







 , Yuping Shao 1,2,*
, Yuping Shao 1,2,* , Rongxiang Liang 1,2,3,*
, Rongxiang Liang 1,2,3,*1 College of Physical Education and Health, Hubei University of Chinese Medicine, 430065 Wuhan, Hubei, China
2 Hubei Shizhen Laboratory, 430065 Wuhan, Hubei, China
3 College of Sports Medicine, Wuhan Sports University, 430079 Wuhan, Hubei, China
Abstract
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disorder characterized by hyperglycemia, hyperinsulinemia, and impaired insulin sensitivity. Although classified as a metabolic disorder, T2DM also contributes to cognitive decline. Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder. T2DM is strongly associated with AD and is considered a major risk factor for its development. AD is therefore recognized as a metabolic disorder mediated by cerebral insulin resistance, often termed “type 3 diabetes”. T2DM and AD exhibit crosstalk, sharing overlapping molecular mechanisms including insulin resistance, mitochondrial dysfunction, oxidative stress, chronic inflammation, autophagy dysregulation, tau hyperphosphorylation, and β-amyloid deposition. Among these, insulin resistance may play a potential role in this interplay. As a non-pharmacological intervention, exercise demonstrates distinct advantages in preventing and managing metabolic and neurological disorders. Exercise maintains glucose homeostasis by mitigating insulin resistance, enhances insulin sensitivity, and concurrently reduces tau hyperphosphorylation and β-amyloid aggregation, thereby improving cognitive function. Building on current literature, this review explores how exercise mitigates insulin resistance to prevent and manage both T2DM and AD. It further proposes that insulin resistance may serve as a potential mechanistic link through which exercise modulates the pathological crosstalk between the two disorders.
Keywords
- Alzheimer’s disease
- exercise
- insulin resistance
- type 2 diabetes mellitus
Type 2 diabetes mellitus (T2DM) and Alzheimer’s disease (AD) are two prevalent
chronic disorders that affect middle-aged and elderly individuals. T2DM is
primarily characterized by pancreatic
IR plays a pivotal role in T2DM pathogenesis. Primarily, it promotes hyperinsulinemia and chronic hyperglycemia [13]. Notably, elevated levels of IR biomarkers have been detected in the hippocampal tissue of non-diabetic individuals with AD, suggesting that IR may be a critical etiological factor in AD pathogenesis [14]. Consequently, AD has been referred to as “type 3 diabetes” [15]. Brain insulin resistance precedes peripheral insulin signaling impairment and is regarded as a potential initiator of T2DM [16]. Current therapeutic strategies for addressing IR in both T2DM and AD involve dietary modifications, weight management, and pharmacological interventions such as metformin, which effectively reduces peripheral IR and may slow the progression of AD [17]. However, most pharmacotherapies target peripheral IR, and the effectiveness of dietary modifications and weight management in mitigating AD remains controversial [18, 19]. There is a notable lack of effective interventions that directly target brain insulin resistance. Exercise, as a non-pharmacological intervention, has been shown to enhance both peripheral and central insulin sensitivity, thereby mitigating IR [20, 21] and holding significant clinical relevance in modulating the pathological crosstalk between T2DM and AD. In summary, exercise holds promise as an effective strategy for mitigating IR and preventing both T2DM and AD. This review discusses the pivotal role of IR in the pathogenesis of both conditions and systematically examines the mechanistic links through which IR contributes to their crosstalk, exploring the therapeutic potential of exercise in mitigating IR to prevent and manage both T2DM and AD. Finally, the review explores the underlying mechanisms by which exercise may alleviate IR, aiming to provide a theoretical foundation for future clinical research and intervention.
Insulin, a 51-amino acid peptide hormone secreted primarily by pancreatic
The relative contributions of peripheral versus brain-derived insulin in modulating central nervous system (CNS) functions remain a topic of debate. Nonetheless, mounting evidence underscores the direct actions of insulin within the CNS, including the regulation of appetite, neuronal glucose uptake, growth hormone secretion, and neuroprotection. For instance, insulin administration into the central amygdala has been shown to reduce food intake and body weight, likely by modulating neuropeptide Y signaling and activity in other neuronal subpopulations involved in feeding regulation (Fig. 1A) [32]. In another study, intranasal insulin significantly enhanced the suppression of endogenous glucose production compared to placebo, suggesting that increased insulin signaling in the hypothalamus and striatum contributes to glucose homeostasis by simultaneously inhibiting hepatic glucose output and promoting peripheral glucose uptake (Fig. 1B) [33]. In addition, neurons in the hypothalamic paraventricular nucleus have been shown to transport newly synthesized insulin via their axon terminals to the anterior pituitary, where it stimulates growth hormone gene expression and secretion, thereby promoting somatic growth (Fig. 1C) [34]. In terms of neuroprotection, cultured-cell studies have demonstrated that insulin promotes neurite distribution and axonal growth in rat fetal neurons through MAPK-dependent phosphorylation [35]. Consistently, the first trial of intranasal insulin administration in healthy subjects reported improvements in attention and memory performance [36], highlighting the essential role of insulin signaling in preserving neuronal integrity and survival (Fig. 1D) [37]. Collectively, these findings highlight the multifaceted and direct roles of insulin in the CNS, which are closely linked to central nervous system homeostasis.
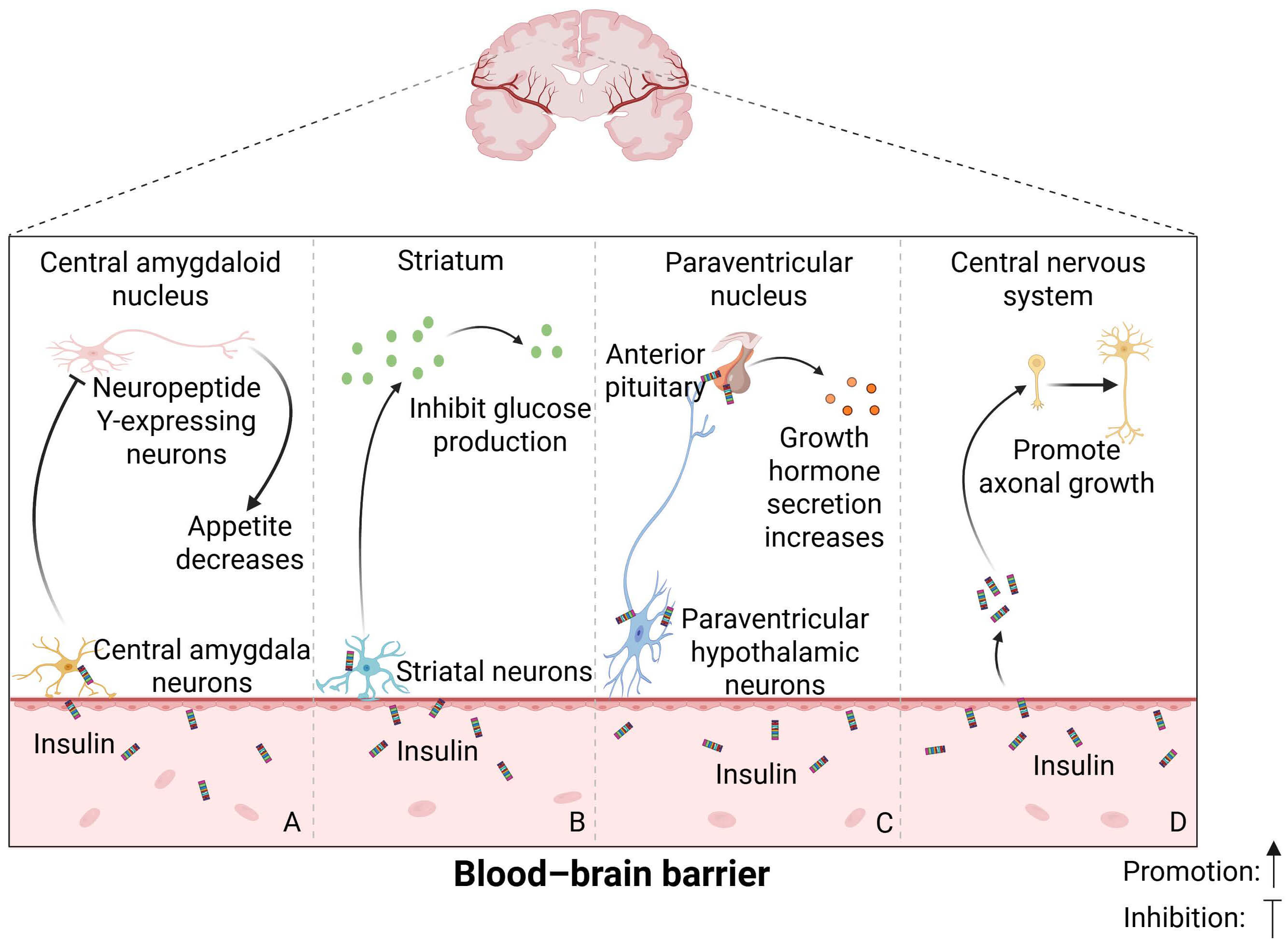 Fig. 1.
Fig. 1.
The role of insulin in the central nervous system. Insulin performs a range of critical functions within the central nervous system, including (A) the regulation of appetite, (B) modulation of neuronal glucose uptake, (C) control of growth hormone secretion, and (D) neuroprotection. Figure created with BioRender (https://www.biorender.com).
The CNS plays a pivotal role in regulating insulin secretion and maintaining glucose homeostasis, primarily through glucose-sensing neurons located in the hypothalamus [38]. For instance, high-fat diet (HFD)-induced obesity has been shown to impair hypothalamic glucose sensing mechanisms, including alterations in AMPK activity [39], thereby disrupting energy and glucose homeostasis. Moreover, recurrent hypoglycemic episodes can desensitize hypothalamic glucose-sensing neurons, impairing the ability of diabetic individuals to suppress insulin secretion and augment glucagon release during hypoglycemia, ultimately leading to a blunted sympathoadrenal response [39]. Insulin interacts with the CNS not only through direct actions but also, seemingly, through indirect pathways.
A recent study using retrograde viral tracing in Ins1-Cre mice demonstrated
direct CNS-to-pancreas connectivity: 72 hours after viral injection into the
pancreatic duct, fluorescent signals were observed specifically in the
paraventricular nucleus of the hypothalamus, but not in other regions such as the
arcuate nucleus, central amygdala, periaqueductal gray, parabrachial nucleus, or
dorsal motor nucleus of the vagus [40]. These findings indicated a discrete
neurocircuit linking a subset of hypothalamic paraventricular nucleus neurons to
pancreatic
The initial (cephalic) phase of insulin secretion is mediated by vagal
stimulation and occurs independently of direct glucose sensing by pancreatic
Notably, insulin functions not only as a regulator of glucose homeostasis but
also as a neurotrophic factor, exerting significant effects in both the central
and peripheral nervous systems [50]. For instance, elevated insulin secretion can
enhance levels of nerve growth factor (NGF), which binds with high affinity to
calmodulin-dependent protein kinase A, promoting the growth and development of
central and peripheral neurons, accelerating repair after neural injury, and
sustaining normal neural function [51]. A number of studies have indicated
considerable overlap between insulin and NGF signaling pathways [52], with
insulin synergistically enhancing NGF-induced neurite outgrowth [53]. Conversely,
NGF can increase Na⁺ current density in pancreatic
Taken together, insulin orchestrates a complex network of signaling mechanisms between peripheral and central compartments, regulating metabolism, inflammatory responses, and neuronal integrity to maintain physiological homeostasis (Fig. 2).
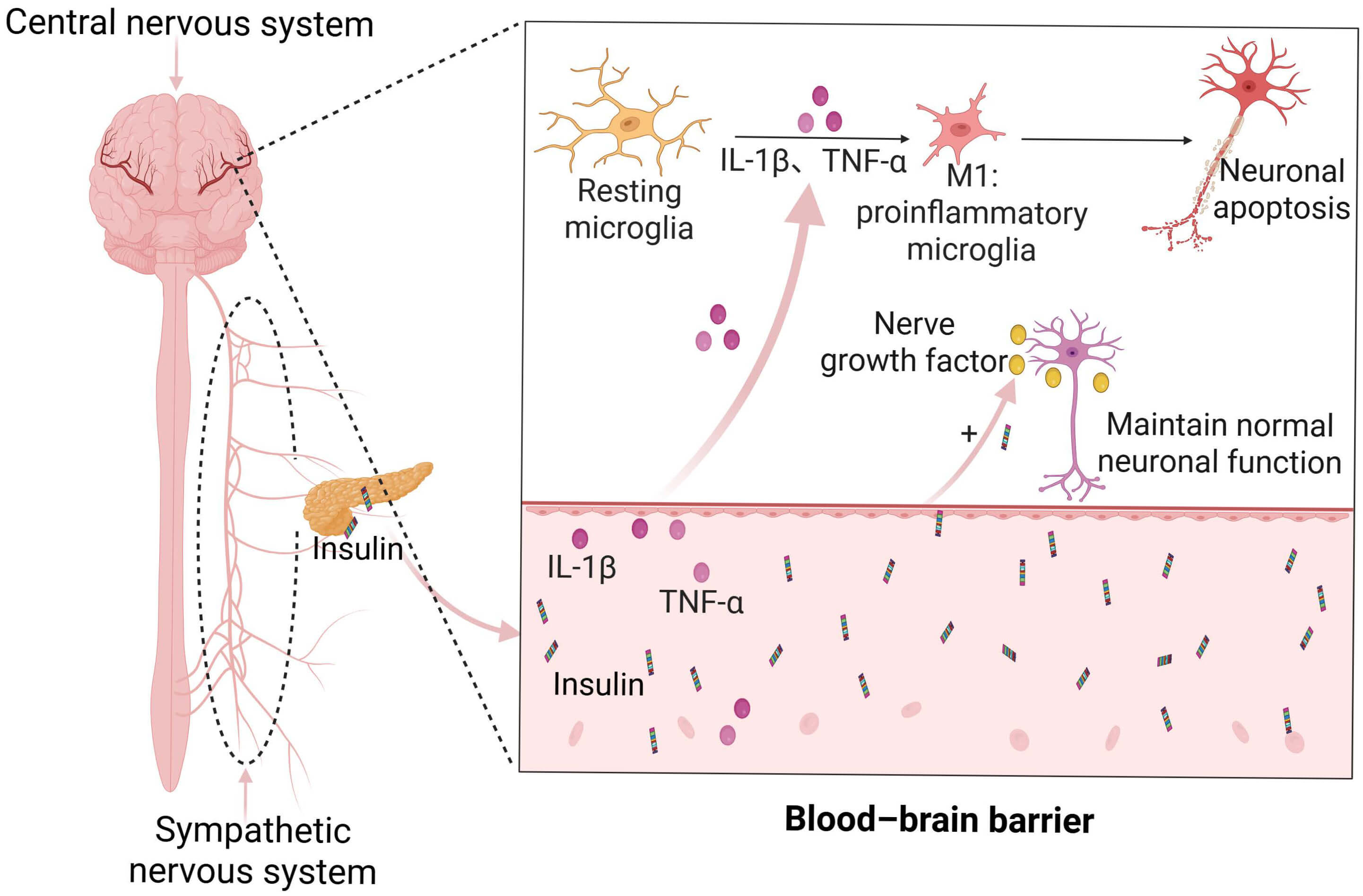 Fig. 2.
Fig. 2.
The indirect relationship between insulin and the central
nervous system. The central nervous system communicates bidirectionally with the
periphery via sympathetic neuronal projections to pancreatic
IR refers to a diminished biological response of insulin-targeted tissues to
physiological levels of insulin, and is also termed reduced insulin sensitivity
[55]. IR represents a central pathological feature of T2DM, affecting multiple
tissues including the liver, skeletal muscle, myocardium, and pancreatic
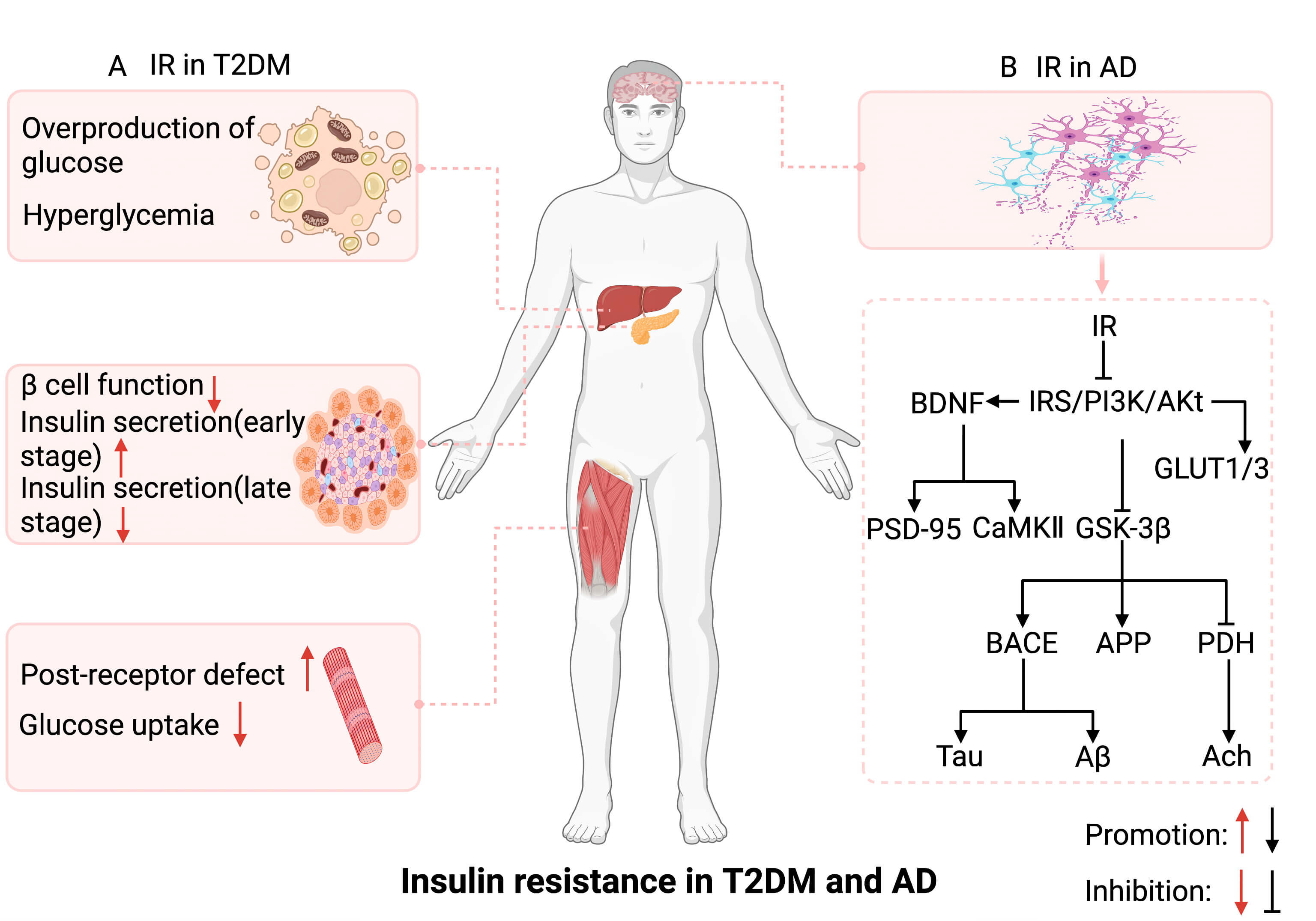 Fig. 3.
Fig. 3.
The potential mechanism of insulin resistance in the
pathogenesis of T2DM and AD. (A) Insulin resistance in T2DM involves multiple
tissues, including the liver, skeletal muscle, and
Despite representing approximately 2% of total body weight, the human brain
consumes nearly 25% of the body’s resting glucose supply [69]. IR impairs
cerebral energy metabolism in AD patients, resulting in neuronal energy
deficiency [70]. Compelling evidence has indicated a strong association between
AD and metabolic dysfunction, with impaired glucose metabolism emerging more than
a decade prior to clinical-symptom onset [71]. As glucose cannot freely cross
cell membranes, its cellular uptake relies on facilitative GLUTs. In the brain,
GLUT1 and GLUT3 serve as the primary transporters mediating glucose entry into
neurons [72]. IR compromises the function of GLUT1 and GLUT3, reducing neuronal
glucose uptake and subsequently lowering levels of uridine diphosphate
N-acetylglucosamine (UDP-GlcNAc), a key glucose-derived metabolite [73].
UDP-GlcNAc is a direct donor substrate for O-linked N-acetylglucosaminylation
(O-GlcNAcylation). Reduced tau O-GlcNAcylation has been implicated in tau
hyperphosphorylation and neurofibrillary degeneration [74], suggesting that
diminished cerebral glucose uptake may actively contribute to AD progression
[75]. Moreover, decreased brain energy metabolism is associated with enhanced
BACE1 activity, promoting the generation and accumulation of A
Notably, brain-derived neurotrophic factor (BDNF) plays a pivotal role in
neuronal growth and the maintenance of synaptic plasticity. Elevated BDNF levels
are critical for mitigating the progression of AD [77]. In the brain, insulin
promotes synaptogenesis by upregulating BDNF expression via activation of the
IRS/PI3K/Akt signaling cascade [78]. BDNF binds to its receptor TrkB and
modulates both postsynaptic density protein 95 and calmodulin-dependent protein
kinase II, thereby enhancing synaptic plasticity. Inhibition of IR elevates
endogenous BDNF levels [79, 80] (Fig. 3B). IR-driven chronic inflammation
exacerbates AD progression, potentially mediated by the release of
proinflammatory cytokines from activated microglia [81, 82]. Furthermore,
mitochondrial dysfunction and endoplasmic reticulum (ER) stress, induced by IR,
contribute to the progression of AD pathology [83]. Specifically, mitochondrial
impairment reduces cellular energy production and elevates reactive ROS,
impairing neuronal function and structure [84]. Concurrently, ER stress
exacerbates the accumulation of misfolded proteins, disrupting intracellular
homeostasis and diminishing neuronal efficiency [85]. Additionally, IR has been
shown to suppress neuronal autophagy, promoting the aggregation of A
T2DM and AD exhibit bidirectional crosstalk, sharing numerous overlapping
molecular mechanisms—including IR, mitochondrial dysfunction, oxidative stress,
chronic inflammation, impaired autophagy, tau hyperphosphorylation, and
A
Mitochondria, as essential organelles, play a central role in energy metabolism,
apoptosis, and the modulation of ROS [90, 91]. Mitochondrial dysfunction can be
characterized by aberrant alterations in biogenesis, fusion-fission dynamics,
mitophagy, and overall mitochondrial function [92]. Mitochondrial biogenesis is
primarily regulated by peroxisome-proliferator-activated receptor-gamma
coactivator 1-alpha (PGC-1
Mitochondrial dysfunction can induce IR and is closely associated with the
development of T2DM [104, 105]. It may also represent a key factor contributing
to cognitive impairment [106]. In states of IR, mitochondrial dysfunction in the
brain is accompanied by impaired cognitive function [107] (Fig. 4A). Intranasal
insulin administration, which ameliorates IR, has been shown to restore
methamphetamine-induced cognitive impairment, likely due to the activation of the
insulin signaling pathway (PI3K/Akt/GSK-3
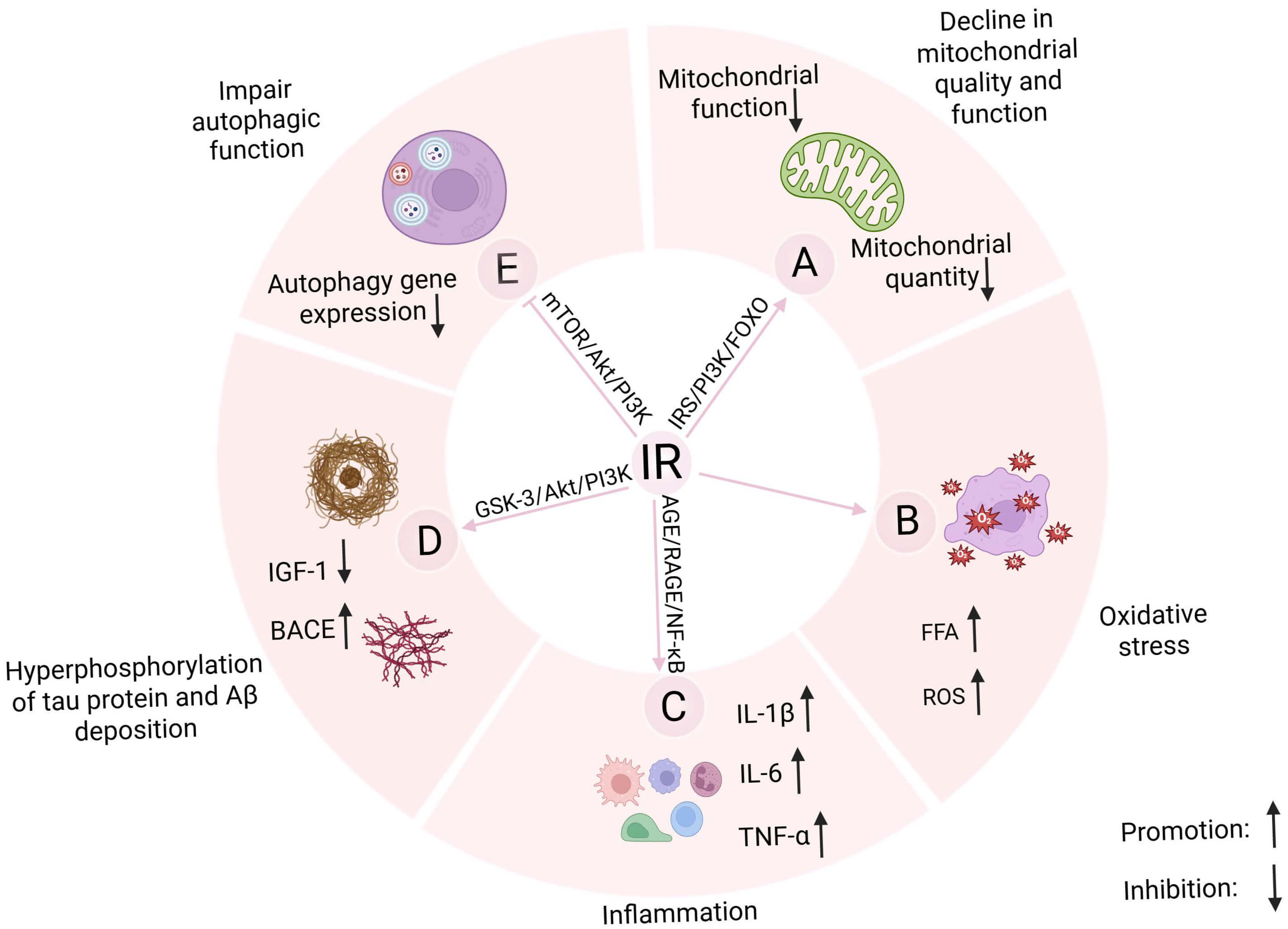 Fig. 4.
Fig. 4.
Effect of insulin resistance on the mechanism of crosstalk
between T2DM and AD. Insulin resistance may engage in a vicious cycle with (A)
mitochondrial dysfunction, (B) oxidative stress, (C) chronic inflammation, and
(E) autophagy dysregulation, mutually exacerbating these processes while (D)
contributing to A
Disruption of redox homeostasis leads to the accumulation of ROS or reactive
nitrogen species, which interferes with normal cellular signaling, induces damage
to biomolecules—including proteins, lipids, and DNA—and alters the expression
of stress-responsive genes, compromising cellular integrity and potentially
triggering cell death [114]. The central mechanism of oxidative stress lies in
the imbalance between ROS production and clearance [115, 116]. Excessive ROS
generation compromises antioxidant defenses and induces oxidative damage, which
serves as a critical contributor to the pathogenesis of both T2DM and AD [117].
In streptozotocin-induced diabetic mice, levels of superoxide, protein oxidation
products, and lipid peroxidation products are markedly elevated in the brain,
accompanied by a significant reduction in the activities of antioxidant enzymes
such as superoxide dismutase (SOD) and catalase (CAT) [118]. In AD patients, the
enzymatic activities of key antioxidants—including glutathione peroxidase
(GPX), CAT, and peroxiredoxins—are also diminished in the superior temporal
gyrus [119]. Moreover, oxidative stress exacerbates T2DM progression [120],
thereby promoting the accumulation of advanced glycation end products (AGEs). The
specific binding of AGEs to their receptor (RAGE) suppresses antioxidant enzyme
activity and reduces glutathione (GSH) levels, thereby amplifying ROS generation
[121]. Notably, persistently elevated levels of AGEs in T2DM patients may
exacerbate amyloid pathology by inhibiting
Studies have shown that oxidative stress contributes to IR, mainly manifested as
an increase in oxidative stress markers and a significant reduction in
insulin-receptor activation [125, 126]. Conversely, IR also exacerbates oxidative
stress [127]. Under IR conditions, increased oxidative stress is primarily
reflected by enhanced NADPH oxidase activity, alongside reduced activities of
antioxidant enzymes such as CAT, GPX, and SOD, as well as decreased GSH synthesis
[128]. The degree of oxidative damage in the brain is positively correlated with
the IR index, suggesting that IR contributes to the development of neuronal
oxidative stress [129]. Under IR conditions, excessive fatty acids crossing the
blood-brain barrier may elevate ROS production in neurons, inducing ER stress and
neuronal toxicity, thereby exacerbating AD pathology [130, 131] (Fig. 4B). In
T2DM, IR impairs
Inflammation is primarily regulated by two categories of cytokines:
pro-inflammatory cytokines such as IL-1
A vicious cycle exists between IR and the inflammatory response, with each
exacerbating the other [144]. Under physiological conditions, insulin is the only
hypoglycemic hormone in the human body exerting anti-inflammatory effects [145].
For example, insulin in combination with antioxidants markedly suppressed
TNF-
Two pathological hallmarks of AD are the aberrant accumulation of insoluble
A
Insulin has been shown to inhibit A
Autophagy is a lysosome-dependent degradation mechanism responsible for clearing
damaged organelles and aberrantly aggregated proteins within cells [178]. During
brain aging, defects in the autophagy-lysosomal pathway increase neuronal
vulnerability, and genetic mutations affecting autophagic or lysosomal function
are causally linked to the development of neurodegenerative disorders, including
AD [179]. Similarly, dysregulated autophagy in pancreatic
Studies have demonstrated that activation of autophagy alleviated IR in both
T2DM and AD [186, 187], further underscoring the bidirectional interplay between
autophagy and IR [86]. Under physiological conditions, insulin exerts anabolic
effects by activating the PI3K/Akt/mTORC1 (mechanistic target of rapamycin
complex 1) pathway, thereby suppressing autophagic flux [188]. Moreover, insulin
has the potential to suppress autophagy in various diseases [189, 190], including
diabetes [191]. In addition, insulin can effectively inhibit astrocytic autophagy
via transcriptional regulation, thereby improving neural function [192]. It is
intriguing that although IR triggers autophagy upregulation in adipose tissue
[193], paradoxical suppression of both autophagic activity and expression of key
autophagy-related genes occurs under conditions of IR and hyperinsulinemia [194]
(Fig. 4E). Transcriptomic analyses of skeletal muscle from individuals with
longstanding and severe IR in the context of T2DM have revealed the
downregulation of autophagy-related genes, suggesting that IR may contribute to
the suppression of autophagy in muscle tissue [195]. In addition, IR impairs
neuronal autophagy, promoting the accumulation of A
IR is a potential pathogenic mechanism in both T2DM and AD, and its inhibition
can effectively delay progression in both conditions. Exercise, a
non-pharmacological intervention, offers significant advantages in preventing and
treating neurological and metabolic disorders. By ameliorating IR, physical
exercise helps maintain glucose homeostasis and stabilize blood glucose levels,
enhancing insulin sensitivity. Moreover, it alleviates the excessive
phosphorylation of tau protein and the aggregation of
As chronic conditions, T2DM and AD profoundly impair patients’ quality of life. Among various therapeutic strategies, exercise emerges as a non-pharmacological intervention that mitigates both conditions by effectively attenuating IR [196]. Studies have confirmed that in patients with T2DM [197], aerobic exercise can significantly improve glycemic control, enhance insulin sensitivity [198], and even upregulate genes critical for maintaining glucose homeostasis and reducing IR [199]. For example, T2DM patients who performed aerobic exercise 4–5 days per week, 60 minutes per day at 75% of VO2 peak for 12–16 weeks, exhibited marked reductions in fasting blood glucose levels and improvements in insulin sensitivity (Table 1, Ref. [198, 199, 200, 201, 202, 203, 204, 205, 206, 207, 208, 209, 210, 211, 212, 213]) [200].
| Disease model | Type of exercise | Exercise dose | Related functional and molecular changes | Source and tests | Reference |
| T2DM patients | Aerobic exercise | 60 min/time, 75% VO2 peak, 4–5 times/week, 12–16 weeks | Fasting blood glucose |
Plasma | [200] |
| Short-term interval training | 2 |
Insulin sensitivity and Brain glucose metabolism |
Brain and plasma | [211] | |
| Short-term exercise training (treadmill walking and stationary cycling) | 50–60 min/time, 80–85% HRmax, 7 days | Insulin sensitivity |
Plasma | [212] | |
| Structured exercise programs (aerobic, resistance, and combined) | 15–45 min/time, 3–5 times/week, 12 weeks | Fasting blood glucose, HbA1c |
Plasma | [204] | |
| T2DM mice | Treadmill running | 10 m/min, 60 min/d, 5 times/week, 4 weeks | Insulin sensitivity and GLUT4 |
Plasma | [198] |
| Aerobic exercise | 45 min/time, 5 times/week, 8 weeks | IRS-1 and Akt |
Liver | [199] | |
| High-intensity interval training | 16–26 m/min, 4 min/time, 5 times/week, 8 weeks | Fasting blood glucose |
Plasma | [202] | |
| 16–26 m/min, 4 min/time, 5 times/week, 8 weeks | IRS1/PI3K/Akt |
Liver and Plasma | [203] | ||
| T2DM rat | Chronic resistance exercise | 3 sets/4 reps daily, 3 times/week, 8 weeks | Muscle musclin protein and fasting blood glucose |
Muscle and Plasma | [201] |
| AD patients | Treadmill running | 4 km/h, 80% HRmax | IGF-1 |
Plasma | [208] |
| Aerobic exercise | 45–60 min/time, 4 times/week, 24 weeks | Insulin sensitivity |
Plasma | [213] | |
| AD rat | Treadmill running | 8–14 m/min, 30 min/times, 5 times/week, 6 weeks | IRS/PI3K/Akt/GSK3 |
Brain | [206] |
| 10–15 m/min, 30–60 min/times, 4 weeks | IGF-1, BDNF, and GLUT4 |
Hypothalamus | [207] | ||
| AD mice | Treadmill running | 3–13 m/min, 30–50 min/times, 1/d, 6 times/week, 12 weeks | IRS/PI3K and Cognitive function |
Hippocampus | [205] |
| 22 m/min, 60 min/times, 5 times/week, 16 weeks | GLUT1 and Cognitive function |
Plasma | [209] | ||
| AD-like model | Aerobic exercise and resistance exercise | 60 min/time, 3 times/week, 6 weeks | IGF-1 and Cognitive function |
Brain and plasma | [210] |
IGF-1, insulin-like growth factor 1; BDNF, Brain-Derived Neurotrophic Factor;
PGC-1
In the context of exercise-mediated suppression of IR, myokines have garnered
increasing attention. Evidence from both animal and human studies suggests that
myokines may contribute to IR in T2DM by impairing glucose metabolism [214, 215].
Resistance training may attenuate IR and hyperglycemia in T2DM rats by
downregulating myokine expression and activating the Akt/GLUT4 signaling pathway
[201]. In HFD mouse models, eight weeks of high-intensity interval training
(HIIT) or moderate-intensity continuous training (MICT) significantly enhanced
hepatic PI3K/Akt/GSK3
Studies in AD mice have shown that physical exercise may mitigate IR and
cognitive deficits by enhancing hippocampal insulin signaling, particularly
through IRS-1, Akt, and GSK-3
IR is a hallmark of T2DM and has been strongly implicated in AD [221]. Insulin
plays a pivotal role in systemic metabolism by activating the PI3K/Akt signaling
pathway. This cascade critically regulates glucose metabolism, neuronal
homeostasis, and mitochondrial function. After IR onset, the PI3K/Akt pathway is
inhibited, exerting detrimental effects on downstream targets, including
GSK-3
Exercise alleviates IR through multiple, convergent mechanisms, with
mitochondrial enhancement and redox regulation as central nodes. By stimulating
mitochondrial biogenesis and boosting respiratory activity, exercise delays aging
and attenuates the progression of metabolic disorders [226, 227]. In T2DM models,
it not only suppresses IR via PI3K/Akt activation [228] but also acts in concert
with the mitochondrial-derived peptide MOTS-c to enhance glucose metabolism
through the AMPK–PGC-1
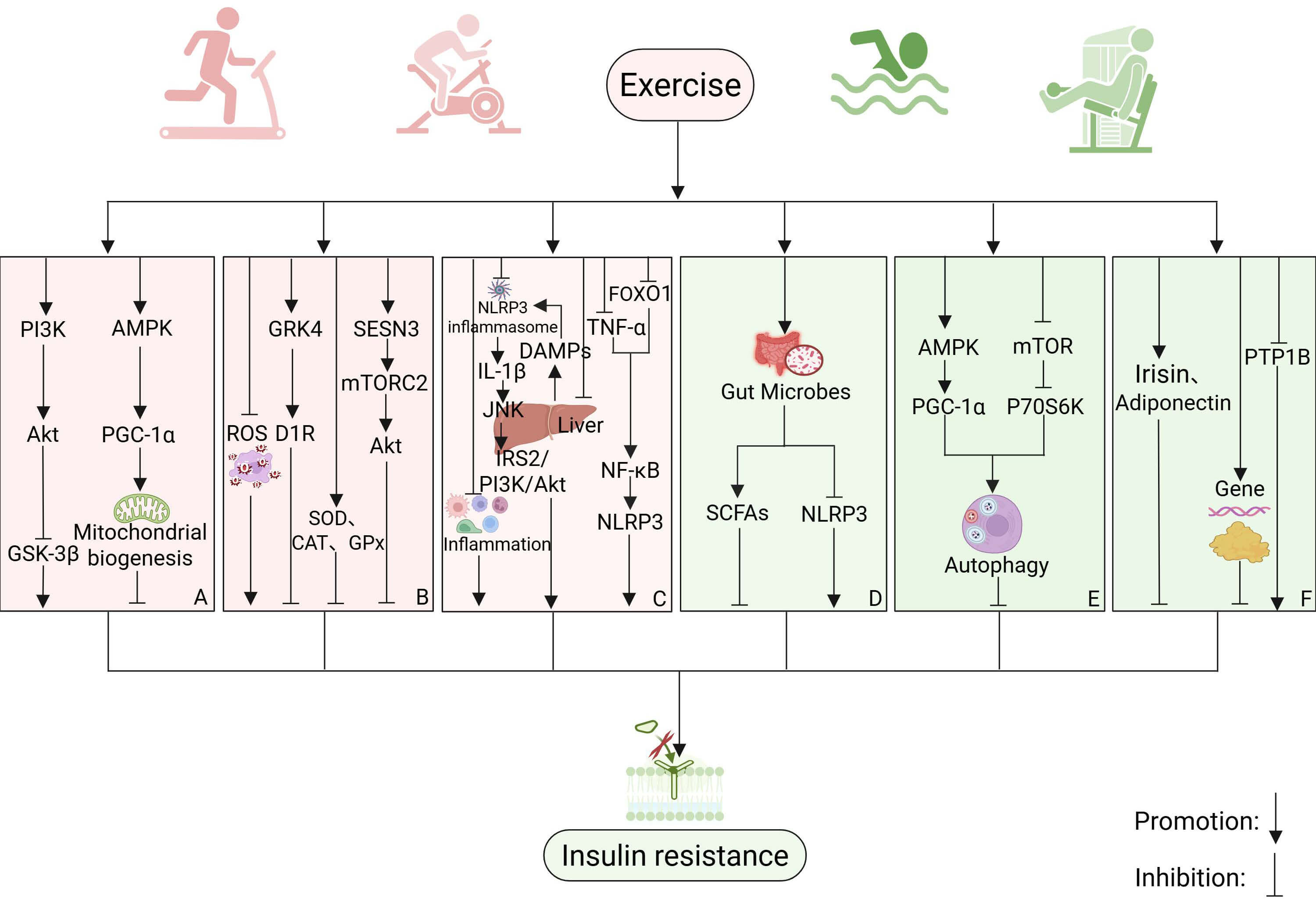 Fig. 5.
Fig. 5.
The potential mechanisms by which exercise alleviates insulin
resistance. (A) Exercise attenuates insulin resistance by targeting the
PI3K/Akt/GSK-3
Exercise also counteracts oxidative stress through complementary pathways. It inhibits ROS accumulation and augments Akt signaling to lower skeletal muscle IR [231], while simultaneously downregulating the ROS/GRK4 cascade, thereby restoring D1 receptor function and improving insulin sensitivity [198]. In parallel, exercise strengthens endogenous antioxidant defenses (SOD, CAT, GPX, GR) [232] and upregulates the stress-responsive protein SESN3, which via the SESN3/mTORC2/Akt pathway enhances GLUT4 expression and prevents obesity-associated hepatic steatosis and IR [233] (Fig. 5B).
Collectively, these findings indicate that exercise may mitigate IR by reinforcing mitochondrial function and antioxidant capacity, offering a promising strategy for the prevention and treatment of T2DM and AD.
IR and inflammatory responses mutually reinforce each other, creating a vicious
cycle [234]. Exercise effectively mitigates IR by suppressing microglial
activation and reducing inflammation [235, 236]. Correlative analyses indicate
that IR is positively associated with NLRP3 inflammasome activity, suggesting
that exercise may alleviate IR through modulation of this pathway [237]. In
skeletal muscle, exercise reduces inflammation and muscle IR by inhibiting the
TNF-
The gut microbiota plays a pivotal role in maintaining physiological homeostasis and has been closely implicated in the pathogenesis of both AD and T2DM through its regulation of inflammation and oxidative stress [241, 242, 243, 244]. IR serves as a potential pathogenic mechanism in both T2DM and AD, engaging in bidirectional crosstalk with the gut microbiota. IR induces dysbiosis of gut microbial composition, whereas its suppression can restore microbial community structure and function, thereby attenuating inflammation [245, 246, 247]. Conversely, modulating the gut microbiota effectively mitigates IR and associated inflammatory responses [248]. For instance, conjugated linoleic acid reduces IR by regulating host-microbe metabolic and immunological interactions [249]. Moderate exercise promotes the proliferation of beneficial bacteria and enhances gut microbiota diversity. These beneficial microbes regulate mucosal immunity, strengthen the gut barrier, and produce metabolites such as short-chain fatty acids (SCFAs), which help prevent gastrointestinal diseases [250]. Studies have shown that SCFAs enhance glucose homeostasis, with elevated levels improving insulin sensitivity [251, 252]. Moreover, the gut microbiota may play a pivotal role in mediating the effects of exercise on the regulation of the NLRP3 inflammasome [253] (Fig. 5D). Animal studies have indicated that exercise can counteract the negative impact of altered gut microbiota on brain function, mitigating the reduction in hippocampal neurogenesis and related behaviors in adult rats [254]. This suggests that exercise may delay the progression of AD and T2DM by modulating the gut microbiota and its metabolic products [255, 256, 257]. Although direct studies on the role of exercise in modulating the gut microbiota to reduce IR are limited, a bidirectional interaction exists between the gut microbiota, exercise, and IR. In summary, exercise may reduce IR through the regulation of gut microbiota, providing a potential therapeutic pathway for the treatment of T2DM and AD.
As a shared mechanism in both T2DM and AD, autophagy is closely linked to IR. In
IR states, autophagic flux and the expression levels of autophagy markers are
significantly increased [258]. Suppression of miR-188, a regulator of the
autophagy-related gene Atg12, exacerbated IR severity [259], indicating
that autophagic dysfunction may initiate the development of IR [260]. Another
study confirmed that induction of aberrant autophagy in granulosa cells increased
the LC3-II/LC3-I ratio, elevated Atg7 levels, and decreased p62, thereby
promoting IR development [261]. Conversely, activating autophagy within
physiological limits helped reduce IR. Studies have shown that autophagy exerts a
protective effect against hyperuricemia-induced hepatic IR [262]. Additionally,
administration of Kun-Dan decoction alleviated IR in high-fat-diet rats by
activating autophagy through the AMPK/mTOR pathway, whereas dihydromyricetin
treatment enhanced autophagy via the AMPK/PGC-1
In addition to the aforementioned mechanisms, several other factors, including
molecular and epigenetic regulation, are believed to contribute to the link
between exercise and IR. Irisin, a myokine produced during exercise, displays
pleiotropic functions, such as metabolic modulation and neuroprotection, and may
play a crucial role in treating metabolic and neurodegenerative diseases. Pilates
and resistance training, as effective exercise interventions, can significantly
reduce IR in overweight women by stimulating irisin secretion [268]. Adiponectin,
a protein hormone secreted by adipocytes, stimulates mitochondrial biogenesis by
activating PGC-1
Numerous studies have demonstrated that exercise plays a critical role in preventing and managing both T2DM and AD by reducing IR. However, the optimal exercise modality, intensity, duration, and frequency for different diseases and disease stages remain to be precisely defined. Some studies indicate that aerobic training alone may not improve systemic IR [273], whereas similar exercise regimens, when matched for intensity and duration, appear to exert differential effects on IR [274]. These discrepancies may stem from variations in sample size, the uncontrollability of human interventions, and differences between human and animal models. Consequently, translating data from animal studies into precise exercise prescriptions for humans remains a major challenge.
Furthermore, critical questions regarding IR as a potential mechanistic link between T2DM and AD remain unresolved. For instance, the temporal sequence through which peripheral and central IR are initiated, and whether a causal relationship exists between them, is still unclear. In addition, given the unique characteristics of the human brain, reliable methods to assess the stage of cerebral IR are lacking, and it is uncertain whether biomarkers could facilitate monitoring of brain IR and disease progression. Notably, much of the current understanding of AD derives from animal models, with limited support from human studies.
Collectively, these limitations point to key avenues for future research, including the investigation of exercise as a modulator of IR in the context of T2DM and AD, and underscore the need for further studies to validate and expand upon the perspectives presented in this review.
A substantial body of animal and clinical studies suggests a bidirectional interplay between T2DM and AD, with numerous overlapping molecular mechanisms, including IR, mitochondrial dysfunction, oxidative stress, chronic inflammation, and cellular autophagy. Among these, IR appears to play a central role. IR impedes insulin signaling, energy metabolism, and cellular autophagy, and simultaneously exacerbates oxidative stress and inflammation, thereby increasing the risk of both T2DM and AD (Fig. 6). Exercise, as a non-pharmacological intervention, has significant potential to mitigate both T2DM and AD by restraining IR. However, the relationship between brain IR and peripheral IR remains controversial, and validation requires further investigation. Additionally, the precise mechanisms by which exercise reduces IR are not yet fully understood, and further evidence is needed in order to elucidate the exact pathways involved. IR may represent the potential mechanism through which exercise attenuates the interplay between T2DM and AD, potentially serving as a critical target for prevention and treatment strategies.
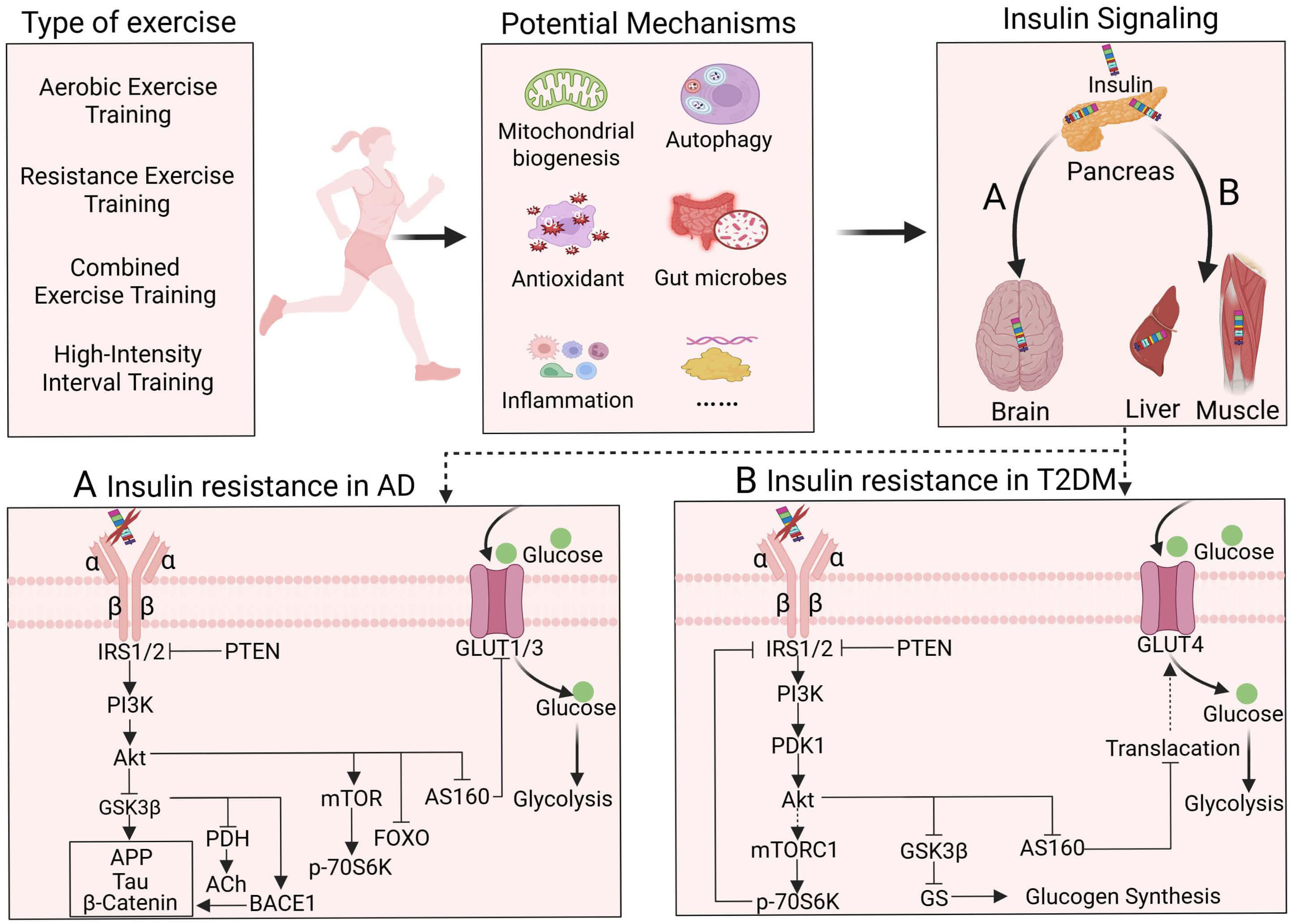 Fig. 6.
Fig. 6.
The potential molecular pathways of exercise to alleviate insulin resistance and the pathways of insulin resistance in T2DM and AD. (A) The induction pathway of insulin resistance in AD. (B) The induction pathway of insulin resistance in T2DM. Standard arrows indicate activation, bar-headed lines indicate inhibition, and dotted arrows indicate indirect or potential links. Figure created with BioRender (https://www.biorender.com).
T2DM, Type 2 diabetes mellitus; AD, Alzheimer’s disease; IR, insulin resistance; NFTs, neurofibrillary tangles; IRS, insulin receptor substrates; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B; GLUTs, glucose transporters; MAPK, mitogen-activated protein kinase; BBB, blood–brain barrier; CNS, central nervous system; HFD, high-fat diet; IL, Interleukin; TNF, Tumor Necrosis Factor; ROS, reactive oxygen species; NGF, nerve growth factor; GSK-3, glycogen synthase kinase-3; BACE1,
BW contributed to the study conception and design, collected and sorted references, designed and drew the figures and tables, and wrote the manuscript. YPS and RXL contributed to the study conception, design, and critical revision of the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We express our gratitude to all those, excluding the authors, who helped during the writing of this manuscript.
This work was supported by the Natural Science Foundation of Hubei Province of China (2023AFB978).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.