



 , Qi Li 1, Zhijun Zhong 1, Qian Ouyang 1, Xueliang Zou 3, Kaiyu Yue 4, Dongyuan Yao 1,*
, Qi Li 1, Zhijun Zhong 1, Qian Ouyang 1, Xueliang Zou 3, Kaiyu Yue 4, Dongyuan Yao 1,*
1 Neurological Institute of Jiangxi Province and Department of Neurology, Jiangxi Provincial People’s Hospital, The First Affiliated Hospital of Nanchang Medical College, and Xiangya Hospital of Central South University at Jiangxi, 330038 Nanchang, Jiangxi, China
2 Joint Program of Clinical Medicine/Biomedical Sciences, Queen Marry College, Nanchang University, 330006 Nanchang, Jiangxi, China
3 Department of Psychology, Jiangxi Metal Hospital, Nanchang University, 330029 Nanchang, Jiangxi, China
4 Department of Clinical Medicine, The Second Clinical School, Tongji Medical College, Huazhong University of Science and Technology, 430021 Wuhan, Hubei, China
Abstract
Sleep paralysis, colloquially known as “ghost pressing” is a state of momentary bodily immobilization occurring either at the onset of sleep or upon awakening. It is characterized by atonia during rapid eye movement (REM) sleep that continues into wakefulness, causing patients to become temporarily unable to talk or move but possessing full consciousness and awareness of their surroundings. Sleep paralysis is listed in the International Classification of Sleep Disorders, 3rd Edition (ICSD-3) as a parasomnia occurring during REM sleep that be classified as either isolated or narcolepsy-associated. Several brain areas, including the forebrain, hypothalamus, and brainstem, as well as several neurotransmitters and modulators, are involved in the control of REM sleep. The primary brain region responsible for inducing muscle paralysis during REM sleep is the subcoeruleus nucleus, also known as the sublaterodorsal (SLD) nucleus in rats. Sleep paralysis results from the inability to immediately restore muscle tone during the transition from sleep to wakefulness. In this article, we systematically review the neural circuit that controls REM sleep and the underlying mechanisms, predisposing factors, clinical characteristics, and treatments for sleep paralysis. We also compare isolated sleep paralysis (ISP) and narcolepsy-associated sleep paralysis and speculate upon the role of microsleep in sleep paralysis.
Keywords
- sleep paralysis
- narcolepsy
- parasomnia
- REM sleep
- sleep disorder
- sleep wake disorder
- mechanism
Sleep is one of the most important physiological phenomena in mammals. It can be categorized into rapid eye movement (REM) and non-rapid eye movement (NREM) sleep [1, 2]. NREM sleep can be further divided into NREM stages 1, 2, and 3. Contrary to popular belief, sleep is not a passive state but one that involves dynamic cellular functions that are controlled and regulated by precise neural structures and networks.
REM sleep, also called active sleep, was first discovered in humans by sleep research pioneers Aserinsky and Kleitman in 1953 [3]. Alternating with NREM sleep (quiescent sleep periods), it is characterized by rapid eye movements, low-amplitude and high-frequency electroencephalography (EEG) waves, and muscle atonia, which was first described in cats by Jouvet et al. [4]. If muscle tone is not suppressed during REM sleep, REM sleep behavioral disorder can occur and cause injury to the patient and their sleep partners [5, 6].
Although sleep paralysis can occur with narcolepsy (i.e., narcolepsy-associated sleep paralysis) [7], it more commonly occurs without it (isolated sleep paralysis, ISP) [8, 9], and many people experience it several times in their lives.
Narcolepsy is a chronic sleep disorder characterized by excessive daytime sleepiness, cataplexy (partial or complete collapse due to generalized muscle weakness), pre-sleep/first awakening hallucinations. Sleep paralysis and narcolepsy can occur in the same patients, and approximately 30–50% of patients with narcolepsy experience sleep paralysis [7]. Whether or not sleep paralysis occurs in narcolepsy patients might be related to the type of narcolepsy, lifestyle, and psychological factors (stress, anxiety, etc.). Sleep paralysis occurs more often with type 1 narcolepsy (NT1)—due to lack of orexin in the central nervous system—than NT2 [10].
Sleep paralysis itself is an innocuous condition, but it is frequently viewed with concern and even dread. If there are frequent episodes of sleep paralysis accompanied by excessive daytime sleepiness, cataplexy, or other symptoms, further investigation is necessary [10]. The state of physical immobility of sleep paralysis is often accompanied by “ghost-like” hallucinations and feelings of fear [11]. These hallucinations typically fall into three categories: (1) a sense of an evil presence, fear, and hallucinations (intruder hallucinations); (2) chest pressure feeling and breathing difficulties (incubus hallucinations); and (3) vestibular–motor (VM) hallucinations, which are illusory sensations of movement and can include flying and out-of-body experiences [8, 12]. The content of hallucinations during sleep paralysis is likely a continuation of dream content from REM sleep, as it occurs during the transition phase between REM sleep and wakefulness, where brain activity associated with REM sleep persists [13]. In many countries, sleep paralysis is intertwined with folklore and attributed to supernatural phenomena, such as witchcraft and demons, which can make it even more terrifying and distressing [8].
Although they share similar symptoms, isolated and narcolepsy-associated sleep paralysis differ in several aspects (Table 1).
| Isolated sleep paralysis | Narcolepsy-associated sleep paralysis | |
| Definition | Occurring alone without narcolepsy | A common symptom of narcolepsy |
| Pathological mechanism | REM sleep abnormalities | Orexin deficiency and REM sleep abnormalities |
| Frequency | Low and episodic | High |
| Severity | Less severe | More severe |
| Concomitant symptom | Anxiety and fear | Excessive daytime sleepiness and cataplexy |
| Hallucinations | Pre-sleep/first awakening hallucinations | |
| Treatment | Primarily non-pharmacological interventions | Primarily pharmacological interventions for narcolepsy |
REM, rapid eye movement.
In recent years, an increasing number of studies have explored the mechanisms underlying sleep paralysis and associated hallucinations [14, 15]. In this article, we systematically review the epidemiology, predisposing factors, pathogenesis, and mechanisms of sleep paralysis, along with the brain circuit that regulates REM sleep.
There is controversy regarding the reported prevalence of sleep paralysis, with estimates varying widely from 2% to 60%, which may be due to heterogeneity in terms of clinical presentation and/or the specific diagnostic tools used [13, 16]. In a systematic review study involving 36,533 participants, the prevalence of lifelong sleep paralysis in the general population was reported as 7.6% [9] and varied in different populations, with higher prevalences in students (28%) and rural [17] and psychiatric patients (32%) [9]. However, a systematic review and meta-analysis involving a total of 167,133 participants from 25 countries in 2024 showed a global prevalence of sleep paralysis of 30%, with subgroup analyses similarly revealing a high prevalence in psychiatric patients (35%) and nonpsychiatric students (34%), while meta-regression showed no association with gender [16]. In addition, according to one previous report, roughly 75% of sleep paralysis episodes were reported to be accompanied by a wide variety of horrifying hallucinations [18]. However, a recent review and meta-analysis reported that 3.8% of patients with sleep paralysis experienced only visual hallucinations, 0.04% experienced only auditory hallucinations, and 24.25% reported both visual and auditory hallucinations; i.e., the vast majority (71.88%) of patients did not have hallucinations [16]. Furthermore, sleep paralysis was found to be accompanied by incubus experiences in 11% of cases in a meta-analysis study [19].
Sleep paralysis was reported to cause clinically significant distress in about 10% of patients and interfere with their lives in 7% of cases [20]. However, not all sleep paralysis attacks were reported to be fearful. One survey showed that up to 20% of sleep paralysis episodes were pleasant [21]. Cultural diversity and different lifestyles across regions greatly influence the incidence and experience of sleep paralysis.
Sleep paralysis can occur in both males and females, with onset usually during adolescence and without any obvious gender preference [22]. Many factors have been demonstrated to be associated with the intensity and/or frequency of sleep paralysis, and several medical conditions other than narcolepsy have also been implicated, including idiopathic hypersomnia, insufficient sleep syndrome, hypertension, Wilson’s disease, and obstructive sleep apnea [14, 16, 23, 24, 25]. The multifactorial nature of sleep paralysis demands a more detailed review of sleep-related predisposing factors, which are presented in the following sections.
Sleep hygiene, a determining factor of sleep quality, is closely related to the presence and onset of sleep paralysis [13]. Sleep deprivation, irregular sleep–wake schedules, jet lag, etc., can all lead to sleep paralysis, which might explain the relatively higher incidence of sleep paralysis in the young and student populations [26]. A study of predominantly Hispanic and female undergraduates at a large university in the southwestern United States showed that students with a history of sleep paralysis had poorer sleep quality and greater levels of mental stress than students without a history of sleep paralysis [27]. Moreover, the incidence of most stress-related psychiatric disorders, such as anxiety and impulse control disorders, peaks during the school-age years, which in turn increases the risk of sleep paralysis [28, 29]. The negative effects of academic burnout are another proven cause of the higher prevalence of sleep paralysis among medical students, especially females [30, 31]. Besides, cross-sectional studies on engineering students in India have also shown a strong correlation between academic stress and poor sleep hygiene with sleep paralysis, which suggests that similar outcomes can also occur in other high-pressure academic fields [32, 33]. Furthermore, the higher prevalence of sleep paralysis among female students may be partially explained by gender-specific stress responses since females are more likely to experience internalizing disorders (e.g., anxiety and depression), heightened emotional reactivity to stress, and increased rumination, all of which are known to disrupt sleep quality and increase vulnerability to parasomnias such as sleep paralysis [34, 35]. Sociocultural expectations and gender-role stress may further exacerbate this risk among female students in competitive academic environments [34].
These psychological and behavioral responses may be rooted in sex-based neurobiological differences that shape how females perceive and physiologically response to stress. Sex differences in the hypothalamic-pituitary-adrenal axis have been well-documented, with females exhibiting heightened activation, slower negative feedback, and prolonged cortisol exposure due to sex-specific patterns of corticotropin-releasing factor receptor signaling and trafficking [36, 37]. These biological differences can lead to difficulty in downregulating the stress response in females, ultimately increasing the risk of stress-related sleep disturbances such as sleep paralysis. Additionally, sex differences in neurotransmitter systems, including serotonin, dopamine, and gamma-aminobutyric acid (GABA), contribute to gender-specific variations in the stress response and sleep regulation. These factors, along with the influence of gonadal hormones like estrogen, increase the vulnerability of females to sleep paralysis [38].
Recent research increasingly suggests a complex reciprocal feedback loop among circadian misalignment, mood disorders, and sleep paralysis [39, 40]. Understanding the interplay among the factors that contribute to circadian disruption is essential for elucidating the underlying mechanisms of sleep paralysis and developing effective intervention strategies.
Melatonin secretion and light exposure are two principal factors linking the circadian clock with sleep regulation, particularly in modulating the timing of the sleep–wake cycle and the structure of REM sleep [41]. Under typical conditions, melatonin is secreted during the night to promote sleep and maintain circadian rhythmicity. However, exposure to light (especially short-wavelength blue light) potently suppresses melatonin secretion and delays its onset in a dose-dependent manner, thereby shifting circadian phase and degrading sleep quality [42, 43]. Irregular light exposure patterns, including excessive nocturnal screen media use or insufficient daytime light exposure, have been shown to impair melatonin excretion and circadian regulation, leading to an increased likelihood of sudden transitions from REM sleep to wakefulness without full restoration of motor control [44]. Furthermore, disrupted melatonin rhythms and circadian dysregulation have been implicated in mood disorders such as depression and anxiety, which are themselves established risk factors for sleep paralysis [45]. In short, disruption of melatonin secretion due to inappropriate light exposure destabilizes circadian rhythm regulation and sleep integrity during REM sleep, increasing the risk of sleep paralysis.
The season of birth determines the photoperiodic environment to which neonates are exposed, potentially influencing circadian preferences and susceptibility to sleep disorders such as sleep paralysis [46]. A study involving 1942 Chinese patients diagnosed with narcolepsy with cataplexy revealed a peak in births during November and a trough in April [46]. This suggests that individuals born during shorter photoperiods (typically winter) experience reduced natural light exposure in early life, which may disrupt melatonin secretion and delay circadian phase alignment, leading to long-term alterations in sleep–wake timing. These findings are supported by a systematic review and meta-analysis encompassing 3776 patients across eight regions, which identified consistent seasonal birth patterns associated with narcolepsy risk [47]. Although these studies focused on narcolepsy, the underlying mechanism involving early-life short photoperiods shaping the circadian system may similarly increase the risk of sleep paralysis.
Irregular breakfast consumption has been known to be associated with disruptions in circadian rhythms, potentially influencing sleep quality and increasing the risk of sleep paralysis [48]. One study found that skipping breakfast for six days induced a significant phase delay in the circadian rhythm of the body temperature in healthy young adults [49]. Additionally, analyses using big data have shown that unfavorable mealtime and meal skipping were associated with a higher risk of circadian syndrome, a condition characterized by concurrent metabolic, mood and sleep disturbances [50]. Given that both circadian misalignment and comorbid mood disorders are well-established risk factors for sleep paralysis, irregular breakfast habits can also contribute to sleep paralysis.
Chronotype refers to an individual’s natural preference for specific activity periods throughout the day, categorized into morning, evening, and intermediate types [51]. In a study involving 432 Italian students, participants who reported experiencing sleep paralysis exhibited higher levels of anxiety and poorer sleep quality, and these individuals were more likely to have an evening chronotype (more active at night) [33]. In other words, individuals with an evening chronotype generally have poorer sleep quality, which is associated with an increased susceptibility to sleep paralysis. Moreover, evening-type individuals are more prone to circadian misalignment, especially in the context of modern lifestyles that often favor late-night schedules, which in turn disrupts the integrity of REM sleep [52]. Evening chronotype appears to negatively impact both sleep quality and circadian rhythm regulation, thereby increasing the risk of sleep paralysis. Further research is needed to explore the underlying mechanisms linking evening chronotype to sleep paralysis and to evaluate potential interventions targeting evening chronotype to mitigate the risk of sleep paralysis.
Sleep paralysis has also been linked to psychiatric disorders, personality, intelligence, bizarre beliefs, sleep disorders, and post-traumatic stress disorder (PTSD) [29]. Patients with psychiatric disorders have the highest prevalence of sleep paralysis due the vicious cycle that exists between psychiatric disorders and sleep disorders, and because they are at higher risk for sleep deprivation, substance abuse, and other factors that are positively correlated with sleep paralysis [16, 29]. Interestingly, psychiatric patients may provide exaggerated accounts of their sleep behavior as an emotional response, subjectively considering themselves conscious yet unable to move their bodies. This may also explain the high prevalence of sleep paralysis in patients with depression and anxiety disorders, although this requires further verification. In addition, a higher prevalence has been demonstrated in people who have experienced stressful and traumatic events (childhood sexual abuse, disasters, death of loved ones, etc.) [53, 54].
Based on recent findings that suggest a potential genetic basis for sleep paralysis susceptibility, ethnic differences in prevalence may provide additional clues to its underlying heritable components [9, 55]. The systematic review encompassing over 36,000 participants revealed significant ethnic differences in the lifetime prevalence of sleep paralysis [9]. Specifically, Africans in the general population exhibited the highest prevalence of sleep paralysis (40.2%) among all ethnic groups, followed by Asians (31.0%). Among the student populations, Asians showed the highest prevalence (39.9%), whereas in psychiatric samples, Hispanics had the highest prevalence (57.1%). Overall, the highest total prevalence was found in Asians (38.7%), followed by Caucasians (36.9%), Africans (34.1%), and Hispanics (18.5%). These findings suggest that variations in gene frequencies across ethnic groups may influence the expression of genes involved in sleep regulation, thereby contributing to the observed ethnic disparities in the prevalence of sleep paralysis. However, the underlying genetic mechanisms responsible for these differences remain underexplored. With the advancement of modern genetic technologies, it is now feasible to conduct large-scale investigations that could improve our understandings of the genetics involving sleep paralysis and its interaction with environmental factors.
In a study based on a community sample of twins from England and Wales, genetic factors were found to have a moderate influence (estimated at 53%) on sleep paralysis [56]. The study identified a significant association between the rs2304672 single-nucleotide polymorphism in the period circadian regulator 2 (PER2) circadian rhythm gene and sleep paralysis in both additive and dominant models of inheritance, suggesting that circadian rhythm disruptions mediated by PER2 may cause individuals more susceptible to sleep paralysis. However, further work is needed to determine specific polymorphisms and their precise role in circadian rhythm regulation and sleep homeostasis.
Another prominent gene associated with sleep paralysis is the human leukocyte antigen (HLA) class II allele DQB1*06:02, which has been identified in approximately 90% of patients with NT1 [57]. This allele is widely regarded as a hall mark of NT1 and is believed to act via T-cell-mediated autoimmune mechanisms that target and destroy orexin-producing neurons in the lateral hypothalamus [10, 58]. The resultant orexin deficiency leads to dysregulation of REM sleep, a core feature of both NT1 and sleep paralysis. The shared REM-related dysregulation underscores the potential utility of narcolepsy as a model for exploring the heritable components of sleep paralysis. Therefore, further investigation into the genetic basis of narcolepsy may provide critical insights into the etiology and inheritance patterns of sleep paralysis.
While some physical health indicators have been examined in relation to sleep paralysis, there is no consensus in the current literature. An association of higher body mass index (BMI) with lifelong and recurrent episodes of fearful sleep paralysis has been observed, but no such link has been found in the majority of relevant studies [59, 60]. Additionally, chronic pain and insomnia were also found to be associated with the experience of sleep paralysis [61]. A systematic review designed to assess the effects of vitamin D supplementation on the mental health of healthy adults suggests that while vitamin D is not directly linked to sleep paralysis, a severe deficiency may lead to poor sleep quality, fatigue, and mood disorders, which can exacerbate the symptoms of sleep paralysis [62]. As vitamin D3 has been linked to neuromuscular function, sleep regulation, and mental health, its role in sleep paralysis warrants further study [63].
Sleep paralysis occurs mainly in the transition between REM sleep and wakefulness. Therefore, it is imperative to understand the brain circuitry that controls REM sleep in order to elucidate the mechanism of sleep paralysis. REM sleep, which occurs in nearly all mammals and birds [64], arises in the brainstem and is facilitated by “REM-on” neurons and inhibited by “REM-off” neurons [65]. “REM-on” neurons are a class of neurons that are highly active during REM sleep but less active or even inactive during NREM sleep and wakefulness. They primarily function in triggering and maintaining REM sleep and coordinating REM sleep features such as rapid eye movements, muscle atonia, and desynchronized EEG activity [66]. In contrast, “REM-off” neurons are highly active during NREM sleep and wakefulness but almost completely inactive during the REM sleep, and they serve to prevent REM sleep from occurring [66]. Both types of neurons are widely distributed in brain regions such as the medulla oblongata, pons, midbrain, and hypothalamus. “REM-on” neurons inhibit “REM-off” neurons and vice versa, analogously to an electronic flip-flop switch. This serves as the foundation for a model of REM sleep generation [67], where mutual inhibition ensures there is rapid transition from one state to another [68].
The regulation of REM sleep directly or indirectly involves several anatomical nuclei primarily located in the lower brainstem, limbic system, thalamus, hypothalamus, and cortex. These are involved in the generation and maintenance of the sleep–wake cycle, of which REM sleep is a major component. In addition, neurotransmitters such as glutamate (Glu), GABA, glycine, histamine, acetylcholine, serotonin, and norepinephrine as well as neuropeptides such as orexin (OX) and melanin-concentrating hormone (MCH) are involved in the induction and regulation of REM sleep [69, 70, 71] (Fig. 1).
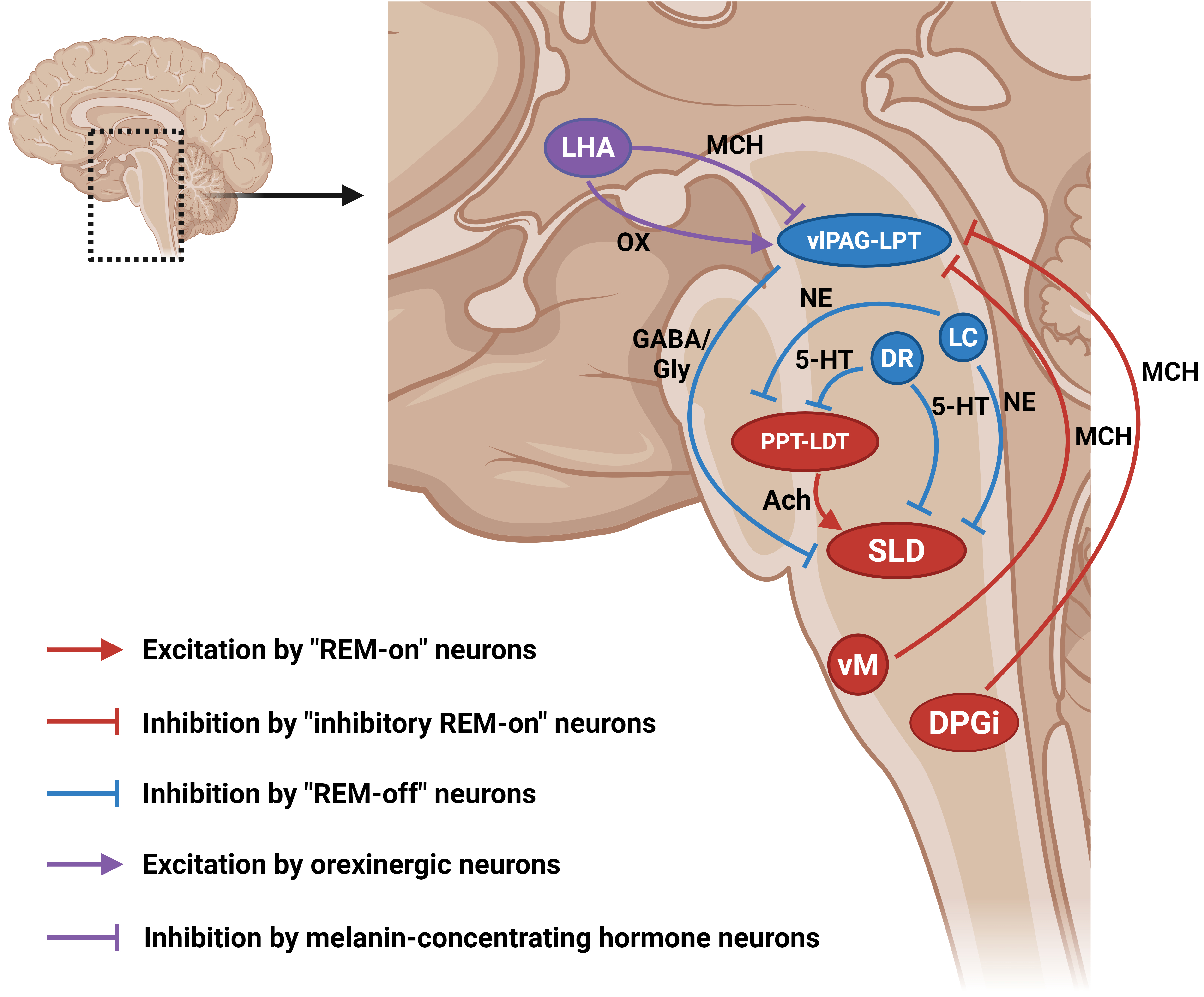 Fig. 1.
Fig. 1.
Schematic diagram of the brain circuitry controlling REM sleep. Brain regions in red (i.e., “REM-on” structures) promote REM sleep, while brain regions in blue (i.e., “REM-off” structures) inhibit REM sleep. Brain regions in purple (i.e., neuropeptide-producing structures) regulate REM sleep by releasing different neuropeptides with different mechanisms. Arrows, excitation; “T” shaped line, inhibition; 5-HT, serotonin; Ach, acetylcholine; DPGi, dorsal paragigantocellular reticular nucleus; DR, dorsal raphe; GABA, gamma-aminobutyric acid; Gly, glycine; LC, locus coeruleus; LDT, laterodorsal tegmentum; LHA, lateral hypothalamus; LPT, lateral pontine tegmentum; MCH, melanin-concentrating hormone; NE, norepinephrine; OX, orexin; PPT, pedunculopontine tegmentum; SLD, sublaterodorsal nucleus; vlPAG, ventrolateral periaqueductal gray matter; vM, ventromedial medulla. Created with BioRender.com (Agreement Number: RM286L4OH2).
Understanding the mechanism of REM sleep is challenging not only because of its complexity and the ever-changing nature of the field but also variations in anatomical atlases and the terminology used to describe the same structures in different species [72, 73, 74]. For example, the subcoeruleus nucleus in humans is equivalent to the peri-locus coeruleus alpha nucleus (peri-LCa) in cats and the sublaterodorsal (SLD) nucleus in rodents [70]. For clarity, only the term SLD is used in this review.
The brain circuitry responsible for regulating REM sleep was first investigated by Michel Jouvet, who discovered an area in the dorsal pontine that is essential for muscle atonia during REM sleep [75]. With the advancement of experimental tools, decades of subsequent experimental work have further pinpointed the position of this “atonia generator” to the sublaterodorsal (SLD) nucleus of the pons [4, 67, 76, 77, 78]. In addition, caudal laterodorsal tegmental (cLDT) neurons, which are located dorsally adjacent to the SLD nucleus, are also thought to be associated with atonia production [67]. The SLD nucleus has now been shown to be the most active region in REM sleep [79], with many spinally projecting glutamatergic neurons in its ventral portion that function to produce muscle atonia in both direct and indirect ways. They directly synapse with inhibitory interneurons in the spinal ventral horn (SVH), which inhibit spinal motoneurons either directly via GABA or glycine or indirectly by projecting to GABAergic and glycinergic neurons in the ventromedial medulla [67, 79, 80, 81]. In addition, many studies have highlighted the importance of the ventromedial medulla in developing muscle atonia, including the medially located ventral gigantocellular reticular nucleus (GiV) and a-gigantocellular reticular nuclei (GiA) [82, 83, 84]. The glutamatergic neurons in the ventromedial medulla are connected to interneurons in the spinal ventral horn, which in turn inhibit motoneurons in the spinal cord (Fig. 2) [85, 86]. Furthermore, the pedunculopontine tegmentum (PPT) and the laterodorsal tegmentum (LDT) in the pontine reticular formation (PRF) release acetylcholine to activate glutamatergic neurons in the SLD nucleus to promote REM sleep.
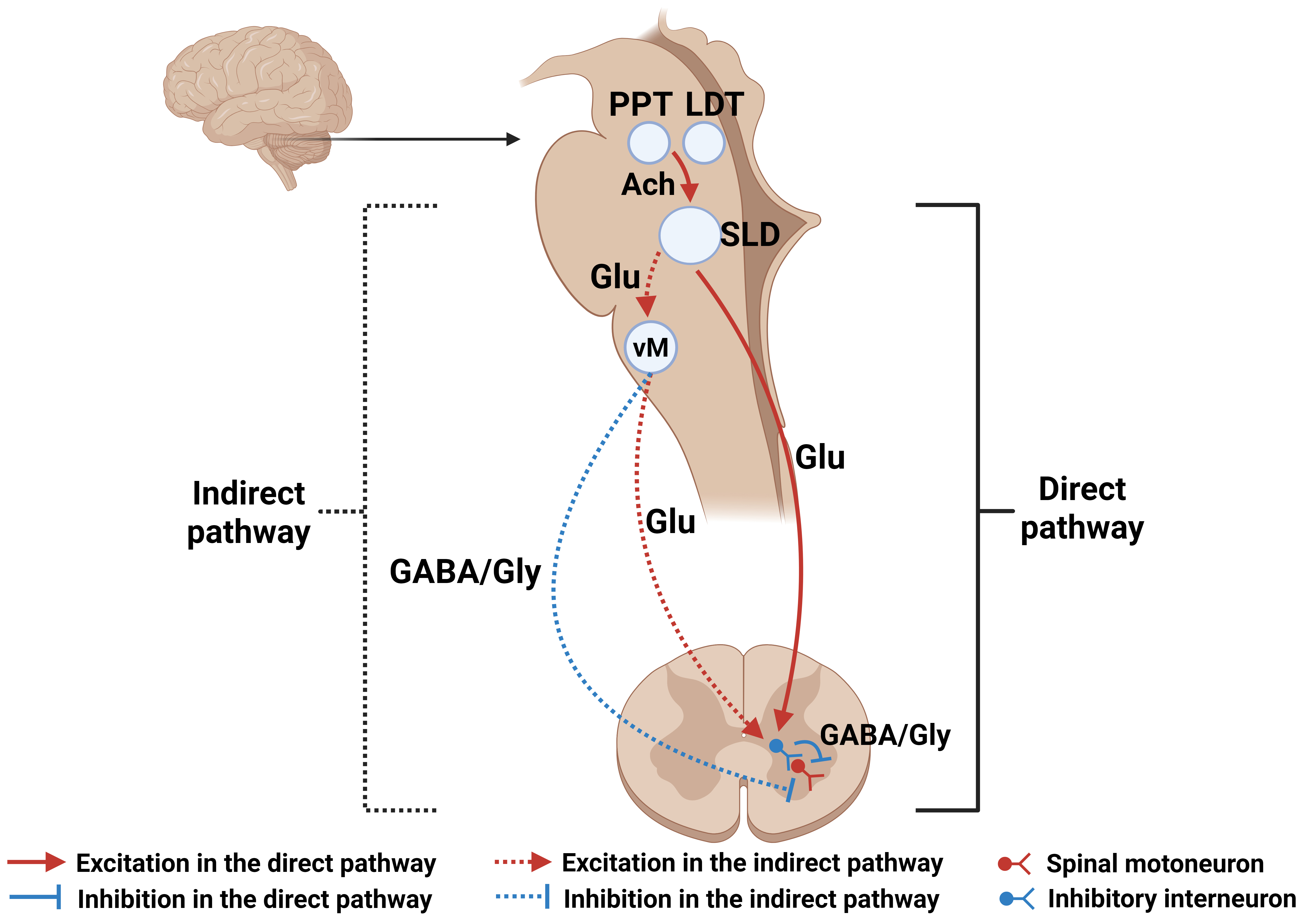 Fig. 2.
Fig. 2.
Schematic diagram of structures in the central nervous system involved in the production of atonia during REM sleep. The SLD nucleus is the core region responsible for REM sleep. It induces muscle paralysis during REM sleep, either directly, by inhibiting spinal motoneurons through inhibition of interneurons in the spinal ventral horn, or indirectly, by inhibition of spinal motoneurons through the ventromedial medulla. Solid line, direct pathway; dashed line, indirect pathway; red color, excitation; blue color, inhibition; Glu, glutamate. Created with BioRender.com (Agreement Number: UF286L4YVE).
Two functionally distinct types of glutamatergic neurons are found in the SLD nucleus: (1) corticohippocampal activation-promoting neurons, which act to increase consciousness through the parabrachial nucleus and medially adjacent precoeruleus region, and (2) reticulospinal REM sleep atonia-generating neurons, which cause atonia during REM sleep [4, 67, 87, 88]. Glutamatergic projections from the parabrachial nucleus and precoeruleus, and cholinergic projections from the pedunculopontine tegmentum and the laterodorsal tegmentum, reach the intralaminar and reticular thalamic nuclei, lateral hypothalamus, and basal forebrain, which in turn reach the cortex and hippocampus to produce cortical activation [71, 88, 89]. Therefore, these two kinds of functionally separated glutamatergic SLD “REM-on” neurons might provide a pathological basis for separating changes in cortical activation and muscle tone associated with REM sleep observed in various REM sleep disorders, including sleep paralysis.
Another class of neurons, “inhibitory REM-on” neurons, is involved in REM sleep regulation. Although both “inhibitory REM-on” and “REM-on” neurons are closely related to the initiation and maintenance of REM sleep, they regulate REM sleep through different mechanisms. “REM-on” neurons are activated at the onset of REM sleep, and they activate the neural network primarily using glutamate as an excitatory neurotransmitter, whereas “inhibitory REM-on” neurons regulate the onset and termination of REM sleep by inhibiting the activity of other brain regions, particularly those associated with non-REM sleep and arousal. These neurons primarily use glycine and GABA to maintain stable REM sleep.
The “inhibitory REM-on” neurons can in turn inhibit the aforementioned brain region containing “REM-off” neurons (ventrolateral periaqueductal gray matter (vlPAG)/lateral pontine tegmentum (LPT)), resulting in the disinhibition of SLD neurons. Studies over the past few years have supported this framework, with neurons in those brain regions referred to as “inhibitory REM-on” neurons [90, 91]. The locations of these neurons have been discovered, with MCH neurons found in the hypothalamus, SLD nucleus, ventromedial medulla (vM), and dorsal paragigantocellular reticular nucleus (DPGi) in the dorsal medulla [92]. These “inhibitory REM-on” neurons inhibit “REM-off neurons” in the vlPAG/LPT, a common postsynaptic target, to facilitate the induction of REM sleep. As microinjection of MCH into the locus coeruleus nucleus has been shown to promote REM sleep [93], activation of the melanin-concentrating hormone system in the locus coeruleus nucleus is thought to be involved in the promotion of REM sleep. Similar neurons are also found in the preoptic area of the hypothalamus [94], with axons projecting to the ventrolateral periaqueductal gray matter, locus coeruleus, and dorsal raphe nuclei [95, 96]. In addition, GABAergic REM sleep-promoting neurons can also be found in the vlPAG/LPT [90, 92], but it remains unclear whether such neurons interact with local “REM-off” neurons.
In summary, “inhibitory REM-on” neurons have been found in many brain regions and share a common mechanism for promoting REM sleep, that is, inhibiting “REM-off” neurons in inhibitory brain regions, thereby disinhibiting the SLD nucleus and initiating a series of reactions downstream.
“REM-on” neurons receive input from a variety of neurons that suppress REM sleep (“REM-off” neurons). Of the various “REM-off” neurons, specific GABAergic neurons in brain regions that inhibit REM sleep are undoubtedly the most important.
REM-off GABAergic neurons in the vlPAG and adjacent (LPT; also called the deep mesencephalic nuclei, DpMe) project to the SLD nucleus and strongly inhibit REM sleep [92, 97]. Before the discovery of “REM-off” neurons in the ventrolateral periaqueductal gray matter, it was proposed that monoaminergic neurons in the nearby dorsal raphe (DR) and locus coeruleus (LC) nucleus suppress REM sleep [98]. Serotonergic neurons in the dorsal raphe nucleus and noradrenergic neurons in the locus coeruleus nucleus are connected to both the SLD nucleus and pontine reticular formation (PRF), inhibiting the generation of REM sleep [65]. Interestingly, however, another subpopulation of GABAergic neurons in the ventrolateral periaqueductal gray matter projects to neurons of the dorsal raphe and locus coeruleus nuclei [92], inhibiting the release of monoamine neurotransmitters and thus promoting REM sleep, but it remains unclear whether such neurons interact with local “REM-off” neurons [90]. Under normal physiological conditions, the function of “REM-off” neuron-related circuits is to prevent inappropriate activation of neurons that produce atonia during the transition from REM sleep to wakefulness, while ensuring that muscle tone is rapidly restored upon awakening [66, 99].
Orexin (OX) and MCH, are two crucial peptides involved in the regulation of REM sleep. In general, orexin promotes wakefulness and suppresses REM sleep by activating neurons in the brain regions that inhibit REM sleep. In contrast, the melanin-concentrating hormone promotes REM sleep by inhibiting “REM-off” neurons in the brain regions that inhibit REM sleep [100].
Orexin-producing neurons are mainly found in the lateral hypothalamus (LHA) and regulate physiological processes, such as arousal and the sleep–wake cycle, to maintain wakefulness and inhibit REM sleep [101, 102]. They also project to neurons that express orexin receptors in many nuclei with a role in regulating REM sleep, including in the ventrolateral periaqueductal gray matter, locus coeruleus nucleus [103], dorsal raphe nucleus [104], and basal forebrain [105], and activate these receptors in these brain regions, which enhances suppression of the SLD nucleus. Conversely, orexin-producing neurons also receive projections from nuclei containing neurons that express orexin receptors. For example, orexin-producing neurons in the lateral hypothalamus are inhibited by GABAergic neurons in the preoptic area [106], and by 5-HT neurons in the dorsal raphe nucleus and noradrenergic neurons in the locus coeruleus nucleus [107, 108, 109]. Conversely, cholinergic neurons in the basal forebrain also project to orexin-producing neurons in the lateral hypothalamus and have a positive effect on wakefulness [110].
MCH-producing neurons in the lateral hypothalamus with opposing functions to the orexin-producing neurons also project to neurons that express MCH receptors in many nuclei, including the ventrolateral periaqueductal gray matter, lateral pontine tegmentum, dorsal raphe nucleus, locus coeruleus nucleus, pontine reticular formation, and SLD nucleus, promoting REM sleep [111, 112].
MCH activates MCH receptors on neurons in the ventrolateral periaqueductal gray matter/LPT to facilitate REM sleep by inhibiting the “REM-off” neurons in these nuclei. In addition, activation of the MCH receptors in the locus coeruleus nucleus is thought to be involved in promoting REM sleep, as microinjection of MCH into the locus coeruleus nucleus has been shown to evoke REM sleep [93].
However, the precise mechanisms underlying the regulation of REM sleep by these neuropeptides, as well as the downstream pathways through which MCH regulates REM sleep, are still unclear and require further investigation.
The pathogenesis of sleep paralysis involves complex neurophysiological processes and is related to abnormalities in the transition from the REM sleep stage to wakefulness. However, isolated and narcolepsy-associated sleep paralysis involve different pathogenic mechanisms [57, 113]. Isolated sleep paralysis results from disturbances in the physiological regulation of the sleep transition process, particularly imbalances in the SLD nucleus and related neurotransmitters [69]. In contrast, narcolepsy is primarily associated with the degeneration of orexinergic neurons, leading to deficiency in both orexin A and B neuropeptides [114, 115]. Notably, these neuropeptides activate two distinct receptor subtypes, OX1R and OX2R, which mediate sleep-related symptoms differently. OX2R dysfunction leads to abnormal discharge of the SLD nucleus, resulting in impaired transition from REM sleep to wakefulness, and therefore its reduced activation is the primary mechanism for the development of sleep paralysis in narcolepsy [116, 117]. In contrast, OX1R deficiency is more relevant to emotionally triggered cataplexy through limbic circuit dysregulation, while its role in sleep paralysis is limited [118]. The dual deficiency of orexin A and B results in abnormal REM sleep regulation and impaired transitions to wakefulness, thereby triggering various narcolepsy symptoms including sleep paralysis [7, 66]. Overall, the mechanisms of narcolepsy are more complex, and the pathogenesis of isolated sleep paralysis may also be present in narcolepsy-associated sleep paralysis.
In narcolepsy patients, the lack of orexin A and B signaling in the circuit for REM sleep regulation can be caused by either a loss of orexin-producing neurons in the hypothalamus or a significant reduction or complete lack of orexin in patients [119, 120, 121]. This reduction results in orexin failing to activate orexin receptors (mainly OX2R) in the ventrolateral periaqueductal gray matter/LPT regions, leading to inappropriate disinhibition of the SLD nucleus during the transition from sleep to wakefulness, which in turn results in loss of muscle tone, i.e., sleep paralysis. In addition, some orexin-producing neurons are also found in the perifornical area, and their absence or hypofunction may further exacerbate the disruption of the sleep–wake cycle due to reduced levels of systemic orexin [122, 123]. However, there is relatively little research on their specific role in and impact on narcolepsy, and more scientific evidence is needed to further validate this point.
Recent evidence suggests a strong link between the brain structures involved in emotional processing and muscle atonia during REM sleep. The amygdala, especially its central nucleus (CeA), has long been recognized as a core brain region for emotional processing and is now understood to exert a direct influence on REM sleep regulation and sleep paralysis [124, 125]. GABAergic neurons in the CeA have been shown to send descending projections to key areas of the REM sleep regulating circuit, including the locus coeruleus nucleus, the lateral pontine tegmentum, and ventrolateral periaqueductal gray matter. Strong emotions, especially negative emotions such as fear and anxiety, activate GABAergic neurons in the central nucleus of the amygdala, which inhibits the locus coeruleus nucleus, the lateral pontine tegmentum, and ventrolateral periaqueductal gray matter and in turn disinhibits the SLD nucleus [126, 127, 128, 129, 130]. However, this only occurs and results in sleep paralysis in the case of orexin deficiency, because CeA-mediated inhibition can be counteracted by inputs of excitatory orexin from the hypothalamus. Thus, in narcolepsy patients lacking orexin, this modulatory balance is disrupted. Emotions such as fear, laugh, surprise, or emotional stress leads to an excessive increase in GABAergic neuronal firing in the central nucleus of the amygdala, which may be an important mechanism underlying sleep paralysis. This abnormal discharge disrupts normal REM sleep regulation and muscle inhibitory mechanisms, leading to immobilization of the individual during the sleep–wake transition stage.
Moreover, the insula acts synergistically with the amygdala to exacerbate the episodes of sleep paralysis by integrating interoceptive and emotional information [131]. It forms a broader regulatory circuit with the anterior cingulate cortex and the parabrachial nucleus [132]. Within this circuit, glutamatergic neurons in the anterior cingulate cortex send excitatory projections to the parabrachial nucleus, where GABAergic neurons are subsequently activated, enhancing GABAergic inhibition of brainstem “REM-off” nuclei [133, 134]. This mechanism further destabilizes REM sleep regulation and contributes to pathological muscle atonia during the sleep–wake transition.
All of the above are mechanisms by which sleep paralysis occurs in the presence of orexin deficiency; pathogenesis unrelated to orexin will be discussed below (i.e., occurring in both isolated and narcolepsy-associated sleep paralysis) (Fig. 3).
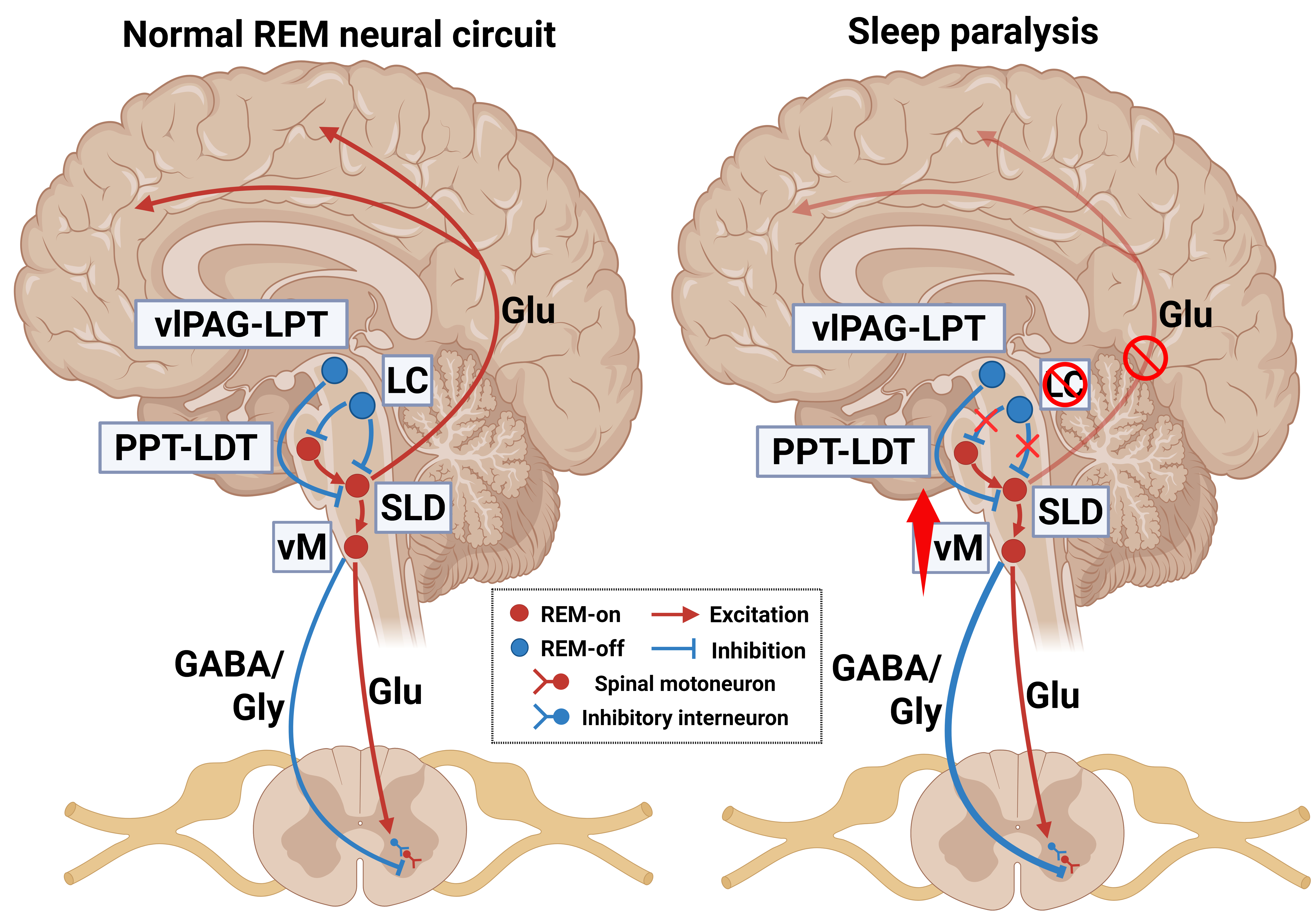 Fig. 3.
Fig. 3.
Pathogenesis of sleep paralysis unrelated to orexin. Synergistic dysregulation of the two groups of glutamatergic neurons in the SLD, sudden cessation of locus coeruleus nucleus firing, and excitation of GABAergic and glycinergic neurons in the ventromedial medulla are responsible for sleep paralysis. Arrows, excitation; “T” shaped line, inhibition; brain regions in red, “REM-on” structures; brain regions in blue, “REM-off” structures; vlPAG, ventrolateral periaqueductal gray matter. Created with BioRender.com (Agreement Number: IW286L53AE).
There are two groups of glutamatergic neurons in the SLD nucleus, with one promoting cortical activity during REM sleep and contributing to REM sleep maintenance and the other enabling muscle atonia. During wakefulness, the two groups of glutamatergic neurons in the SLD nucleus are inhibited, and consciousness is restored along with muscle tone. However, when these two groups of neurons are dysfunctional, “REM-on” neurons that promote cortical activity during REM sleep are inhibited, while neurons that promote muscle atonia continue to be activated, resulting in the loss of muscle tone while conscious [70, 99, 135].
During wakefulness, noradrenergic neurons in the locus coeruleus nucleus are highly active. However, when cataplexy occurs, noradrenergic neurons in the locus coeruleus nucleus suddenly stop firing and cause a loss of muscle tone [136]. Evidence suggests that the mechanisms of cataplexy and sleep paralysis overlap in some aspects, particularly in those involving noradrenergic neuron inactivation and muscle atonia [137, 138]. Thus, neuronal inactivation in the locus coeruleus nucleus may also lead to sleep paralysis when consciousness is normally restored during REM sleep–wake transitions. However, unlike locus coeruleus neurons, dorsal raphe neurons do not cease firing during sleep paralysis. At the same time, the ventromedial medulla is a key area for generating muscle atonia, and it has been confirmed that the activity of the GABAergic and glycinergic neurons in the ventromedial medulla increases during cataplexy [139]. Thus, inhibition of spinal motor neurons due to increased ventromedial medulla activity may be another mechanism promoting sleep paralysis.
The mechanism underlying sleep paralysis has been established. However, relatively little is known about the pathological causes of its complications (hallucinations). One reason that patients feel very scared during sleep paralysis is incubus hallucination, i.e., the feeling of a heavy weight on their ribs or that someone is pressing on their chest. This is because when sleep paralysis occurs, the diaphragm (the primary respiratory muscle) continues to function normally, while the activity of striated muscles, including some of the auxiliary respiratory muscles, is inhibited [140]. However, recent studies have shown that the state of skeletal muscle paralysis leads to significant decreases in alveolar ventilation and tidal volume, which can lead to hypercapnia during REM sleep. When patients with sleep paralysis suddenly become aware of these physiological changes, they experience dyspnea and suffocation [8, 16, 23]. In addition, decreased excitability due to the hyperpolarization of motor neurons in the respiratory muscles affects the sensation of respiration, resulting in the patient’s perception of chest pressure and dyspnea.
Hallucinations can be classified as serotonergic or dopaminergic depending on the neuronal transmitters involved. Serotonergic hallucinations (e.g., those caused by lysergic acid diethylamide), also known as nonpsychotic hallucinations, are similar in nature to dreams, and the experiencer is able to clearly perceive that they are not reality [141, 142]. In contrast, dopaminergic hallucinations are psychotic hallucinations (common in schizophrenia) that the experiencer cannot distinguish them from real life [143].
Hallucinations during sleep paralysis often resemble dreams to the extent that the event itself is sometimes interpreted as a dream, and the patient is able to fully recognize it as a hallucination [140]. Therefore, hallucinations accompanying sleep paralysis are often considered to be serotonergic. Serotonin is functionally involved in the production of hallucinations and fear during sleep paralysis, mainly through its action on serotonin 2A receptors (5-HT2AR), which are widely distributed in the visual cortex and limbic system [11, 144, 145]. Activation of the serotonergic arousal system during the transition between REM sleep and wakefulness can facilitate the wakefulness process, which leads to serotonin overactivity in the visual cortex and limbic system of sleep paralysis patients and ultimately to hallucinatory experiences via 5-HT2AR activation [146]. 5-HT2A receptor activation in the visual cortex elicits visual hallucinations by increasing cortical excitability and altering visually evoked cortical responses, yet 5-HT2A receptors in the limbic system play a very important role in the regulation of fearful emotions [147, 148]. Their joint action profoundly modulates circuits in which hallucinatory experiences and fear are intertwined. This also provides treatment for hallucinations and fear symptoms of sleep paralysis, as detailed in the Treatment section below.
Current research suggests that an increasing number of road traffic accidents are related to microsleep in drivers [149] including professional drivers such as long-haul truck drivers and those working in logistics, who are required to drive vehicles for extended periods of time with irregular schedules [150]. Despite regulatory mandated rest periods, these drivers often sleep in their vehicles under suboptimal conditions such as noise, lack of space, poor ventilation, and temperature fluctuations [151]. In addition, modern behavioral factors such as excessive smartphone use at night also exacerbate poor sleep quality [152, 153]. These environmental and behavioral stressors severely impact sleep quality and sleep continuity, leading to increased daytime sleepiness and microsleep. Because microsleep also occurs during the sleep–wake transition and shares some similarities with the occurrence of sleep paralysis, we propose that microsleep may arise during the transition between REM sleep and wakefulness, leading to sleep paralysis. Microsleep is a sleep-like condition of short duration that occurs when people are overtired or lack sleep [154, 155, 156]. Typically, this phenomenon involves a brief shutdown of the brain’s activation system with parallel activation of the sleep-promoting centers [157]. In one study, these microsleeps were found to be associated with decreased neural activity in arousal-related brain regions, including the thalamus, midbrain, and posterior cingulate cortex, but with increased activity in the frontoparietal, insula, parahippocampal, and temporo-occipital cortices [158]. Severe sleep deprivation, poor sleep quality, and excessive fatigue are all causes of microsleep, which greatly overlap with the predisposing factors of sleep paralysis. Therefore, we speculate that sleep paralysis may be produced when microsleep occurs in the transition between sleep and wakefulness. Its occurrence is more likely when people are in a microsleep state, resulting in the recovery of people’s consciousness and inability to move. Although microsleeps are common and pose significant risks in daily life [159, 160], a comprehensive, systematic understanding of the underlying brain mechanisms is still lacking [158], and this speculation needs to be further verified by scientific research.
In conclusion, disruptions of REM sleep regulation (premature activation of either motor inhibitory pathways or the brain’s arousal system) resulting in a state of muscular paralysis misaligned with conscious arousal is central to the pathogenesis of sleep paralysis.
There is no specific treatment for sleep paralysis, and the main principles of treatment are to improve sleep, optimize mood, and manage related psychiatric disorders [16, 113, 161]. Due to the specific characteristics of ISP, therapeutic interventions are unnecessary in most cases, as patients usually return to normal for some time after several episodes. The management of ISP focuses on prevention, and medications are used only in patients who have frequent attacks and exhibit severe symptoms for which non-pharmacological interventions have been unsatisfactory [14]. Narcolepsy-associated sleep paralysis patients usually have more severe symptoms than ISP, and treatments are directly focused on narcolepsy.
Examples of non-pharmacologic management include sleep hygiene improvement, lifestyle modification, cognitive-behavioral therapy (CBT), and the induction of out-of-body experiences (OBEs).
A recent online cross-sectional study reported on three of the most common preventive interventions for sleep paralysis, namely adjustments to the sleep environment, sleep patterns, and sleep position [113]. It also found that the overall effectiveness of disruptive interventions is low, with contextualization (e.g., focusing on an object in the room or telling oneself that one is now sleeping) and breathing (e.g., rapid breathing) being more effective, but that most patients attempted to move limbs or smaller body parts as well as make noise as counteractive measures [20, 113]. Since sleep hygiene is a crucial cause of sleep paralysis [162], improving sleep hygiene such as through a suitably adjusted bedroom temperature, a stable sleep–wake routine, adequate sleep, and so on, can promote sleep initiation and maintenance [29]. Moreover, sleep paralysis has been reported to occur mostly during sleep in a supine [163] rather than sitting [140] position, and maintaining a lateral or prone sleeping position is also an effective preventive measure [113, 164].
In 2024, Sawant and Kamble [63] reported a case of an ISP patient with osteoporosis, finding that lifestyle changes such as practicing yoga, meditation, and chanting may have an important role in the recovery of sleep paralysis by reducing stress, enhancing sleep quality, and promoting psychological well-being. Meditation is particularly effective and has been shown to stabilize REM sleep patterns and reduce the frequency of sleep paralysis [63, 165, 166].
CBT is also recommended [25, 167]. This psychotherapeutic approach improves the mood and psychological well-being of patients by optimizing their negative thinking and behavioral patterns associated with sleep paralysis, thereby effectively reducing the impact of hallucinations and fears during episodes of sleep paralysis and reducing the incidence [168, 169]. As such, it may be the best strategy for patients with depression, anxiety, and other psychiatric disorders.
In a recent study, it was observed that individuals with isolated sleep paralysis who experienced OBEs (a type of vestibular–motor hallucination) had more positive emotions (e.g., happiness, excitement, etc.) and fewer negative emotions (e.g., fear, anxiety) than people with isolated sleep paralysis who did not experience OBEs and those with other types of hallucinations. It was also found that subjects experienced auditory, visual, and tactile sensations prior to sleep paralysis episodes, which are known as “aura”. As a result of this insight, a clinical approach was proposed for the treatment of recurrent sleep paralysis by which OBEs are induced in patients by training them to recognize “aura”, combined with psychoeducation to reduce the negative emotions associated with sleep paralysis [21, 170].
Since sleep paralysis often co-occurs with narcolepsy, the medications used for treating sleep paralysis are also used for narcolepsy.
(1) Serotonin and noradrenaline reuptake inhibitors (e.g., venlafaxine), tricyclics, selective serotonin reuptake inhibitors (SSRIs), and other antidepressants are known to reduce the number of sleep paralysis episodes [14, 25, 171, 172]. They can enhance the activity of serotonin and norepinephrine, which in turn increases the effects of two REM sleep-inhibitory brain regions, locus coeruleus and dorsal raphe nuclei. Therefore, they have a pharmacological role in suppressing sleep paralysis. However, while most patients experience improved sleep with SSRIs, a recent case report found that sertraline (a type of SSRIs) may cause isolated sleep paralysis as a potential side effect [173, 174]. Therefore, further research is necessary to elucidate the effects of SSRIs on sleep, and practice guidelines should clarify their role. Moreover, when prescribing depressants to patients, doctors must remain alert to the possibility of sleep paralysis and consider substituting medications, such as trazodone, if necessary [174, 175].
(2) The selective 5-HT2AR inverse agonist pimavanserin [11] is a drug that selectively targets hallucination and fear responses in sleep paralysis. It was first proposed to use for the treatment of sleep paralysis because of its ability to inhibit 5-HT2A receptors in the visual cortex and limbic system, which effectively relieves hallucination and fear responses in sleep paralysis.
(3) Pitolisant, an N-piperidinyl derivative [176], is an inverse agonist of the histamine H3 receptor with wake-promoting effects [177]. Pitolisant blocks the self-inhibitory effect of histamine on its release, leading to increased histamine release [178]. Additionally, Pitolisant affects other neurotransmitter systems, enhancing the release of acetylcholine and dopamine in the cerebral cortex [178]. Therefore, it is used in the European Union for the treatment of narcolepsy in adults with or without cataplexy, and it has recently been shown to be effective for the treatment of sleep paralysis [179]. Solriamfetol, a phenylalanine derivative, is a dopamine and norepinephrine reuptake inhibitor, so it should theoretically have an effect on sleep paralysis [180, 181] and has undeniable advantages over Pitolisant in terms of withdrawal symptoms and drug abuse [179, 182].
(4) At present, FT218 (a controlled-release sodium oxybate) and JZP-258 (a novel low-sodium oxybate) [179, 183], AXS-12 (reboxetine, a selective norepinephrine reuptake inhibitor) [179], and THN102 (the combination of modafinil and flecainide) [184] are being developed and tested. Both FT218 and JZP-258 are in Phase III clinical trials; the former is being tested for the treatment of excessive daytime sleepiness associated with narcolepsy and sudden collapse and has been designated an orphan drug by the FDA, whereas the latter is being tested for the treatment of narcolepsy. AXS-12 was first developed for the treatment of depression and is now being developed for the treatment of sudden collapse and excessive daytime sleepiness associated with narcolepsy. It has been designated an orphan drug by the FDA. THN102 is in Phase II clinical trials for the treatment of excessive daytime sleepiness associated with narcolepsy [179]. All have demonstrated some anti-narcolepsy effects and can be used as potential drugs for sleep paralysis [182].
(5) Supplementation of orexin could be a potential treatment for narcolepsy-associated sleep paralysis caused by orexin deficiency [113, 185]. However, there is a lack of sufficient data to demonstrate its therapeutic efficacy [16].
(6) In the case report of the ISP patient with osteoporosis mentioned above, it was also suggested that oral or injectable vitamin D can be used to correct severe vitamin D deficiency and thereby help improve mood and energy levels, which indirectly aids in the recovery from sleep paralysis while addressing bone problems [63]. In addition, a systematic review found that vitamin D supplementation significantly improves sleep quality and thus mitigates sleep paralysis [186].
Most current pharmacologic managements for sleep paralysis have primarily been studied and implemented in adults, and their applicability to younger individuals remains unclear [187]. One study indicates that only sodium oxybate has data supporting its use in pediatric patients and has received approval from the European Medicines Agency, whereas other medications are used off-label based on expert opinion and clinical practice [188]. Additionally, Pitolisant has been approved by the European Medicines Agency for the treatment of narcolepsy with or without cataplexy in children and adolescents aged 6 years and older [188, 189]. Therefore, currently available research on medications approved for use in minors is limited to the management of narcolepsy, and future research is essential to evaluate the safety and efficacy of these medications for minors with sleep paralysis.
Sleep is a crucial restorative state that is dependent on precise coordination of the production of numerous biological and chemical structures and their proper function. Knowledge of the multiple interactions between neuromodulators, brain regions, and the whole organism provides insights into REM sleep mechanisms and the pathogenesis of many sleep disorders.
This article focuses on the neural network of REM sleep and the mechanisms involved in the pathogenesis of sleep paralysis. The REM arousal transition and REM muscle atonia are regulated due to the actions of “REM-on” and “REM-off” neurons throughout the brain. It is generally recognized that the SLD nucleus is the core region for generating REM sleep muscle atonia. Neuropeptides (i.e., orexin and MSH) are also widely distributed in the brain and are responsible for regulating many physiological functions, including sleep. Their presence facilitates the coupling of the aforementioned sleep behaviors to neurotransmitter regulatory systems. Sleep paralysis is a dissociative state, with REM sleep muscle atonia continuing into the waking state. It is important to note that although sleep paralysis typically causes fear and interferes with the daily lives of those affected, the vast majority of sleep paralysis episodes are benign and not associated with serious pathology. Therefore, sleep paralysis does not require treatment in most cases, only when there is repetition and fear.
The current studies have some clear limitations. A large number of variables related to sleep paralysis have been proposed. However, it is worth noting that there is a great deal of variation in the methods used to assess sleep paralysis across studies, which is a limitation. An important area for future research is the use of methods that more accurately assess predisposing factors for sleep paralysis and the development of more standardized and uniform diagnostic criteria to allow differentiation from other sleep disorders (nightmares, night terrors, etc.). Although there has been some research in recent years into the role of the various components that control REM sleep, how these components work together through different brain regions in regulating sleep remains unclear. For example, melanin-concentrating hormone is a substance that acts on the REM sleep network and regulates REM sleep. It has only recently been discovered, and its mechanism of action is still far from clear. Further studies on neuropeptides can reveal more details of the complete sleep network in all its complexity, which can help to explain the pathogenesis of various sleep disorders. Therefore, future efforts should be directed toward understanding the regulatory mechanisms of different neuropeptides. It is only in recent years that our understanding of REM sleep control circuits has expanded to include numerous brain regions, and it is important to further investigate the relationship between sleep paralysis and different regions of the brain, particularly how the conflict arises between muscle atonia and consciousness during the transition between REM sleep and wakefulness. A deeper understanding of this circuit will certainly help us to further explore the mysteries of REM sleep and sleep disorders. In addition, it is important to further clarify the differences between REM sleep, isolated sleep paralysis, and narcolepsy-associated sleep paralysis, such as the subtle differences in eye movements and EEG and electromyography (EMG) data. Moreover, there is some evidence demonstrating that although ISP does not always progress to narcolepsy, its frequent early occurrence during the course of narcolepsy suggests that it may be a precursor. This has important clinical implications, i.e., in the early identification and intervention of ISP in patients to potentially prevent progression to more severe narcolepsy, but validation is required through future studies with large sample sizes.
Arguably, the most pressing issue in understanding the REM sleep network is to study and treat the associated human diseases. Currently, there are no specific medications for sleep paralysis. Future research should continue to focus on why REM sleep-controlling circuits are susceptible to pathological recruitment, as well as more subtle neurological action mechanisms. In addition, more in-depth exploration of the interactions between other sleep disorders and sleep paralysis, as well as continued probing of the effects of genetic polymorphisms on the prevalence of sleep paralysis, would be of great help in developing additional therapeutic strategies targeting the pathogenesis of sleep paralysis. Whether various hormones have an effect on the pathogenesis of sleep paralysis is also a question for further research. Therefore, in the future, we recommend establishing a multidisciplinary collaborative treatment model—integrating expertise from sleep medicine, neurology, psychiatry, genetics, endocrinology, and other fields—to develop and formulate more effective and targeted individualized treatment plans for normalizing activity of the disrupted neural pathways mediating sleep paralysis.
5-HT, Serotonin; 5-HT2AR, Serotonin 2A receptor; Ach, Acetylcholine; BMI, Body mass index; CBT, Cognitive-behavioral therapy; CeA, Central nucleus of the amygdala; cLDT, Caudal lateral dorsal tegmentum; DPGi, Dorsal paragigantocellular reticular nucleus; DpMe, Deep mesencephalic nuclei; DR, Dorsal raphe; EEG, Electroencephalography; EMG, Electromyography; GABA, Gamma-aminobutyric acid; GiA,
YW and DY conceptualized the review and revised the manuscript. YW, QL, ZZ, QO and XZ conducted the literature research and wrote the first draft of the manuscript. YW and KY created figures. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank BioRender.com for providing the platform used to create the figures.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.