Genes such as TARDBP, FUS and others are associated with impaired DNA repair and impaired RNA metabolism in ALS. They encode DNA/RNA-binding proteins that interfere with normal function when aggregation occurs. Loss of nuclear TDP-43 leads to the accumulation of double-stranded DNA breaks, impairing genomic stability [55, 56, 57]. Mutations in the SOD1 gene have been found in some familial ALS cases [47]. Most of the mutations occur in exons and result in malfunction of the SOD1 protein, leading to mitochondrial dysfunction. This in turn increases oxidative stress and neuronal damage, with some of the mutant proteins acting like prions to induce normal proteins in the cell to aggregate. Cells with abnormal SOD1 aggregates can also infect other normal cells with intracellular SOD1 aggregates [17, 19, 57, 58]. Double amplification of the C9orf72 gene, mutated FUS, and aggregation of TDP-43 have all been shown to impair nucleoplasmic transport [55, 59, 60], while mutations in some genes have been linked to dysfunctional DNA repair [23].
The development of SCOs has opened new possibilities for ALS research and provided a more realistic model for studying the pathogenesis of ALS and testing new therapeutic approaches [61]. Although more studies have been conducted on the brain organs of ALS patients in recent years, most of these have been on cortical brain organs, with spinal cord organs still needing further research [62, 63].
To investigate the pathogenic mechanism of the C9orf72 mutation in ALS,
researchers have constructed a large number of animal models, including rat,
mouse and zebrafish models with mutant C9orf72, as well as C9orf72 knockout
models in rats, mice, zebrafish and the nematode Caenorhabditis elegans [64, 65].
However, due to species variability, some of these animal models did not show
pathological manifestations associated with ALS [66]. To address this issue, Gao
et al. [67] studied spinal cord-like organs derived from induced
pluripotent stem cells (iPSCs), observing pathological changes in neurons and
glial cells similar to ALS. These researchers established C9orf72
gene-silenced iPSCs (C9-iPSCs) by lentiviral transfection, and then induced
their differentiation to mimic ALS disease [61]. Pro-inflammatory factors were
found to be up-regulated in C9-iPSC-derived 2D cells and in 3D-cultured SCOs, and
mature astrocytes were detected by qRT-PCR. The mRNA expression levels of
inflammatory factors such as IL6, IL-1
In the 3D model, NMPs self-organize to form long-term, viable neuromuscular
organoids (NMOs) that persist for over 100 days and gradually mature. Schwann
cells (S100
This model effectively simulates the ALS disease. In an iPSC-derived model of spinal muscular atrophy (SMA), the muscle fibers are disorganized, NMJ numbers are reduced by 60%, and there is arrow poison-resistant muscle contraction. In a myasthenia gravis (MG) patient model, antibody treatment leads to NMJ structural breakdown and loss of contraction function. These findings provide a novel in vivo simulation platform for studying the pathological mechanisms of NMJ dysfunction in ALS.
Protein aggregation (primarily TDP-43), disruption of the nuclear membrane, and
nuclear pore complex homeostasis are considered common pathological mechanisms in
ALS/Frontotemporal Dementia (FTD) [70, 71]. The Linker of Nucleoskeleton and
Cytoskeleton (LINC) complex is the second largest transmembrane complex in the
nuclear membrane and plays a key role in maintaining nuclear homeostasis [72].
Sirtori et al. [73] used SCOs to investigate whether alterations in the
LINC complex play a role in the pathogenesis of ALS/FTD [74]. They quantified the
distribution and nuclear membrane abundance of the spindle aberration deficient 1 (Sad1) and UNCordinated locomotion protein 84 (UNC84)
domain-containing protein 1 (SUN1) and SUN2 proteins by immunofluorescence and
analyzed whole protein extracts with western blotting. The nuclear levels of SUN1
and SUN2 proteins were markedly reduced in C9orf72 motor neurons and showed an
abnormal distribution. This demonstrates impairment of the LINC complex in the 3D
environment of SCOs [74, 75]. Reconstruction of 3D microenvironmental dynamic
systems is clearly a core feature of SCOs [76]. Extracellular matrix (ECM)
components play a key neuroprotective role in ALS through dynamic remodeling
[77]. Using a C9orf72 gene mutation model, Milioto et al. [78]
found the spinal cord motoneuron microenvironment was significantly enriched for
ECM proteins, and that post-polyGR directly activates TGF-
The use of SCOs to screen potential therapeutic drugs is an important direction in ALS research [80, 81]. The efficacy of drugs can be assessed by observing pathological changes associated with the introduction of ALS in spinal cord organ tissues [82], while the mechanism of drug action can also be investigated [61, 74, 83]. Chooi et al. [84] tested the therapeutic effects of unfolded protein response (UPR) inhibitors on ALS pathological changes in SCOs. UPR inhibitors were found to reduce the pathological symptoms of ALS by modulating protein folding and metabolic processes in the cell, thereby reducing the aggregation of dipeptide repeat proteins and autophagy phenomena, and suggesting a potential therapeutic effect by these inhibitors [84]. Using alginate hydrogel as an alternative, Chooi and Chew [85] found that matrix gel fixed the shape of the SCOs with similar growth-promoting efficiency to Matrigel, observing mature myelinated neurons at day 120 [86]. Homozygous, iPSC pairs carrying the ALS-causing mutation TDP43 (G298S) showed increased mis-localization of TDP43 in mutant organoids, thus demonstrating the suitability of alginate hydrogels for modeling organoid disease [87]. Together, these findings indicate the great potential of spinal organ tissues for drug development and evaluation of efficacy [88, 89].
Using two IPSC cell lines to create receptor brain organoids, Tamaki et al. [90] demonstrated the cell-to-cell spreading mechanism of pathogenic TDP-43 in human ALS tissues. Pathogenic TDP-43 from ALS patient-derived protein extracts was shown to spread in brain organoids and induce TDP-43 pathology, astrocyte proliferation, cell apoptosis, and DNA double-strand breaks, thereby revealing one of the pathogenic mechanisms of ALS [91]. The results of this study are consistent with previous findings on the role of TDP-43 in ALS [92, 93], suggesting the brain organoid model can better simulate the pathophysiological processes of ALS. However, a limitation of this study was that it did not consider the spinal cord, another key site of TDP-43 pathology in ALS. It is hoped that a multi-system scheme for the organ-tissue relationship of the spinal cord can be established in the future, and the anatomical structure and physiological function of human spinal cord tissue can be reproduced [94].
SCOs have shown great potential in the field of ALS research and treatment [95]. The generation of SCOs using patient-derived iPSCs can mimic the pathogenesis of ALS in a highly realistic manner, providing an important model for in-depth investigation of disease mechanisms [61, 88, 96]. Such models can accurately reflect the disease characteristics of ALS and help to achieve personalized medicine [97]. SCOs generated from the patient’s cells can provide a feasible pathway for personalized treatment [98]. Furthermore, based on the response of SCOs to drugs, personalized treatment plans can be developed to improve the choice and efficacy of therapy [99].
The effects of the microgravity environment on neurological functions offer new perspectives for the study of neurodegenerative pathologies under extreme conditions [100]. Sharma et al. [100] used 3D models of the human cerebral cortex and motor neuron-like organ models to simulate and study neuropathological processes in Alzheimer’s disease, FTD, and ALS in a microgravity environment. Their study provides mechanistic insights into the onset and progression of degenerative spinal cord lesions such as ALS by simulating the effects of different environments on neurons (e.g., microgravity, oxidative stress). It also provides a comprehensive reference for the application of SCOs in neurodegenerative diseases, from the construction of disease models to the simulation of environmental factors. Fig. 3 provides an overview of the direction of future research in this field.
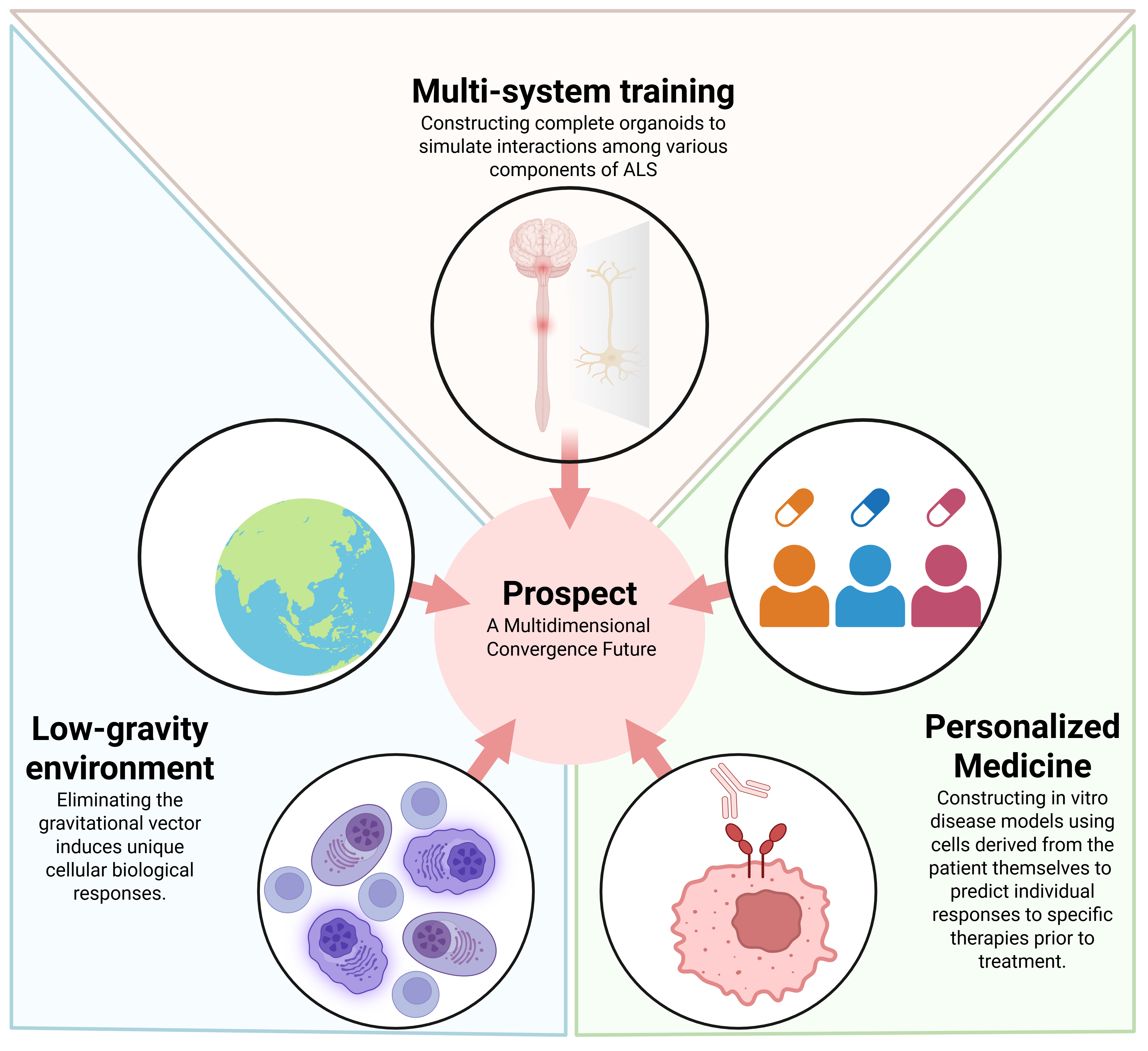 Fig. 3.
Fig. 3.
Prospects for the application of spinal cord organoids in ALS. Three possible directions for future development are shown: from multi-person healthcare to personalized healthcare, from single-system research to multi-system joint research, and from constructing disease models to simulating environmental factors. Created in https://BioRender.com.
SCOs offer a crucial platform for research on ALS and are a key tool for
overcoming species differences. They can reconstruct the human spinal cord
microenvironment and accurately mimic the mitochondrial dysfunction and axonal
transport defects of
However, SCOs still face three major technical challenges. First, at the cellular level, it is difficult to completely reproduce the demyelination and vascular microenvironment caused by oligodendrocyte Monocarboxylate Transporter 1 (MCT1) deletion. Second, in terms of technological maturity, the 120-day culture cycle for SCOs is much longer than the 60-day standard process for forebrain organoids, and the scattered distribution of motor neurons hinders the precise localization of axon transport deficits. Third, in terms of pathology, SCOs neglect the role of the downward inputs of corticospinal tracts, which may regulate low-extremity drugs, and only weakly model low-expression genes. These limitations contrast with forebrain organoids, which efficiently model TDP-43 trans-neuronal propagation and FTD co-morbidity mechanisms through standardized protocols. However, the inability of forebrain organoids to construct intrinsic defects of the NMJ means that studies of motor end-pathology must rely on complementary support from SCOs.
Currently, the breakthrough pathway still focuses on multi-dimensional
integration, i.e., the use of microfluidic chip-coupled forebrain organoids, SCOs
and muscle organoids to construct the entire pathological chain of ‘cortical
initiation
The ultimate goal is to synergize the different advantages offered by forebrain and spinal cord-like organs - forebrain models to solve “disease origins”, and of spinal cord models to address “motor terminal collapse”. These two models are integrated in 3D to advance ALS research from a single pathology simulation to a fully actionable and humanized strategy.
YXZ made substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data; TZ, YXL, BHC, and DZK were involved in drafting the manuscript, reviewing it critically for important intellectual content, and analyzing data; BHC gave final approval of the version to be published. All authors contributed to editorial changes in the manuscript. All authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by grants from the National Natural Science Foundation of China (No.82301543), Youth Scientific Research Project of Fujian Provincial Health Commission (No.2021QNA025), Technology Platform Construction Project of Fujian Province (2021Y2001), and Technology Platform Construction Project of Fujian Province (2020Y2003).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.