


 , Samaneh Ghorbani Shirkouhi 2,†, Seyed Sepehr Khatami 3, Farzin Kamari 4, Sarvenaz Ghaedi 5, Morten Blaabjerg 6,7, Sasan Andalib 6,7,*
, Samaneh Ghorbani Shirkouhi 2,†, Seyed Sepehr Khatami 3, Farzin Kamari 4, Sarvenaz Ghaedi 5, Morten Blaabjerg 6,7, Sasan Andalib 6,7,*
1 School of Medicine, Guilan University of Medical Sciences, 41937-13111 Rasht, Iran
2 School of Medicine, Shahroud University of Medical Sciences, 36147-73943 Shahroud, Iran
3 Department of Medicine, Valley Health System, Las Vegas, NV 89118, USA
4 Department of Neurophysiology, Institute of Physiology, Eberhard Karls University of Tübingen, 72074 Tübingen, Germany
5 Department of Neurology, University of California Irvine, Irvine, CA 92617, USA
6 Research Unit of Neurology, Department of Clinical Research, Faculty of Health Sciences, University of Southern Denmark, 5230 Odense, Denmark
7 Department of Neurology, Odense University Hospital, 5000 Odense, Denmark
†These authors contributed equally.
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that leads to a decline in cognitive function, including memory. The exact causes of AD are not fully understood, and to date no treatments are available that can stop the progression of this neurocognitive disorder. AD is associated with progressive loss of neurons, synaptic connectivity, and disruption of neuroplasticity in the brain. Neuroplasticity is the nervous system’s ability to adapt and recover in response to experiences, injuries, or a pathological change. Synaptic dysfunction and impairment of neuroplasticity are important elements of AD progression and cognitive decline. Studies have demonstrated that enhancement of neuroplasticity effectively improves cognition and memory, preventing the progression of AD. In this narrative review, we discuss the role of various pathophysiological explanations regarding the impairment of neuroplasticity in the pathogenesis of AD. We also highlight neuromodulation approaches, such as exercise, neurotrophic factor mimetics, pharmacological drugs, light therapy, and diet therapy that can promote neuroplasticity and have the potential for use in the prevention and treatment of AD.
Keywords
- Alzheimer’s disease
- neuroplasticity
- cognitive function
- neurotrophic factors
Alzheimer’s disease (AD), a neurodegenerative disease, is the most common form of dementia. AD presents with different and multiple clinical symptoms, mainly affecting cognitive domains. The early manifestation of AD is impairment in short-term memory. As the disease progresses, it affects attention, expressive speech, visuospatial processing, and executive functions [1, 2]. AD gives rise to significant cognitive impairment and severely limits daily functioning, ultimately making patients dependent on family members or caregivers.
The exact cause of AD has not yet been fully uncovered, but it is characterized
by the changes in the brain, including the accumulation of amyloid beta
(A
The amyloidogenic pathway of the cleavage of amyloid precursor protein (APP)
forms neurotoxic A
Neuroplasticity is characterized by the capability of the nervous system to adapt and recover, structurally and functionally, in response to novel experiences, injuries, or pathological changes [11]. On another reading, structural neuroplasticity refers to brain structural changes following learning and experiences, while functional neuroplasticity is the remodeling of synapses in response to brain injury or dysfunction. Neuroplasticity is a vital brain process that includes several mechanisms. It induces neurogenesis, synaptogenesis, and nerve sprouting through physiological and structural mechanisms that originate primarily at the cellular and molecular levels within synapses [12]. In this respect, synaptic dysfunction and impaired synaptic plasticity have emerged as principal contributors to the progression of AD and its associated cognitive decline [13].
Herein, we aim to review literature investigating neuroplasticity and its relevant mechanisms within the complex pathophysiology of AD. We also focus on the potential roles of neuroplasticity in the prevention and treatment of AD via pharmacological and non-pharmacological interventions.
A basic explanation of neuroplasticity is initiated from the Hebbian plasticity
theory, describing how synaptic connections are shaped, strengthened, and
weakened [14]. Long-term potentiation (LTP) and long-term depression (LTD) are
the two key elements of synaptic plasticity contributing to improved synaptic
transmission and neuronal connections [15]. LTP and LTD are important for
learning and memory. LTP is classically induced by repetitive high-frequency
input, strengthening more efficient information flow, possibly through synaptic
refinement, sprouting, and further synaptogenesis [16]. In contrast, LTD is
typically associated with repetitive low-frequency input, which deteriorates the
less efficient synaptic connections through synaptic pruning [17]. However, in
recent years, the classic definition of LTP and LTD has changed. LTP and LTD are
associated with the activation or reduction of glutamate receptors in the
synapses, promoting a biochemical process in the postsynaptic terminals, and
further enhancing the synaptic activity [18]. The excitatory neurotransmitter,
glutamate, plays a role in synaptic plasticity. In most cases, both LTP and LTD
induce their neuroplastic effects by N-methyl-D-aspartate (NMDA) and
Synaptic loss and disruption of neuroplasticity are the hallmarks of AD [24].
A
Neuroplasticity plays an important role in learning and memory. Memories are encoded as spatio-temporal dynamic patterns of coordinated cellular activities of a neuronal engram, and the dynamics may be gradually altered to accommodate new information [27]. An engram is defined as a lasting physical change in the brain after an experience that leads to the storage of a memory [28]. In other words, an engram is the physical trace of the memory in the brain. It has been shown that silencing engram neurons prevents memory expression [29]. Neuronal representations are neural activities correlated with task-related stimuli, actions, and cognitive variables, while representational drift indicates ongoing changes in these representations [30]. It has been shown that the hippocampus facilitates learning and information recalling through two computational processes, which are known as pattern completion and pattern separation [31]. Pattern completion is a network’s ability to respond to a degraded input pattern with the entire previously stored output pattern [32]. Pattern separation makes the stored representations of two similar input patterns more different, reducing the probability of errors in memory recall [32].
A study on male B6/129 F1 hybrid mice investigated the mechanism of altered plasticity following behavioral training [33]. It was tested in this study that alterations in intrinsic excitability, which are induced by learning, facilitate the encoding of new memories via metabotropic glutamate receptor (mGluR) activation. The hippocampal neurons showed increases in intrinsic excitability following learning, lasting for several days [33]. When animals were trained on a new task during this period, excitable neurons were reactivated, and memory formation depended on the activation of mGluRs rather than NMDARs. It was concluded that increases in intrinsic excitability may serve as a metaplastic mechanism for memory formation [33].
Aging is accompanied by memory decline [34], a reduction in attentional efficiency [35], and decreased task performance [36]. Age-related volume changes of the brain involve widespread white matter depletion [37] and region-specific gray matter changes, such as in the hippocampus [37, 38], leading to ventricular enlargement. Since the normal cognitive function is generally associated with neuroplasticity, an age-related reduction of neuroplasticity may lead to cognitive decline [39]. Although the exact mechanism involved in this process is not fully known, a potential role for brain-derived neurotrophic factor (BDNF) has been suggested [40]. Aging negatively affects BDNF-involved cascades by reducing its gene transcription. Moreover, Other effects include disrupting BDNF protein synthesis and processing, along with desensitizing its receptor, tyrosine receptor kinase B (TrkB) [40].
The nicotinamide adenine dinucleotide (NAD)-dependent deacetylases, sirtuins,
can regulate lifespan through inhibiting genomic instability and chromatin
modification [41]. There is evidence about the anti-aging effects of sirtuins
[42]. A study analyzed sirtuin1 [silent mating type information regulation 2
homolog 1 (SIRT1)] gene polymorphisms (rs7895833 A
SIRT1 may be considered a predictive marker of AD in early stages [43]. Kumar
et al. [43] evaluated the alterations in serum SIRT1 concentration in
healthy individuals and patients with AD and mild cognitive impairment (MCI). A significant reduction in
SIRT1 concentration was observed in patients with AD and MCI, compared to that in
healthy elderly individuals. A previous study established a time point model for
the clearance of A
Furthermore, it has been shown that a reduction in SIRT1 and BDNF levels changes synaptic plasticity and neuronal excitability in older mice [45]. SIRT1 is also expressed in neurons of the hippocampus [46]. In a study using a combination of behavioral and electrophysiological paradigms, the effects of deficiency and overexpression of SIRT1 on mouse learning and memory and synaptic plasticity were assessed [46]. The results of the study showed cognitive abilities impairment in the condition of SIRT1 deficiency. It also has been found that the cognitive deficits in SIRT1 knock-out mice lead to defects in synaptic plasticity [46].
The immune system is involved in the regulation of memory, learning, neurogenesis, and neuroplasticity, predominantly mediated by inflammatory cytokines, neurotrophic factors, and glial cells [47].
Interleukin-1 beta (IL-1
Besides immune components, the glial cells, and astrocytes in particular, modulate synaptogenesis, neuroplasticity, learning, and memory [51]. Astrocytes are key elements of the regulation of neurotransmitter concentration in the synaptic cleft, modulating the release and removal of neurotransmitters in the tripartite synapses [52]. Tripartite synapses refer to the existence of bidirectional communication between neurons and astrocytes [53]. Through their signaling activity, astrocytes maintain and modulate the balance between excitatory and inhibitory synapses, thereby playing a role in the regulation of neuronal activity and synaptic plasticity [54]. In addition, astrocyte glycolysis provides support for the neuronal activity, positively contributing to neuroplasticity and cognitive function [55]. Regarding these studies, astrocytes contribute to synaptic transmission, remodeling, plasticity, and normal brain function. Consequently, impairment in their normal functioning may lead to complications in synaptic function and neuroplasticity.
Disruption of synaptic transmission and neuroplasticity is one of the hallmarks
of AD and cognitive decline [56]. It has been shown that a disruption in the
physiological function of astrocytes favors A
Mitochondria are the main organelles involved in adenosine triphosphate (ATP) production, alongside the regulation of metabolism and apoptosis in cells. The cells’ energy demands are primarily met by oxidative phosphorylation of sugar through the tricarboxylic acid cycle. Neurons are energy-intensive cells.
A
Mitochondria are plastic and dynamic organelles in the neurons. Mitochondria are capable of adaptation and remodeling to supply neuronal energy demands in response to different neuronal energy states. Such adaptation leads to structural and functional changes in mitochondria, which are involved in neuroplasticity and AD [61]. Mitochondrial dynamics occur in both pre- and post-synaptic neurons during synaptic transmission [62, 63].
Proper mitochondrial distribution and transport are important in providing support to synaptic transmission [64]. BDNF, through the activation of the TrkB receptor at the presynaptic terminal, increases the Ca2+ levels, halting mitochondrial transport, and induces presynaptic mitochondrial accumulation in the terminal axon [65]. This process promotes the stationary mitochondrial population and mitochondrial motility arrest, leading to the gathering of more mitochondria in the presynaptic area [65]. The effect of BDNF on mitochondrial halting promotes synaptic transmission, neurotransmitter release, and further neuroplasticity [65].
LTP induces mitochondrial fission in the postsynaptic dendrites, an essential process in maintaining synaptic plasticity [62]. LTP, through the activation of postsynaptic NMDARS, increases the influx of Ca2+ into the postsynaptic neuron, which further enhances mitochondrial localization and fission at the postsynaptic dendrites [62]. Mitochondrial fission is mediated by dynamin-related protein 1 (Drp1), which is a key guanosine triphosphatase (GTPase) protein [66]. Eventually, as a result of this process, LTP, by inducing synaptic activity and further mitochondrial fission, results in synaptic plasticity [62]. In contrast, impaired mitochondrial fission possibly reduces dendritic mitochondrial fission, disrupting synaptic plasticity in the nervous system [62, 67].
Acetylcholine (ACh) is an important neurotransmitter associated with wakefulness, attention, learning, and memory. Nucleus basalis of meynert (NBM) is a structure that accommodates the basal forebrain cholinergic neurons (BFCNs). NBM has been identified as the primary and principal source of cholinergic innervation in the brain [68]. Tauopathy neurodegeneration of NBM is involved in neuronal loss and synaptic dysfunction of cholinergic innervation neurons, further leading to memory impairment and progression of AD [69]. Baskerville et al. [70] observed the effects of depleted ACh input from the NBM on cortical plasticity in young adult male rats. In this study, in the absence of ACh, no remarkable cortical plasticity was demonstrated. However, in the presence of ACh, notable enhancement of cortical and synaptic plasticity was detected.
Glutamate is the major excitatory neurotransmitter that mediates excitatory
signal transmission on postsynaptic neurons, which is mediated through two
important receptors, AMPA and NMDA receptors [19]. In AD, A
Gamma-aminobutyric acid (GABA) is the primary inhibitory neurotransmitter in the CNS. Phosphorylated tau accumulation in GABAergic interneurons of the dentate gyrus of the hippocampus is related to disrupted hippocampal neurogenesis and cognitive decline in AD [74]. These findings indicate the important role of the GABAergic system in neurogenesis and synaptic plasticity, making it a therapeutic target for AD.
Norepinephrine (NE) or noradrenaline is implicated in modulating cognitive
functions, including memory, attention, and arousal [75]. In AD, degeneration of
the locus coeruleus (LC), the principal source of NE within the brain, is among
the earliest pathological events [75, 76]. Disruption of NE aggravates AD
pathogenesis, including A
Dopamine (DA) is essential for movement, motivation, and memory [79]. The dopaminergic system is also affected in AD [80]. Using repetitive transcranial magnetic stimulation (rTMS) revealed impaired dopamine-modulated synaptic plasticity in AD patients [81]. The affected plasticity was possibly improved by a dopamine agonist [81]. Ventral tegmental area (VTA) dopaminergic neuron degeneration is related to impaired synaptic plasticity and memory decline in AD mice [82, 83, 84].
BDNF is a key neurotrophic factor signaling via the TrkB receptor, which plays
an important role in synaptic outgrowth and neuroplasticity [85]. Exercise and
training have been identified to promote neurogenesis and neuroplasticity through
the upregulation of neurotrophic factors such as BDNF [86]. Reduced levels of
BDNF in the brain tissue samples of postmortem AD patients favor the AD pathology
[87]. Decreased BDNF/TrkB signaling is associated with
Nerve growth factor (NGF) is another key neurotrophic factor with a high
affinity to TrkA and a low affinity to P75 neurotrophin receptors (p75NTR).
NGF modulates cholinergic neurons projected to the hippocampus to induce
hippocampal LTP [89]. Dysfunction of the NGF signaling pathway in hippocampal
neurons has been linked to the activation of the amyloidogenic pathway of APP and
A
There is no curative treatment for AD. Approaches improving neuroplasticity can be potential therapies that could improve function or slow down cognitive decline in AD.
Donepezil is an acetylcholinesterase inhibitor medication that reduces the
breakdown of ACh and increases ACh levels in the synapses, promoting cognitive
function [92]. Donepezil has been shown to prevent A
Memantine, an NMDAR antagonist, is also used to improve cognitive
function in AD [93]. Caneus et al. [94] examined the effectiveness of
several AD therapeutic drugs on an LTP system, a human-based platform that was
designed to mimic the clinical manifestation of AD and also to serve as a
screening system for monitoring responses to therapeutic agents. LTP was induced
in mature human induced pluripotent stem cell (iPSC)-derived cortical neurons
cultured on microelectrode arrays [94]. The authors concluded that Donepezil,
Memantine, Saracatinib, and Rolipram can prevent A
Nygaard et al. [96] performed a 4-week phase Ib randomized, double-blind, placebo-controlled clinical trial on 24 patients with mild to moderate AD to evaluate the safety, tolerability, and CNS availability of oral AZD0530 (Saracatinib), a Fyn kinase inhibitor. Findings suggested that Saracatinib is safe and well-tolerated in different doses and can be a potential therapeutic agent for AD. van Dyck et al. [97] conducted a phase IIa randomized clinical trial in 159 patients with mild AD to examine the efficacy, safety, and tolerability of Saracatinib. The authors observed that Saracatinib is generally safe and well-tolerated in patients with mild AD. Remarkable effects in AD treatment were not shown; however, they did not exclude the potential role of Saracatinib as a therapeutic agent for AD.
Cong et al. [98] studied the efficacy of Rolipram, a
phosphodiesterase-4 (PDE-4) inhibitor, in the improvement of cognitive function
and depression in 3xTg-AD mice. Behavioral tests related to learning, memory,
anxiety, and depression were compared, and different neurochemical measurements
were administered. The authors showed that Rolipram suppressed A
P021 is a neurotrophic and neurogenic peptide mimetic compound that inhibits
leukemia inhibitory factor (LIF) signaling pathway and increases BDNF expression
[85]. P021 reduces A
LM11A-31 is a small molecule that modulates p75NTR signaling and can
prevent A
Zhang et al. [103] studied the efficacy of sulforaphane (SFN), a
metabolite enriched in cruciferous vegetables, in the prevention of AD
progression in an AD mouse model. Treatment with SFN was found to attenuate
A
In a study by Yao et al. [105], P75 Ectodomain (P75ECD) was shown to be
largely reduced in the CSF and the brain of the AD mice. Restoration of
physiological levels of P75ECD via injection of adeno-associated virus
(AAV)-P75ECD-Fc in the lateral ventricles of A
The beneficial effect of exercise on improving cognitive function has been well demonstrated [106, 107]. Exercise improves cognitive function and neuroplasticity while enhancing BDNF immunoreactivity [108]. Following aerobic exercise, BDNF directly influences neuroplasticity by activating the Akt (protein kinase B) and cyclic adenosine monophosphate (cAMP)-response element binding protein (CREB) signaling pathways in the rat hippocampus [109]. A study on 3xTg-AD mice examined the effect of the intravenous injection of plasma extracted from the exercised mice on the improvement of hippocampus-dependent cognitive functions [110]. The results showed an improvement in mitochondrial function, neuroplasticity, and cognitive function, in addition to a suppression of apoptosis. A randomized controlled trial evaluated the effect of 6-month cycling on the cognition of AD patients using the AD assessment scale-cognitive subscale (ADAS-Cog) test, demonstrating a significant reduction in the scores, i.e., cognitive improvement, compared with the controls [111].
Nigam et al. [112] assessed the effect of exercise-induced upregulation
of BDNF expression on AD pathogenesis. Their findings demonstrated that increased
BDNF signaling, through enhanced
In addition, it has been shown that exercise can increase SIRT1 expression
levels [113]. Shi et al. [114] investigated the effects of
8 weeks of aerobic exercise, administration of chlorogenic acid,
and a combination of both on A
Light therapy has been suggested as a non-invasive and promising intervention
for cognitive function and neuroprotection in AD, influencing through
neuroplasticity mechanisms [115]. A study in an AD mouse model indicated that
transcranial light therapy improved synaptic plasticity [115]. In this study,
synaptic plasticity using electrophysiological parameters, including field
excitatory postsynaptic potential (fEPSP), paired pulse facilitation (PPF), LTD,
and LTP, was evaluated. The treated group showed higher levels of LTP than the
control group. In another study using the 3xTg-AD mouse model, the combined
effects of exercise with 40-Hz light flickering on cognitive function were
investigated by evaluating the neuroinflammation, mitochondrial function, and
neuroplasticity [116]. The results showed a significant decrease in A
The effect of a healthy diet on cognition has been demonstrated [119]. The
results of a 4-year randomized controlled trial in 1401 men and women aged 57–78
years at baseline showed that a combination of moderate-intensity aerobic
exercise and a healthy diet may improve cognition in older individuals [119]. In
this regard, the role of SIRT1 has been considered [120]. The association between
diet containing lipopolysaccharides/patulin and SIRT1 inactivation, cellular
aging, and delayed hepatic A
Transcranial magnetic stimulation (TMS) is a noninvasive method that generates a magnetic field over the scalp using a wired coil probe [121]. The magnetic field produces an electric impulse that travels down the skull directly to induce neuronal depolarization in the targeted brain region [121]. TMS can be used with low- or high-frequency stimulation modes to modulate LTP and LTD in the CNS [122]. Repetitive low-frequency TMS is associated with the weakening and inhibition of the synapses, resulting in LTD, while repetitive high-frequency TMS is related to the strengthening and excitation of the synapses, leading to LTP [122]. The LTP and LTD induced by rTMS can lead to neuroplasticity; however, the exact neurobiological mechanisms are not completely clear [122].
rTMS has been shown to promote synaptic plasticity and AD rehabilitation [123].
Findings suggest that rTMS enhances the efficiency of A
Regarding these findings, rTMS can enhance synaptic transmission, neurogenesis, and neuroplasticity in the neuronal network connection, thereby improving cognitive and memory function. However, the exact mechanism beyond the neuroplastic effects and cognitive improvements is not well-known.
Vagus nerve stimulation (VNS) is a neuromodulation technique applying electrical stimulation to the vagus nerve [128]. The surgical or invasive VNS (iVNS) was initially used to treat refractory epilepsy cases [128]. This method requires a VNS device to be implanted in the body for direct stimulation, which may follow surgical complications or device malfunction [129]. These challenges hampered the feasible use of iVNS in the routine management of nervous system disorders [129].
The non-invasive VNS method has similarly been used to modulate the nervous system activity, while preventing the invasive method complications [130]. The non-invasive VNS, clinically known as transcutaneous VNS (tVNS), provokes the auricular branch of the vagus nerve, Arnold’s nerve, on the ear, or the cervical vagus nerve on the neck [130].
The tVNS exerts its effects through catecholamine release, especially NE from LC in response to afferent stimulation of the vagus nerve [131]. The NE released from the LC enhances the gene transcription of anti-inflammatory molecules and suppresses the proinflammatory cytokine signaling pathways in astrocytes and microglia [132]. Consequently, the anti-inflammatory effects induced by tVNS prevent neuroinflammation and neurodegeneration and further may favor neuroplasticity [133]. Besides its beneficial effects through the catecholamine release, VNS regulates the synaptic and memory function via the dopaminergic pathways [134]. The activation of LC causes dopamine release in the hippocampus, which modulates LTP, thereby enhancing synaptic transmission and memory [135].
Moreover, VNS has been identified to promote hippocampal phosphorylation of TrkB receptors and BDNF release, possibly promoting neuroplasticity and memory function in a rat brain [136]. Altogether, VNS may seem to be a promising neuromodulation technique for retaining cognitive function and memory in AD through enhancement of synaptic transmission and neuroplasticity [137].
Fig. 1 summarizes the potential therapeutic approaches contributing to the enhancement of synaptic plasticity and neurogenesis in AD.
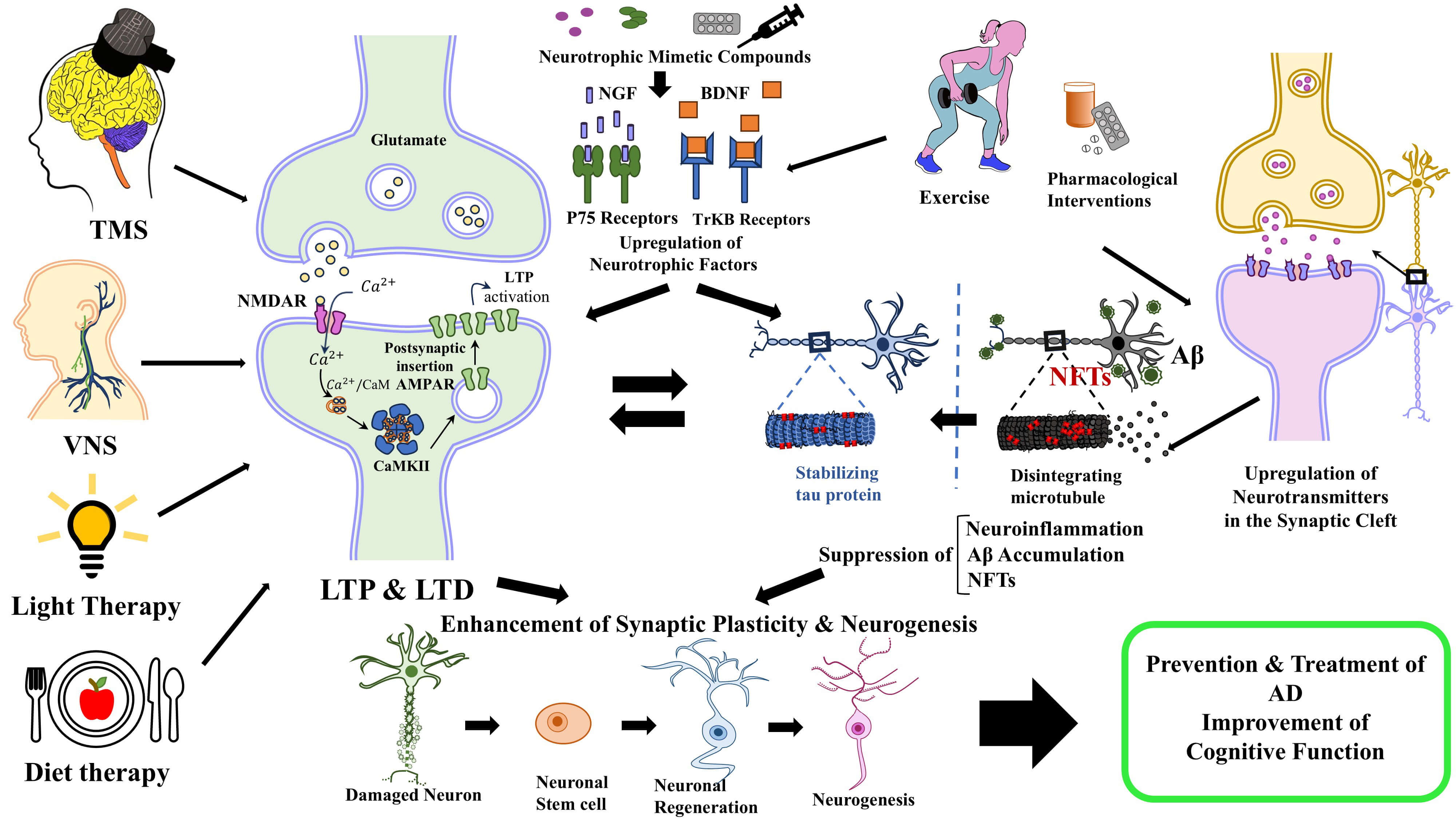 Fig. 1.
Fig. 1.
The potential therapeutic approaches contributing to the
enhancement of synaptic plasticity and neurogenesis in AD. Pharmacological
interventions through upregulation of neurotransmitters and neurotrophic factors
in the synaptic cleft, suppress the neuroinflammation, A
Cognitive decline in AD is associated with impairment of synaptic plasticity and
neuroplasticity. Accumulation of A
SA had the idea for the narrative review article. All authors contributed to the literature search of the article. AAG and SGS drafted the article. SSK, FK, SG, MB, and SA critically reviewed and revised the article. SG drew the figure. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.