







 , Wenbin Duan 1, Xiaomin Pu 1, Changdi Ma 1, Huai Huang 2, Zhenghu Xu 3,*
, Wenbin Duan 1, Xiaomin Pu 1, Changdi Ma 1, Huai Huang 2, Zhenghu Xu 3,*
1 Department of Neurosurgery, Baoshan Municipal People’s Hospital, 678000 Baoshan, Yunnan, China
2 Department of Neurology, Hebei PetroChina Central Hospital, 065000 Langfang, Hebei, China
3 Department of Neurosurgery, The Second Affiliated Hospital of Kunming Medical University, 650000 Kunming, Yunnan, China
Abstract
Mitochondrial dysfunction is closely associated with the pathogenesis of Parkinson’s disease (PD). Lutein has been shown to exert protective effects in neurological disorders. This study aimed to investigate the ameliorative effects of lutein on mitochondrial function in PD and its underlying molecular mechanisms.
Animal and cellular PD models were established by intraperitoneal injection of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in mice and treatment of SH-SY5Y cells with 1-methyl-4-phenylpyridinium ion (MPP+), respectively. Motor function was assessed using the rotarod, adhesive removal, and pole tests. Mitochondrial function was evaluated using MitoSOX Red staining, JC-1 staining, and adenosine triphosphate (ATP) content measurement. Western blotting and reverse transcription-quantitative polymerase chain reaction (RT-qPCR) were used to measure the levels of relevant proteins and mRNA.
Lutein significantly ameliorated MPTP-induced motor dysfunction in PD mice, increased the number of tyrosine hydroxylase (TH)-positive neurons, and alleviated damage to striatal brain tissue. At the cellular level, lutein significantly suppressed MPP+-induced apoptosis of SH-SY5Y cells, upregulated the expression of B-cell lymphoma-2 (Bcl-2), and downregulated the expression of Bcl-2-associated X protein (Bax) and cleaved caspase-3. Additionally, lutein significantly reduced reactive oxygen species (ROS) levels, restored mitochondrial membrane potential, increased ATP levels, and increased the activity of mitochondrial respiratory chain complex I. At the molecular level, lutein promoted the ubiquitination of dynamin-related protein 1 (Drp1), whose degradation was impaired in the PD model. This effect was mediated by the E3 ubiquitin ligase Tripartite Motif-containing protein 31 (TRIM31), whose expression was downregulated in the disease state. Functional experiments confirmed that overexpression of TRIM31 enhanced Drp1 ubiquitination and improved mitochondrial function, whereas TRIM31 knockdown partially attenuated the therapeutic effects of lutein.
In summary, this study revealed, for the first time, that lutein alleviates PD progression by increasing Drp1 ubiquitination and degradation via TRIM31 transcription and translation, ultimately improving neuronal mitochondrial function. These findings not only elucidate a novel mechanism underlying lutein’s neuroprotective effect but also identify a potential therapeutic target and offer a new strategy for PD treatment.
Keywords
- Parkinson’s disease
- lutein
- mitochondrial function
- neurons
- TRIM31
- Drp1
Parkinson’s disease (PD) is the second most common neurodegenerative disorder
[1]. Its incidence increases significantly with age, and the number of PD
patients is projected to exceed 12.9 million by 2040 [2]. This disease is
characterized by motor symptoms, including bradykinesia, rigidity, resting
tremor, and postural instability [3]. Pathologically, PD is defined by the loss
of dopaminergic (DA) neurons in the substantia nigra and the accumulation of
Dynamin-related protein 1 (Drp1) plays a crucial role in controlling mitochondrial division and is localized to the mitochondrial outer membrane [7]. Previous studies have established that excessive Drp1-mediated mitochondrial fission is a key driver of DA neuron apoptosis and that its inhibition can protect against cell death [8]. In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD models, inhibition of Drp1 expression was shown to improve mitochondrial integrity via the PTEN-induced putative kinase 1 (PINK1)/Parkin pathway [9]. Separately, the E3 ubiquitin ligase Tripartite Motif-containing protein 31 (TRIM31), which is highly enriched in brain mitochondria, has been implicated in neurodegenerative processes through substrate ubiquitination [10, 11]. TRIM31 is critical for maintaining mitochondrial function in DA neurons, and its deficiency leads to mitochondrial impairment [12, 13]. Notably, TRIM31 can regulate Drp1 levels during mitochondrial stress [11]. Building on this evidence, we hypothesize that TRIM31 plays a significant role in PD by modulating Drp1 to alleviate mitochondrial dysfunction.
Lutein is a naturally occurring carotenoid abundant in dark green vegetables such as spinach and kale [14]. Beyond its well-established antioxidant and anti-inflammatory properties, accumulating evidence indicates that lutein can directly modulate mitochondrial function and ubiquitination processes. For instance, in a neurodevelopmental model, lutein was shown to prevent abnormalities induced by mitochondrial dysfunction [15]. In neuronal cells, lutein not only enhances mitochondrial function but also promotes neuronal differentiation via the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT) pathway [16]. Furthermore, a recent study revealed that lutein directly binds to mouse double minute 2 (MDM2), facilitating mitochondrial translocation of the key respiratory complex protein NADH: ubiquinone oxidoreductase core subunit S1 (NDUFS1), thereby improving mitochondrial function and suppressing ferroptosis [17]. Additionally, lutein has been reported to upregulate the expression of HMG-CoA reductase degradation protein 1 (HRD1), an endoplasmic reticulum-associated E3 ubiquitin ligase, suggesting its potential to regulate ubiquitination pathways [18]. However, whether lutein can modulate TRIM31, a mitochondria-associated E3 ubiquitin ligase, to influence Drp1-mediated mitochondrial fission and mitigate PD pathology remains unclear.
Therefore, this study aimed to systematically investigate whether lutein regulates mitochondrial dynamics through the TRIM31/Drp1 pathway in both cellular and animal models. Our work seeks to elucidate the neuroprotective mechanisms of lutein in PD from the novel perspective of ubiquitination regulation, providing a theoretical foundation for its potential therapeutic application.
Fifteen 8-week-old male C57BL/6 mice (20–22 g) were obtained from the Animal
Experiment Center of Kunming Medical University and housed under standard
conditions (23
Human neuroblastoma cells (SH-SY5Y) were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (SCSP-5014; Shanghai, China). All cell lines used in this study were rigorously authenticated and confirmed to be free of mycoplasma contamination. Authentication was performed by short tandem repeat (STR) profiling, and mycoplasma testing was conducted regularly using the polymerase chain reaction (PCR)-based method; all results were negative. Cells were cultured in DMEM/F12 (11320033; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) (A5256701; Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin (15070063; Gibco, Grand Island, NY, USA) at 37 °C under 5% CO2. To establish a PD cellular model, cells were treated with 1 mM MPP+ (D048, Sigma-Aldrich, St. Louis, MO, USA) for 24 h. For lutein pretreatment, cells were incubated with 10 µM lutein for 2 h prior to MPP+ exposure.
TRIM31 overexpression (OE-TRIM31) and knockdown (si-TRIM31) plasmids, along with their corresponding negative controls (OE-NC, si-NC), were purchased from GenePharma (Shanghai, China). SH-SY5Y cells were seeded in 6-well plates and transfected at 70–80% confluence using Lipofectamine 3000 (L3000150; Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Transfection efficiency was confirmed by Western blotting.
Rotarod test: Mice were placed on an accelerating rotarod (5–40 rpm), and the latency to fall was recorded. If a mouse did not fall within 5 min, it was recorded as 300 s. Each mouse was tested five times at 30 min intervals. Adhesive removal test: A small adhesive label was placed on each mouse’s forepaw, and the time to remove it was recorded. Three trials were performed per mouse at 30 min intervals. Pole test: Mice were placed on a vertical pole (1 cm in diameter, 60 cm in height), and the time to descend to the floor was recorded. The average of three trials was calculated.
Brain tissues were fixed, paraffin-embedded, and sectioned at 5 µm. For hematoxylin and eosin (H&E) staining, sections were deparaffinized, stained with hematoxylin and eosin (G1120; Solarbio), and imaged under a light microscope. For immunohistochemistry, antigen retrieval was performed using citrate buffer. Endogenous peroxidase activity was blocked with 3% H2O2, and nonspecific binding was blocked with 5% goat serum (C0265; Beyotime). Sections were incubated overnight at 4 °C with anti-TH antibody (1:500, ab137869; Abcam, Cambridge, UK), followed by an horseradish peroxidase (HRP)-conjugated secondary antibody (1:500, ab97051; Abcam) and 3,3’-diaminobenzidine (DAB) development (DA1010; Solarbio). Finally, the samples were observed using an optical microscope (BX53; Olympus, Tokyo, Japan) and photographed for record.
Cell viability was assessed using the Cell Counting Kit-8 (CCK-8) assay (C0038; Beyotime). SH-SY5Y cells were seeded in 96-well plates, treated as indicated, and incubated with CCK-8 reagent for 1 h. Absorbance was measured at 450 nm. Apoptosis was detected by flow cytometry using a fluorescein Isothiocyanate-conjugated Annexin V (Annexin V-FITC)/propidium iodide (PI) staining kit (556547, BD Biosciences, San Jose, CA, USA). After treatment, the cells were incubated in the dark at 25 °C for 15 min. Finally, the apoptosis rate of the cells was detected using a flow cytometer (FACScan; Becton, Dickinson and Company (BD), Franklin Lakes, NJ, USA).
Proteins were extracted from tissues or cells using radio-immunoprecipitation
assay (RIPA) lysis buffer (P0013, Beyotime), separated by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to
polyvinylidene fluoride (PVDF) membranes (IPVH00010; Merck Millipore, Burlington,
MA, USA). After blocking with 5% skim milk, membranes were incubated with
primary antibodies overnight at 4 °C, followed by incubation with
HRP-conjugated secondary antibodies (1:4000, ab97051; Abcam or
1:5000, ab205719; Abcam). Protein bands were visualized using an
enhanced chemiluminescence (ECL) kit (34075; Thermo Fisher Scientific, Waltham,
MA, USA) and quantified with ImageJ (V1.8.0.112, National Institutes of Health,
Bethesda, MD, USA). Primary antibodies used included Bcl-2-associated X protein
(Bax) (1:1000, ab32503; Abcam), B-cell lymphoma-2 (Bcl-2) (1:1000,
ab196495; Abcam), cleaved caspase-3 (1:1000, #9661S; Cell
Signaling Technology, Danvers, MA, USA), Drp1 (1:1000, ab184247; Abcam,
Cambridge, UK), TRIM31 (1:1000, PA5-40961; Thermo Fisher Scientific, Waltham, MA,
USA), and
Mitochondrial reactive oxygen species (ROS) levels were measured using MitoSOX Red (S0061S; Beyotime). Cells were stained for 25 min at 37 °C and imaged by fluorescence microscopy. Mitochondrial membrane potential was assessed using JC-1 staining (C2006; Beyotime), and the ratio of red to green fluorescence was calculated. ATP content was determined using a commercial ATP assay kit (A095-1-1; Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and a microplate reader. Complex I activity was measured using a spectrophotometric assay kit (BC0515; Solarbio).
Cells were lysed in NP-40 lysis buffer (P0013F; Beyotime, Shanghai, China) supplemented with a protease inhibitor “cocktail” (539134; Merck Millipore). Lysates were then centrifuged at 12,000 rpm for 10 min at 4 °C, and the supernatant was collected and incubated with a Drp1 antibody (1:1000, ab184247; Abcam) at 4 °C for 6 h. After incubation, protein A/G agarose magnetic beads (#78609; Thermo Fisher Scientific) were added, and the mixture was rotated and incubated overnight at 4 °C. The immune complexes bound to the beads were washed three times with immunoprecipitation (IP) buffer. Finally, the attached proteins were extracted by boiling in loading buffer and analyzed by Western blotting.
Cell supernatants were prepared as described previously. Protein samples were incubated overnight at 4 °C with an anti-TRIM31 antibody (1:100, PA5-40961; Thermo Fisher Scientific), followed by incubation for 1 h at 4 °C with protein A/G magnetic beads (30 µL/sample). As a negative control, supernatants were incubated with rabbit IgG (1:500, 98136-1-RR; Proteintech Group Inc., Rosemont, IL, USA). The magnetic beads were subsequently triple-rinsed with lysis buffer, and the immunoprecipitated proteins were eluted using loading buffer for Western blot analysis.
Total RNA was extracted with TRIzol (15596026; Invitrogen, Carlsbad, CA, USA),
and cDNA was synthesized using a reverse transcription kit (P4202; Genenode,
Wuhan, China). qPCR was performed with SYBR Green (SR1110; Solarbio) on a 7900HT Fast Real-Time PCR System. The 2-ΔΔCt
method was used for quantification, with
| Target | Sequence |
| TRIM31 (Human) | F: 5′-AACCTGTCACCATCGACTGTG-3′ |
| R: 5′-TGATTGCGTTCTTCCTTACGG-3′ | |
| F: 5′-CATGTACGTTGCTATCCAGGC-3′ | |
| R: 5′-CTCCTTAATGTCACGCACGAT-3′ |
F, Forward primer; R, Reverse primer; TRIM31, Tripartite Motif-containing protein 31.
All data were analyzed using GraphPad Prism 8.0 software (GraphPad Software, La Jolla, CA, USA) and
are presented as the mean
First, we investigated the potential protective effect of lutein in a PD mouse model. H&E staining revealed marked focal encephalomalacia and neuronal structural damage in MPTP-treated mice, whereas lutein intervention significantly ameliorated these pathological alterations (Fig. 1A). Behavioral tests further demonstrated that MPTP induction led to motor dysfunction characterized by prolonged pole climbing time, shortened retention time, and delayed label removal, all of which were effectively reversed by lutein treatment (Fig. 1B–D). Immunohistochemical analysis revealed a notable decrease in TH-positive neurons in the MPTP group compared with the control group, whereas lutein treatment markedly increased the number of TH-positive neurons (Fig. 1E). Further in vitro validation of lutein’s role demonstrated that compared with the negative control (NC) group, the MPP+ group exhibited significantly reduced cell viability and markedly increased apoptosis. Lutein treatment effectively reversed these changes (Fig. 1F,G). Western blot analysis of apoptosis-related proteins revealed that compared with the NC group, the MPP+ group had significantly lower Bcl-2 expression and markedly higher Bax and cleaved caspase-3 expression. Lutein treatment partially reversed these changes in protein expression levels (Fig. 1H) (The original, uncropped western blot image(s) corresponding to this figure are available in the Supplementary Material). These results suggest that lutein can alleviate PD progression in mice and inhibit neuronal cell damage in vitro.
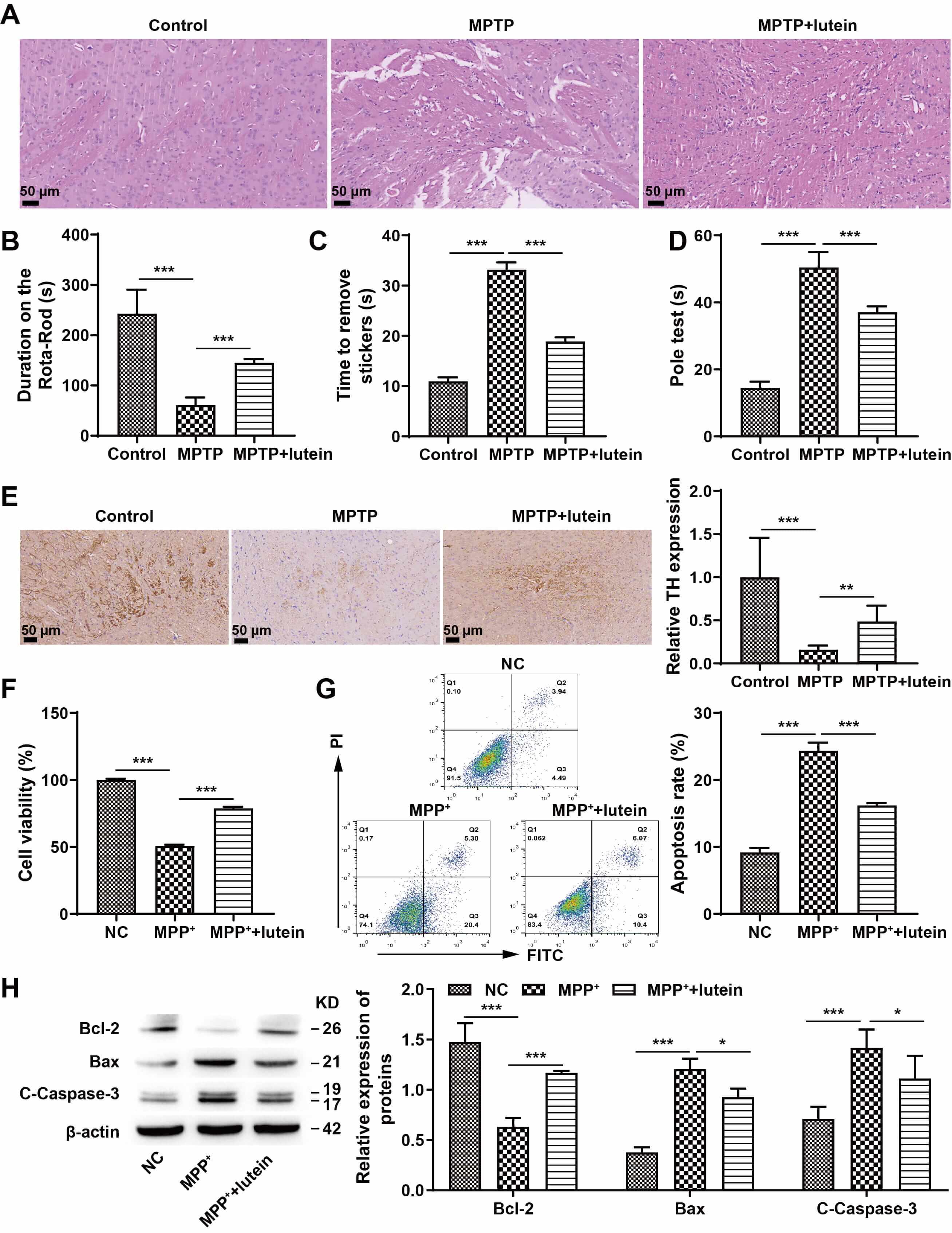 Fig. 1.
Fig. 1.
Lutein attenuated PD progression in mice and inhibited neuronal
injury in vitro. (A) Pathological damage was detected through H&E
staining, scale bar: 50 µm. (B) Rotarod assessment for motor ability. (C)
Adhesive removal test to assess motor function. (D) Pole-climbing test to assess
motor function. (E) IHC analysis of TH expression, scale bar: 50 µm. (F)
CCK-8 assay of cell viability. (G) Flow cytometry identification of apoptosis.
(H) Western blot analysis of apoptosis-associated proteins Bcl-2, Bax, and
cleaved caspase-3. * p
Previous studies have shown that mitochondrial dysfunction is strongly associated with PD [20, 21]. Consequently, we evaluated the effect of lutein on MPP+-induced alterations in mitochondrial function in neurons. Measurements of ROS levels and mitochondrial membrane potential revealed that, compared with the NC group, MPP+ treatment significantly increased mitochondrial ROS levels and significantly decreased the mitochondrial membrane potential. In contrast, lutein treatment reduced ROS levels and increased the mitochondrial membrane potential (Fig. 2A,B). Treatment with MPP+ reduced ATP concentrations, whereas lutein markedly reversed this effect and restored ATP levels (Fig. 2C). In addition, lutein treatment significantly increased the activity of mitochondrial respiratory chain complex I (Fig. 2D). These results suggest that lutein can improve mitochondrial dysfunction induced by MPP+ in neuronal cells.
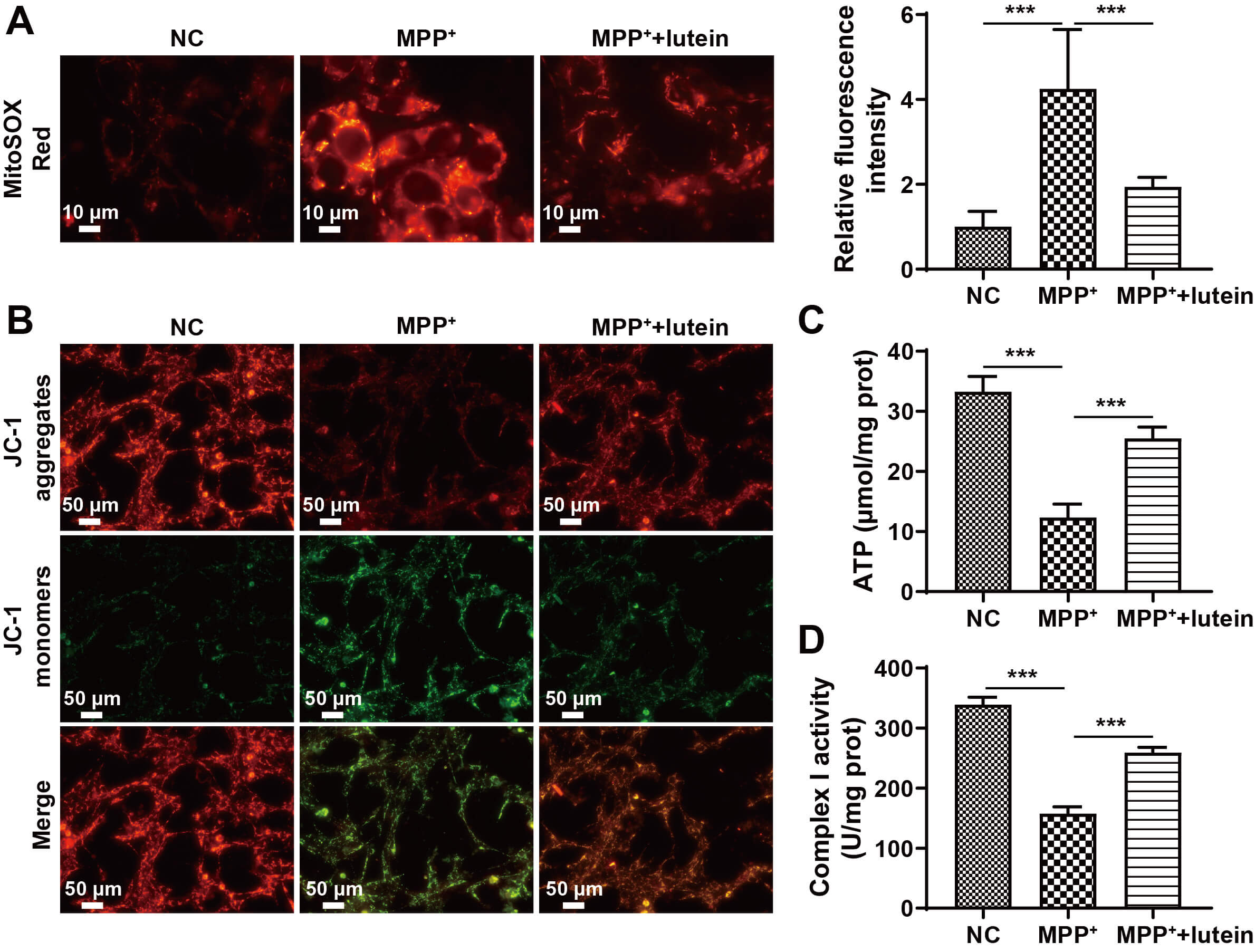 Fig. 2.
Fig. 2.
Lutein improves MPP+-induced mitochondrial dysfunction in
neuronal cells. (A) Mitochondrial ROS identified via MitoSOX Red staining, scale
bar: 10 µm. (B) Mitochondrial membrane potential measured using JC-1
staining, scale bar: 50 µm. (C) ATP concentration in SH-SY5Y cells measured
with a kit. (D) Activity of mitochondrial respiratory chain complex I. *** p
Drp1 plays a crucial role in mitochondrial fission. Previous studies have shown that Drp1 expression is upregulated in PD [22] and that its ubiquitination is crucial for regulating mitochondrial fission [23]. Western blot analysis indicated that compared with NC treatment, MPP+ treatment markedly increased Drp1 expression and decreased its ubiquitination (Fig. 3A,B). Lutein treatment increased ubiquitinated Drp1 levels (Fig. 3C) (The original, uncropped western blot image(s) corresponding to this figure are available in the Supplementary Material). These results indicate that Drp1 ubiquitination is decreased in PD and that lutein can promote its ubiquitination.
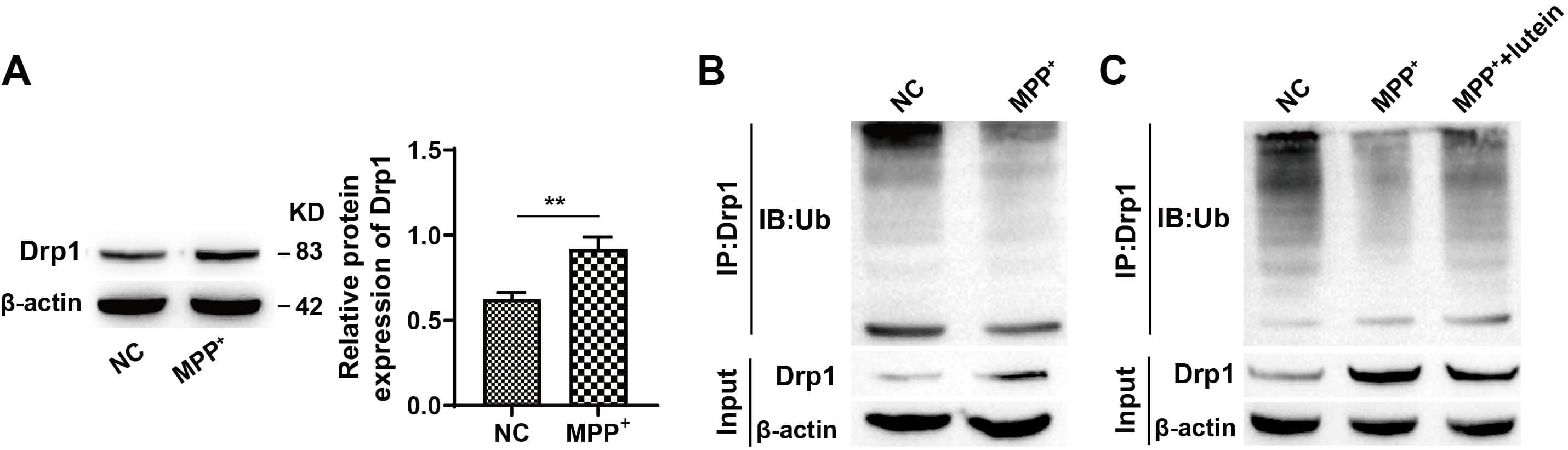 Fig. 3.
Fig. 3.
Lutein promotes the ubiquitination of Drp1. (A) Western blot
analysis of Drp1 expression. (B) Detection of ubiquitinated Drp1 levels. (C) The
effect of lutein treatment on Drp1 ubiquitination. ** p
Previous studies have shown that TRIM31 is enriched in mitochondria [12]. Consequently, our research focused on the role of TRIM31 in controlling mitochondrial activity in neurons. Analysis of the Gene Expression Omnibus (GEO) database (GSE160299) revealed that TRIM31 expression is reduced in PD patients (Fig. 4A,B). Furthermore, TRIM31 expression tended to decrease in MPTP or MPP+-induced animal and cellular models (Fig. 4C,D). As previously noted, TRIM31 can regulate Drp1 expression in response to mitochondrial damage caused by cerebral ischemia [11]. Co-immunoprecipitation experiments further verified the interaction between TRIM31 and Drp1 (Fig. 4E). To investigate whether TRIM31 regulates neuronal mitochondrial function through Drp1, we overexpressed TRIM31. Western blot analysis demonstrated a significant increase in TRIM31 expression (Fig. 4F). Ubiquitination assays showed that TRIM31 overexpression significantly promoted Drp1 ubiquitination (Fig. 4G) (The original, uncropped western blot image(s) corresponding to this figure are available in the Supplementary Material). CCK-8 and flow cytometry assays revealed that overexpression of TRIM31 significantly promoted SH-SY5Y cell proliferation and reduced apoptosis (Fig. 4H,I). Further analysis of indicators related to mitochondrial function revealed that overexpression of TRIM31 decreased mitochondrial ROS levels, increased mitochondrial membrane potential and ATP content, and elevated mitochondrial respiratory chain complex I activity (Fig. 4J–M). These findings suggest that TRIM31 overexpression improves mitochondrial function in neuronal cells by promoting Drp1 ubiquitination.
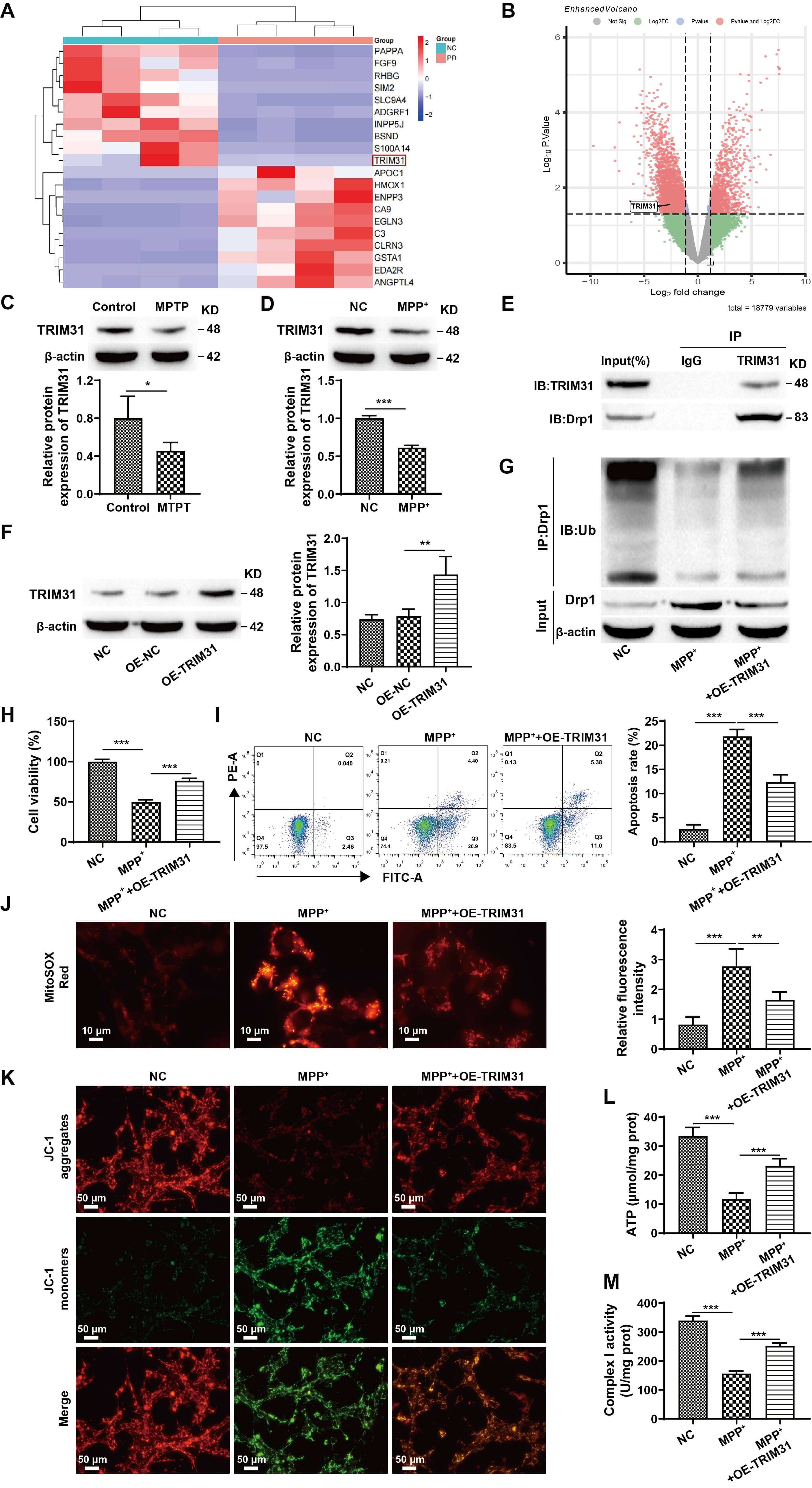 Fig. 4.
Fig. 4.
TRIM31 improves neuronal cell loss and mitochondrial function by
regulating Drp1 ubiquitination levels. (A) Cluster heatmap. (B) Volcano plot.
(C) Western blot detection of TRIM31 expression in tissues. (D) Analysis of
TRIM31 expression in cells using Western blotting. (E) Identification of the
interaction between TRIM31 and Drp1 through co-immunoprecipitation. (F) Analysis
of the efficiency of OE-TRIM31 transfection. (G) Identification of the
ubiquitination level of Drp1. (H) Results of the CCK-8 assay for the
proliferation of SH-SY5Y cells. (I) Flow cytometry for detecting apoptosis in
SH-SY5Y cells. (J) MitoSOX Red staining to detect mitochondrial ROS, scale bar:
10 µm. (K) Measurement of mitochondrial membrane potential using JC-1
staining, scale bar: 50 µm. (L) Assessment of ATP concentration in SH-SY5Y
cells using a kit. (M) Assessment of mitochondrial respiratory chain complex I
activity. * p
These studies demonstrated that both lutein and TRIM31 can regulate ubiquitinated Drp1 levels. To further explore whether lutein affects Drp1 expression via TRIM31, TRIM31 expression was knocked down. Western blot results revealed that TRIM31 knockdown significantly decreased TRIM31 protein levels (Fig. 5A) (The original, uncropped western blot image(s) corresponding to this figure are available in the Supplementary Material). RT-qPCR results showed that lutein treatment significantly increased TRIM31 mRNA levels, whereas further knockdown of TRIM31 led to a decrease in its mRNA level (Fig. 5B). In addition, Western blot results revealed that lutein treatment increased TRIM31 protein expression and decreased Drp1 protein expression, while further TRIM31 knockdown partially reversed the effects of lutein on the expression of these two proteins (Fig. 5C) (The original, uncropped western blot image(s) corresponding to this figure are available in the Supplementary Material). These results suggest that lutein inhibits Drp1 expression by promoting TRIM31 transcription and translation.
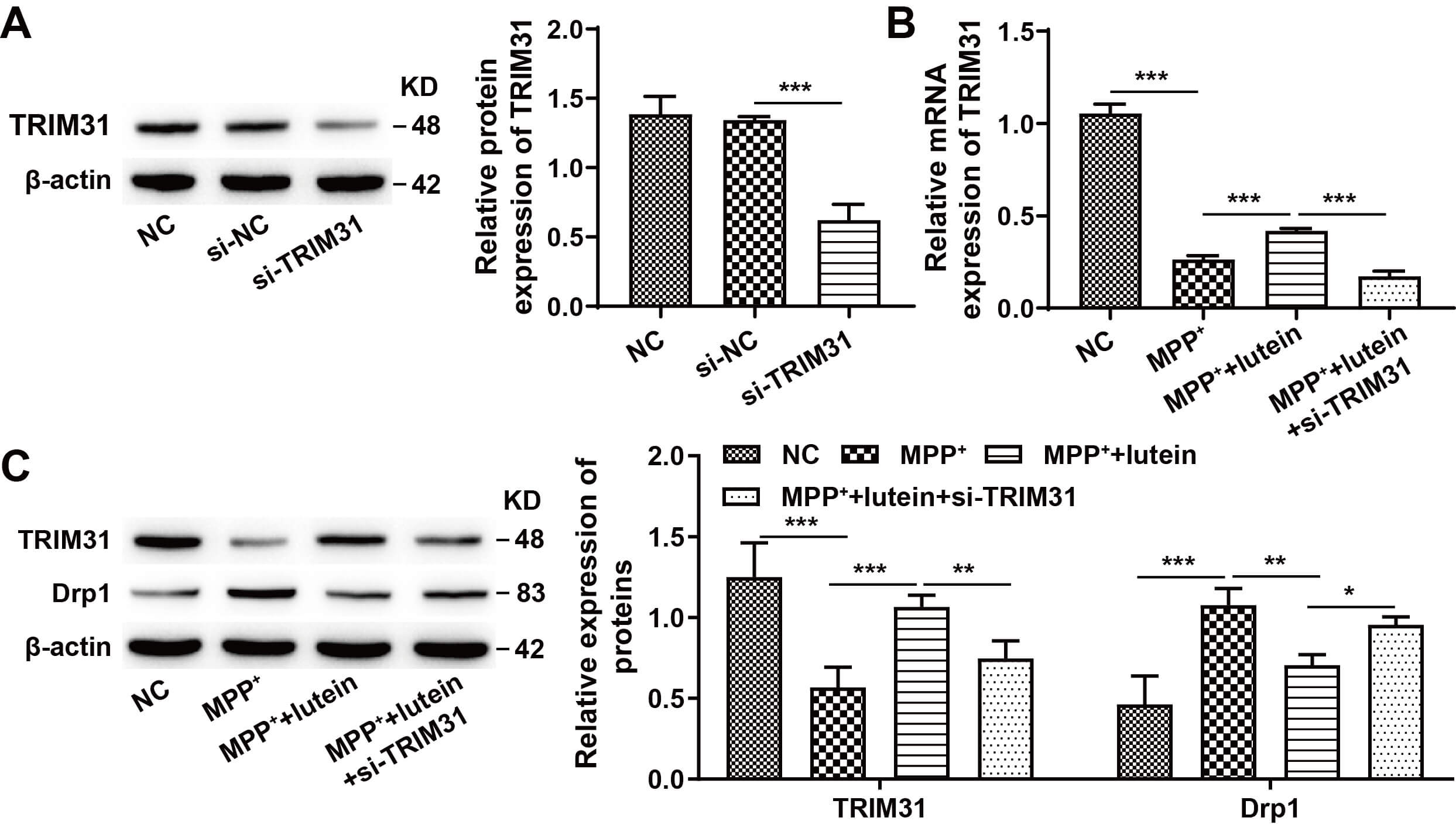 Fig. 5.
Fig. 5.
Lutein indirectly regulates Drp1 expression by promoting
TRIM31 expression. (A) Western blot detection of si-TRIM31 transfection
efficiency. (B) TRIM31 mRNA levels in SH-SY5Y cells measured by RT-qPCR.
(C) TRIM31 and Drp1 protein expression in SH-SY5Y cells assessed by Western
blotting. * p
To determine whether lutein exerts its effects through the TRIM31/Drp1 signaling pathway, Drp1 was overexpressed in SH-SY5Y cells. Western blot analysis revealed that Drp1 expression was significantly upregulated in the OE-Drp1 group compared with the NC-OE group, confirming successful transfection (Fig. 6A) (The original, uncropped western blot image(s) corresponding to this figure are available in the Supplementary Material). Functional assays, including CCK-8 and flow cytometry, revealed that both lutein treatment and TRIM31 overexpression significantly increased cell viability and suppressed apoptosis, whereas further overexpression of Drp1 effectively reversed these protective effects (Fig. 6B,C). In terms of mitochondrial function, additional Drp1 overexpression led to increased mitochondrial ROS levels, decreased mitochondrial membrane potential, reduced ATP content, and decreased respiratory chain complex I activity compared with the MPP+ + lutein group and the MPP+ + OE-TRIM31 group (Fig. 6D–G). Collectively, these results indicate that lutein alleviates MPP+-induced neuronal loss and mitochondrial dysfunction by regulating the TRIM31/Drp1 signaling pathway.
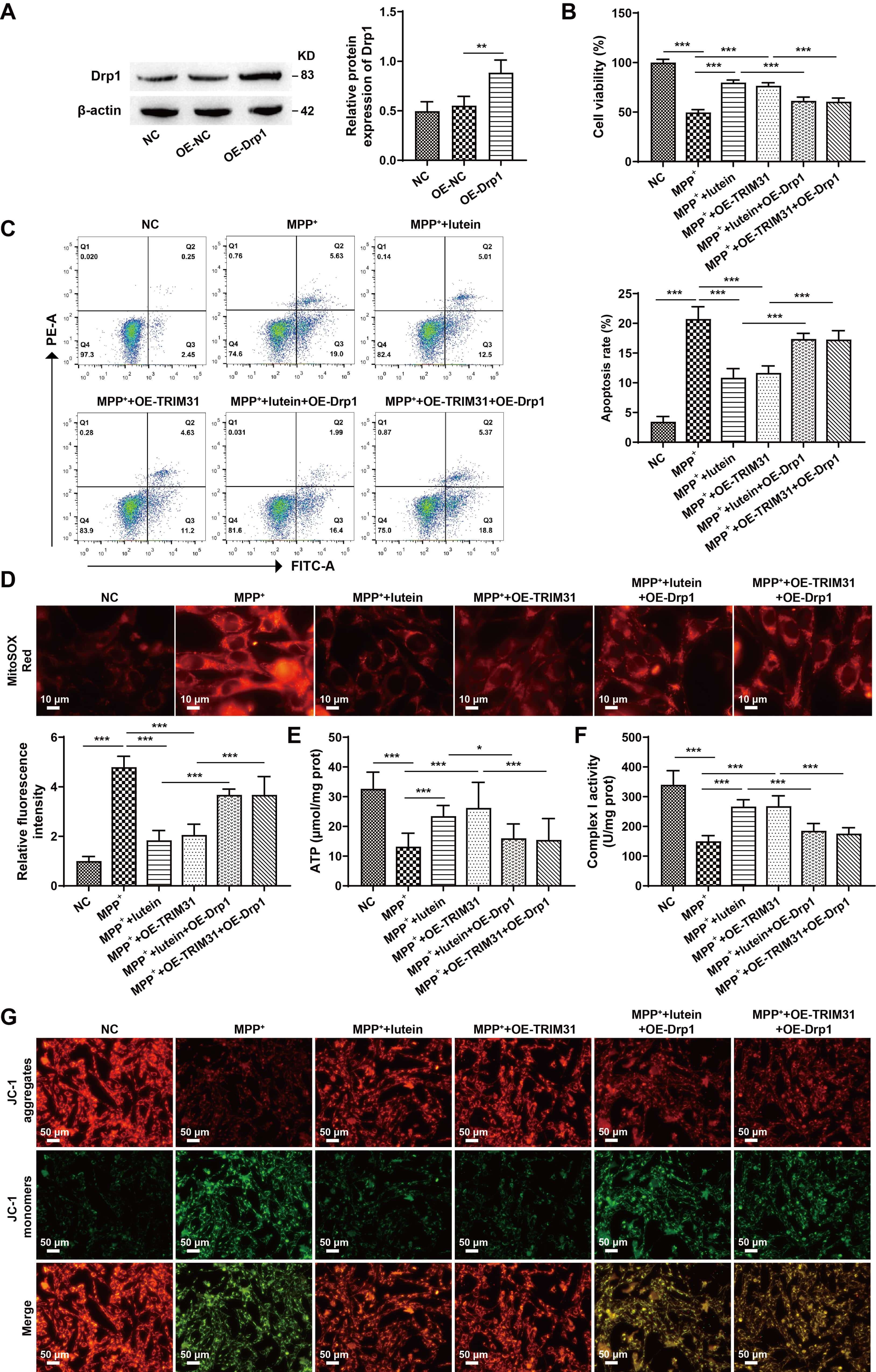 Fig. 6.
Fig. 6.
Lutein ameliorates MPP+-induced neuronal damage and
mitochondrial dysfunction via the TRIM31/Drp1 pathway. (A) Western blot analysis
of Drp1 expression to confirm transfection efficiency. (B) CCK-8 assay for
SH-SY5Y cell viability. (C) Flow cytometry for apoptosis detection in SH-SY5Y
cells. (D) Measurement of mitochondrial ROS levels using MitoSOX Red staining,
scale bar: 10 µm. (E) ATP concentration measured by an ATP assay kit. (F)
Activity of mitochondrial respiratory chain complex I. (G) Mitochondrial membrane
potential assessed by JC-1 staining, scale bar: 50 µm. * p
The pathogenesis of PD remains incompletely understood. Current evidence suggests that mitochondrial dysfunction plays a critical role in its pathology, with impaired mitochondrial complex I function in the substantia nigra potentially being a key contributing factor [20, 21]. In this study, using both in vivo and in vitro PD models induced by MPTP/MPP+, we demonstrated that lutein has significant neuroprotective effects: it ameliorated motor deficits and increased the number of TH-positive neurons in the substantia nigra in animal models, while promoting proliferation and suppressing apoptosis in SH-SY5Y cells. Mechanistic investigations revealed that the protective effects of lutein are closely associated with modulation of the TRIM31/Drp1 signaling pathway and improvements in mitochondrial function.
Lutein, a dietary carotenoid that selectively accumulates in neural tissues, has demonstrated significant potential for the prevention and treatment of neurodegenerative diseases due to its remarkable antioxidant properties [22]. Studies have shown that lutein not only alleviates methylglyoxal-induced mitochondrial damage in models of diabetic encephalopathy [23], but also significantly inhibits neuronal apoptosis and improves mitochondrial dysfunction and motor deficits in PD models by regulating the Bcl-2/Bax ratio and caspase signaling pathways [24]. Consistent with these findings, our study confirms that lutein improves motor dysfunction in PD mice, suppresses neuronal injury in vitro, and reverses MPP+-induced mitochondrial dysfunction in neuronal cells. Notably, our results further substantiate lutein’s neuroprotective effects. Additionally, lutein exhibits excellent blood–brain barrier permeability, as confirmed by previous pharmacokinetic studies, which may facilitate its direct action within the central nervous system [25]. However, while lutein shows specificity for neural targets in our experimental models, potential off-target effects, such as interactions with other carotenoid metabolic pathways or broader antioxidant responses in peripheral tissues, cannot be entirely ruled out and warrant further investigation. Collectively, these findings underscore the considerable potential of lutein as a therapeutic candidate for PD.
Drp1 belongs to the dynamin guanosine triphosphatase (GTPase) family and plays a pivotal role in controlling mitochondrial division [26]. A lack of, or alteration in, Drp1 is intimately linked to neuronal malfunction and atypical brain growth [27]. Recent research indicates that Drp1 plays a role in PD development. For example, in a rat model of PD induced by rotenone (Rot), elevated Drp1 levels correlated with mitochondrial homeostasis imbalances, whereas the neuroprotective effects of Drp1 inhibitors were notable in a PD model [28]. Additionally, several post-translational modifications of Drp1 have been reported, including phosphorylation, ubiquitination, and acetylation [29]. In this study, we found that Drp1 ubiquitination was reduced in the MPP+ cell model. Furthermore, research indicates that certain medications, including andrographolide, can modulate Drp1 in mitochondria [30]. Therefore, we further investigated whether lutein regulates mitochondrial function by influencing Drp1 ubiquitination. The results showed that lutein significantly promoted Drp1 ubiquitination and accelerated its protein degradation. As a key regulator of mitochondrial fission, excessive activation of Drp1 is known to induce ATP depletion, increase ROS production, and trigger the release of apoptotic factors, ultimately leading to cellular damage [31, 32]. These findings demonstrate that the protective effects of lutein on mitochondria are closely associated with its role in promoting Drp1 ubiquitin-mediated degradation.
TRIM31 is an E3 ubiquitin-protein ligase [33] that plays a role in regulating a range of pathological states, including inflammation, viral infections, cancer progression, and neurodegenerative disorders [34, 35, 36]. Previous studies have demonstrated that TRIM31 expression is downregulated in the substantia nigra and striatum of MPTP-induced PD mice and in MPP+-stimulated SH-SY5Y cells, with TRIM31-deficient mice exhibiting mitochondrial dysfunction [12]. Consistent with these findings, our results indicate that TRIM31 is underexpressed in MPTP-induced PD models. Furthermore, TRIM31 overexpression significantly reduced mitochondrial ROS levels, increased mitochondrial membrane potential and ATP content, and enhanced mitochondrial respiratory chain complex I activity, demonstrating its crucial role in ameliorating mitochondrial dysfunction. Furthermore, previous studies have indicated that reducing mitochondrial Drp1 levels can have therapeutic effects in MPTP-induced PD mouse models [22]. TRIM31 has been shown to regulate the ubiquitination of substrate proteins via its distinct recognition role [37]. In this study, we found that TRIM31 overexpression can increase ubiquitinated Drp1 levels. However, these findings contrast with those of Zeng et al. [11], who suggested that TRIM31 deficiency prevents the increase in Drp1 expression, thereby alleviating neuronal mitochondrial damage induced by cerebral ischemia. The reason for this discrepancy may be that TRIM31 has distinct functions in different diseases, resulting in varying regulatory roles under different pathological conditions. Furthermore, this study further indicates that lutein indirectly suppresses Drp1 expression by promoting TRIM31 transcription and translation. Therefore, lutein inhibits Drp1 expression by promoting TRIM31 expression, thereby improving mitochondrial dysfunction and exerting neuroprotective effects. While our study demonstrates that lutein acts through the TRIM31/Drp1 pathway, the precise mechanism by which lutein regulates TRIM31 expression warrants further investigation. Current evidence suggests several potential upstream signaling pathways that might mediate this regulation. Notably, the nuclear factor erythroid 2–related factor 2 (Nrf2) antioxidant pathway, known to be activated by various carotenoids, represents a plausible mechanism, as lutein has been shown to activate Nrf2 in other experimental models [38]. Alternatively, the PI3K/Akt signaling cascade, which plays a crucial role in neuronal survival and mitochondrial biogenesis, may also participate in this regulatory process [16]. Further studies utilizing specific pathway inhibitors or genetic approaches are needed to determine whether lutein directly modulates TRIM31 expression or acts through these upstream signaling mechanisms.
In summary, this study reveals the mechanism by which lutein ameliorates mitochondrial dysfunction and delays PD progression by promoting TRIM31-mediated ubiquitination and degradation of Drp1. However, several limitations should be acknowledged. First, the validation of the TRIM31/Drp1 pathway in animal models remains insufficient, and further in vivo investigations are needed to elucidate its regulatory mechanisms. Second, although the current sample sizes in each experimental group meet statistical requirements, expanding the sample size would enhance the reliability and generalizability of the findings. Despite these limitations, this study provides new evidence supporting the neuroprotective effects of lutein and establishes a theoretical foundation for its future development as a therapeutic agent for PD.
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
JD, WD and ZX designed the research study. JD, WD and XP performed the research. XP, CM and HH provided help and advice on the experiments. CM and HH analyzed the data. JD and ZX wrote the paper. ZX revised the paper for intellectual content. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All the animal experiment protocols were approved by the Laboratory Animal Welfare and Ethics Committee of the Second Affiliated Hospital of Kunming Medical University (Approval time: 2025/7/1), and the animal procedures adhered to the ARRIVE guidelines 2.0. All animal procedures were performed in compliance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Not applicable.
This research was supported by the Doctoral Research Program of the Second Affiliated Hospital of Kunming Medical University (2024BS06).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN45758.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.