






 , Li Chen 2,†, Heren Gao 3, Tao Liu 4
, Li Chen 2,†, Heren Gao 3, Tao Liu 41 College of Integrated Chinese and Western Medicine, Anhui University of Chinese Medicine, 230012 Hefei, Anhui, China
2 Department of neurology, Shanghai Sixth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, 201306 Shanghai, China
3 College of Acupuncture and Massage, Anhui University of Chinese Medicine, 230012 Hefei, Anhui, China
4 Orthopedic Center, Maanshan General Hospital of Ranger-Duree Healthcare, 243000 Maanshan, Anhui, China
†These authors contributed equally.
Abstract
Lamotrigine (LTG) is an antiepileptic drug that stabilizes the presynaptic membrane by blocking sodium channels and inhibiting excessive glutamate release. Its neuroprotective effects have been demonstrated in various pathological states. However, the role of LTG in spinal cord injury (SCI) and its relationship with autophagy, which is essential for cellular homeostasis, warrant further investigation.
We established a mouse model of SCI using complete spinal transection. The neuroprotective effects of LTG were assessed using immunostaining and functional assessments, including Basso Mouse Scale (BMS) scores, lesion site area, and synapse survival. Western blot analyses were also performed to further examine the underlying cellular and molecular mechanisms of autophagy.
LTG treatment promoted the post-traumatic survival of spinal neurons, improved BMS scores, reduced lesion site area, and enhanced synapse survival in a mouse model of SCI. Furthermore, LTG attenuated apoptosis following SCI by activating autophagy during the secondary injury phase. These findings indicate that LTG-enhanced autophagosome formation and autolysosome degradation play a key role in reducing neuronal loss after SCI.
LTG appears to attenuate post-traumatic spinal neural injury by enhancing autophagy flux.
Keywords
- lamotrigine
- autophagy
- spinal cord injuries
- apoptosis
- neuroprotection
Spinal cord injury (SCI) is a severe disease characterized by paralysis and sensorimotor deficits. It involves both primary and secondary injuries, although the mechanisms are still not fully understood [1, 2, 3]. The primary injury happens instantly at the time of trauma, triggering direct neuronal loss. In contrast, the secondary injury evolves over time, driven by prolonged pathological factors such as ischemia, oxidative stress, ionic disturbances, inflammation, and excitotoxicity [4]. Together, these factors intensify a “neurotoxicity storm” that diminishes neuronal viability around the initial lesion site, further impairs motor functions, and delays recovery.
Whereas primary injuries are immediate and unavoidable, significant opportunities exist to mitigate neuronal damage during the secondary phase. In both animal experiments and clinical studies, interventions that target these secondary processes have shown potential for halting the progression of neurological damage, fostering neuronal regeneration, and enhancing functional recovery [5, 6]. Therefore, it is crucial for clinicians and researchers to investigate the pathological mechanisms of secondary injury and identify effective interventions. Understanding these aspects is key to improving the treatment and management of SCI, and offering hope for better patient outcomes.
Autophagy is a crucial cellular mechanism that maintains organelle function and protein quality by removing damaged organelles and protein aggregates through a process known as autophagy flux. This process begins with the formation of double-membrane autophagosomes that capture toxic proteins and damaged cellular components. Autophagosomes subsequently fuse with lysosomes, leading to the degradation of their contents in a process that is essential for cellular homeostasis. However, when autophagy is downregulated below physiological levels in animal experiments, this can result in the accumulation of toxic proteins within neurons, contributing to neurodegenerative disorders such as Alzheimer’s disease (AD) and Parkinson’s disease [7, 8]. Conversely, the upregulation of autophagy in mouse experiments was shown to reduce energy loss and increase the scavenging of damaged protein and organelle aggregates, thus playing a role in the pathology of SCI [9, 10]. Overall, autophagy is essential for promoting neuronal survival and contributing to cell death under stress. These functions highlight its complex involvement in SCI and warrant further investigation.
As we know, within minutes after primary SCI, a combination of direct cellular damage and ischemia/hypoxia triggers a significant rise in extracellular glutamate, the main excitatory neurotransmitter in the central nervous system [11]. This glutamate excess triggers excitotoxicity, resulting in extensive apoptotic and necrotic cell death [12]. Lamotrigine (LTG), a mood stabilizer known for its ability to block sodium channels and reduce excessive glutamate release, has shown promise as a neuroprotective agent in various neurological disorders, as demonstrated in both animal experiments and clinical studies [13, 14]. Studies have shown that LTG inhibits glutamate release in several brain areas, including the medial prefrontal and cortex hippocampus [15, 16, 17]. This characteristic made LTG a candidate for investigation in SCI.
However, beyond its well-documented effects on glutamate regulation, emerging evidence suggests that LTG may exert neuroprotective effects through mechanisms involving autophagy. In animal model of AD, LTG was demonstrated to mitigate abnormal neuronal activity, preserve dendritic spines, maintain synaptic integrity, and prevent neuronal loss, ultimately improving synaptic plasticity and cognitive performance [18]. These benefits are thought to arise, at least in part, from the activation of autophagy, a cellular process vital for degrading damaged organelles and proteins while maintaining neuronal health under pathological stress [19]. Given the shared pathological features of AD and SCI, such as excitotoxicity, mitochondrial dysfunction, oxidative stress, and neuroinflammation, investigating LTG in the context of SCI provides a valuable opportunity to deepen our understanding of its therapeutic potential.
In contrast to chronic neurodegenerative diseases, SCI provides a well-defined model of acute neurotrauma that is uniquely suited for investigating neuroprotective strategies during rapid and severe secondary injury. The clearly delineated temporal and spatial progression of SCI allows focused exploration of mechanisms such as excitotoxicity, oxidative stress, and inflammation. In the present study we examined whether LTG can stimulate autophagy and reduce secondary neural damage in SCI. By focusing on this underexplored therapeutic approach, we hope to gain deeper insights into the underlying mechanisms, thus paving the way for new and more effective treatments for SCI.
Adult C57BL/6 mice (18–20 g, 8–10 weeks old) were purchased from Huachuang Xinnuo Pharmaceutical Technology Co., Ltd. (Taizhou, Jiangsu, China; License No.: SCXK (China, Jiangsu) 2020-0009) and housed in the Key Laboratory of Integrated Traditional Chinese and Western Medicine, Anhui University of Chinese Medicine. The animal experiments were approved by the Ethics Committee of our institute (Approval No.: AHUCM-mouse-2024224). Mice were kept in cages (five per cage) under a 12-hour light/dark cycle, controlled temperature (23–25 °C), and 40–50% humidity, with free access to food and water.
In total, 230 mice were randomly assigned to groups using a randomized digital table. Data collection and analysis were performed by blinded researchers. The experimental groups were as follows: (i) Control group (Control), (ii) SCI-vehicle group (SCI-Veh), (iii) SCI-LTG group (SCI-LTG), (iv) SCI-Bafilomycin A1 (Baf) group (SCI-Baf), and (v) SCI-LTG-Baf group (SCI-LTG-Baf). The Control group underwent laminectomy without spinal cord transection as a sham procedure, while all other groups underwent spinal cord transection. The SCI-LTG group received LTG treatment, the SCI-Veh group received an equivalent volume of DMSO and saline as the vehicle, and the SCI-Baf group was treated with Bafilomycin. The SCI-LTG-Baf group received both LTG and Bafilomycin. The number of mice in each experiment is provided in the figure legend.
LTG (3,5-diamino-6-[2,3-dichlorophenyl]-1,2,4-triazine, L3791, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in DMSO (Dimethyl sulfoxide, 276855, Sigma-Aldrich) and diluted to 30 mg/mL with 0.9% saline. Mice were administered LTG via intraperitoneal injection at a dose of 30 mg/kg immediately following SCI surgery, with daily injections continuing for 4 weeks [18, 20]. Bafilomycin A1 (Baf, 11038, Cayman Chemical, Ann Arbor, MI, USA) was dissolved in DMSO and adjusted to 0.5 mg/mL with 0.9% saline. Baf was injected intraperitoneally at a dose of 1 mg/kg [21], beginning 1 h after the LTG injection and continuing daily for the same 4-week period. The control group received a vehicle solution with the same dilution components to assess any potential effects of the solvent.
Mice (8–10 weeks old, 20–25 g) underwent surgery while under 40 mg/kg pentobarbital sodium (P3761, Sigma-Aldrich) anesthesia (i.p.). For the transection groups, a complete spinal cord transection at T9 was performed using iridectomy scissors and a micro-knife to ensure thorough lesioning. The muscle, fascia, and skin were then sutured [10, 22]. Post-surgery, manual urination was carried out twice daily until bladder function was restored. All procedures were performed by a blinded, independent surgeon.
Following pentobarbital sodium anesthesia (40 mg/kg, i.p.), mice received a saline perfusion, followed by 0.1 M phosphate-buffered saline (PBS, P5493, Sigma-Aldrich) containing 4% paraformaldehyde (PFA, pH 7.4, 441244, Sigma-Aldrich). The spinal cords were post-fixed in 4% PFA overnight at 4 °C, transferred to 20% sucrose (S5016, Sigma-Aldrich) for 12 h, followed by 30% sucrose for another 12 h, and finally embedded in optimum cutting temperature (OCT) compound (4583, Sakura Finetek USA, Torrance, CA, USA). Tissues were sectioned at 25 µm and incubated with primary antibodies overnight at 4 °C (NeuN, a neuronal nuclear-specific protein, ABN78, Millipore, Burlington, MA, USA, 1:500; GFAP-Cy3, glial fibrillary acidic protein-cyanine 3, ab49874, Abcam, Cambridge, UK, 1:1000; ChAT, choline acetyltransferase, AB144, Sigma-Aldrich, 1:200; vGlut1, vesicular glutamate transporter 1, AB5905, Sigma-Aldrich, 1:500; p62, sequestosome-1, P0067, Sigma-Aldrich, 1:500; LC3B, B-light chain 3, NB100-2220, Novus, Centennial, CO, USA, 1:500). The sections were then washed and incubated overnight at 4 °C with Alexa Fluor 488 or 555-conjugated secondary antibodies (A31572, A21206, Thermo Fisher Scientific, Waltham, MA, USA, 1:600), followed by observation with a Zeiss LSM710 confocal microscope (Zeiss, Oberkochen, Germany). Quantification was performed using ImageJ software (1.43, NIH, Bethesda, MD, USA), with researchers blinded to the group allocations during analysis.
To prepare samples for Western blot analysis, 1.5 mm of spinal cord tissue
containing the lesion scar was collected from each mouse after euthanizing with
200 mg/kg pentobarbital sodium (i.p.) and perfusion with ice-cold PBS to clear
blood. Samples were sonicated and lysed in radioimmunoprecipitation assay (RIPA)
buffer (1% nonidet P-40, NP-40, ST2045, Beyotime, Shanghai, China; 50 mM tris
hydroxymethyl-aminomethane hydrochloride (Tris-HCl, B548127, Sangon Biotech,
Shanghai, China); 1 mM disodium ethylenediaminetetraacetate (Na2-EDTA,
E8030, Solarbio Life Sciences, Beijing, China); 0.25% sodium deoxycholate
(Na-deoxycholate, ST2049, Beyotime); 150 mM sodium chloride
(NaCl, A501218, Sangon Biotech)) containing protease and
phosphatase inhibitors (4906837001, Roche, Basel, Switzerland).
After pretreatment, equal protein amounts were separated by SDS-PAGE and
transferred to 0.2 µm polyvinylidene fluoride (PVDF) membranes (ISEQ00010, Millipore), followed by overnight incubation at 4 °C with
primary antibodies (p62, P0067, Sigma-Aldrich, 1:1000; LC3B,
NB100-2220, Novus, 1:1000; vGlut1, 48-2400, Invitrogen,
Carlsbad, CA, USA, 1:500; Bcl-2, B-cell lymphoma-2, ab182858, Abcam, 1:1000; Bax,
Bcl-2-associated x, ab32503, Abcam, 1:1000; Cleaved caspase-3,
ab32351, Abcam, 1:1000;
To observe apoptotic neurons, a terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling (TUNEL) assay was performed using the One Step TUNEL Apoptosis Assay Kit (C1086, Beyotime) according to the manufacturer’s instructions. After tissue preparation and freezing, the sections were post-fixed with 4% PFA for 30 minutes, then treated with PBS containing 0.5% Triton X-100 (X100, Sigma-Aldrich) and incubated at room temperature for 5 minutes. Sections were subsequently incubated with 50 µL TUNEL reaction mixture at 37 °C for 1 h in a dark environment. Finally, the images were captured, and TUNEL+ neurons were manually counted using ImageJ software (1.43, NIH) in a blinded manner.
Hindlimb movement recovery was assessed at 0, 1, 3, 7, 14, 21, 28, 35, 42, 49, 56, and 63 days using the Basso Mouse Scale (BMS) open-field test, as described by Basso and colleagues [23]. One day before SCI surgery, mice were positioned in the open field to familiarize themselves with the environment. BMS scoring began on the first-day post-surgery, with each session lasting 4 minutes. The BMS scale ranges from 0 for complete paralysis, to 9 for normal hindlimb movement. A higher score indicates better recovery. Three mice were excluded from the experiment because their hindlimbs were bitten and injured after SCI. Two researchers, blinded to group allocation, independently evaluated the mice. When the BMS scores varied between hind limbs, the average was calculated.
To quantify the lesion site area, spinal cords were sectioned sagittally, with the midline as the reference point. Three sagittal sections centered around the spinal cord midline were selected for analysis. Each section was immunostained with GFAP-Cy3 to delineate the boundary of the lesion site, defined as the region lacking intact GFAP+ astrocytic networks. The lesion area for each selected section was manually outlined and measured with ImageJ software. The average lesion area across the three sections was calculated, representing the lesion site area for each sample [24].
All quantification was performed blind to group allocation, with sample numbers
provided in the corresponding figure legends. Data are presented as the mean
We conducted a series of experiments to evaluate the neuroprotective effect of LTG (Fig. 1a). Spinal cords were harvested and serially sectioned into coronal slices that spanned 600 µm rostrally and caudally around the lesion epicenter (Fig. 1b). The results showed that LTG treatment (30 mg/mL) significantly improved the survival of spinal neurons 4 weeks after SCI, suggesting it has the potential to mitigate neuronal loss (Fig. 1c,d). Further analyses revealed an increase in ChAT+ motor neurons in the SCI-LTG group compared with the SCI-Veh group at 4 weeks post-SCI, thus reinforcing the neuroprotective effect of LTG (Fig. 1e,f). Additionally, co-immunostaining for 4′-6-diamidino-2-phenylindole (DAPI) and GFAP was performed to assess the lesion area at the injury site (Fig. 1g,h). This revealed a smaller lesion area in the SCI-LTG group compared with the SCI-Veh group. Overall, LTG treatment effectively reduced spinal neural injury after SCI.
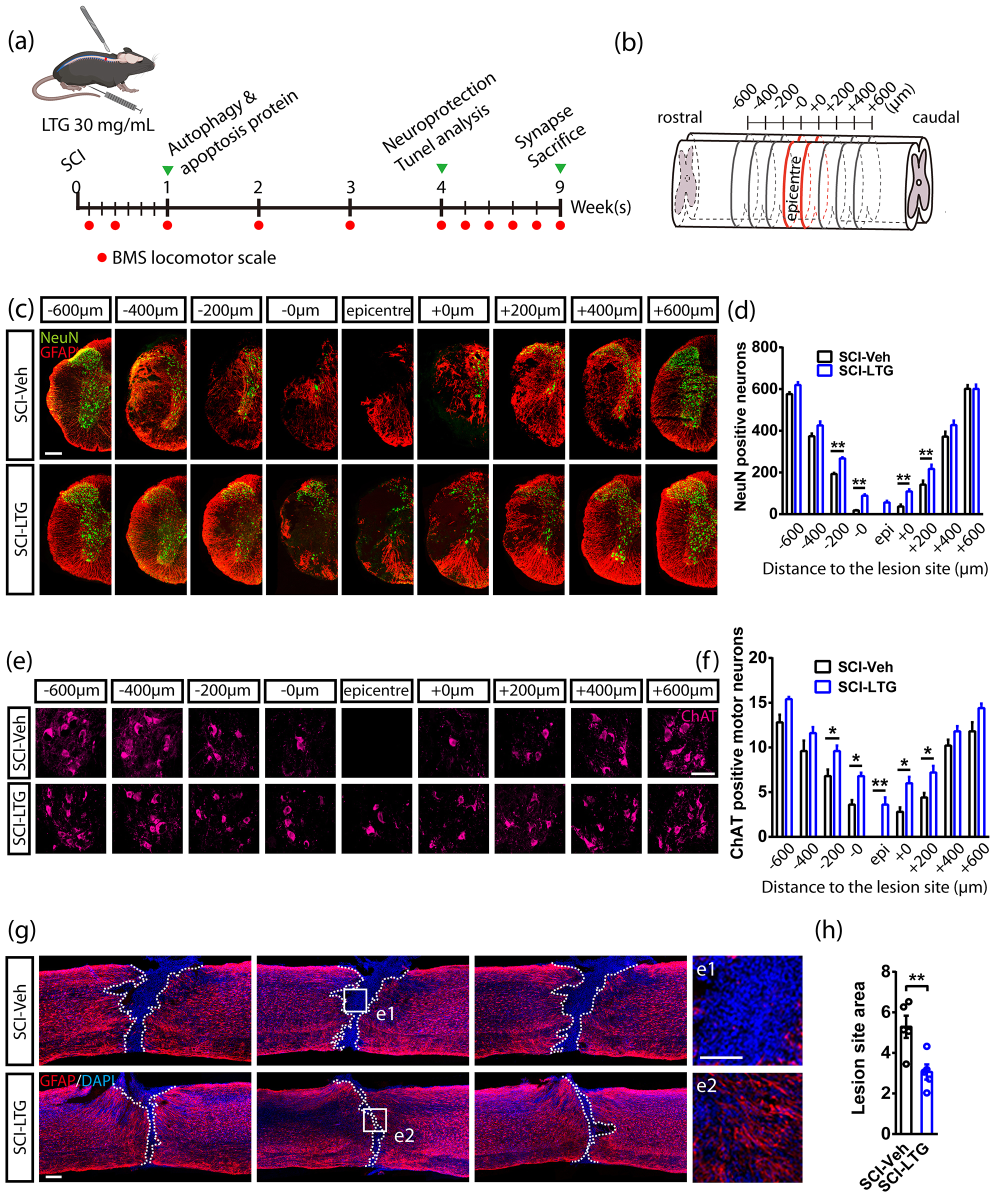 Fig. 1.
Fig. 1.
LTG mitigates post-traumatic spinal neural
injury. (a) Experimental timeline showing SCI surgery, LTG treatment,
neuroprotection assessment, BMS scoring, analysis of apoptosis and
autophagy-related proteins, and synapse survival post-injury. (b) A
representative serial coronal section showing NeuN+ and ChAT+ neurons,
and spanning rostrally (–600 µm) and caudally (+600 µm) from the
lesion epicenter. (c,d) Co-immunostaining (c) and quantification (d) of NeuN and
GFAP from 600 µm rostral to 600 µm caudal around the epicenter in
SCI-Veh and SCI-LTG groups at 4 weeks post-injury. The number of NeuN+
neurons was counted, with data presented as mean
Previous research has indicated that LTG attenuates spinal neural injury after SCI. However, the impact of these neuroprotective effects on locomotor function still requires further investigation. We assessed the open-field locomotor function in mice with a complete T9 transection by using a double-blinded BMS scoring system. Initially, both SCI-LTG and SCI-Veh groups exhibited severe impairment in hind limb locomotion, which showed gradual improvement over time. On days 35, 42, 49, 56, and 63 post-injury, BMS scores were significantly higher in LTG-treated mice compared to the Veh group (Fig. 2a,b).
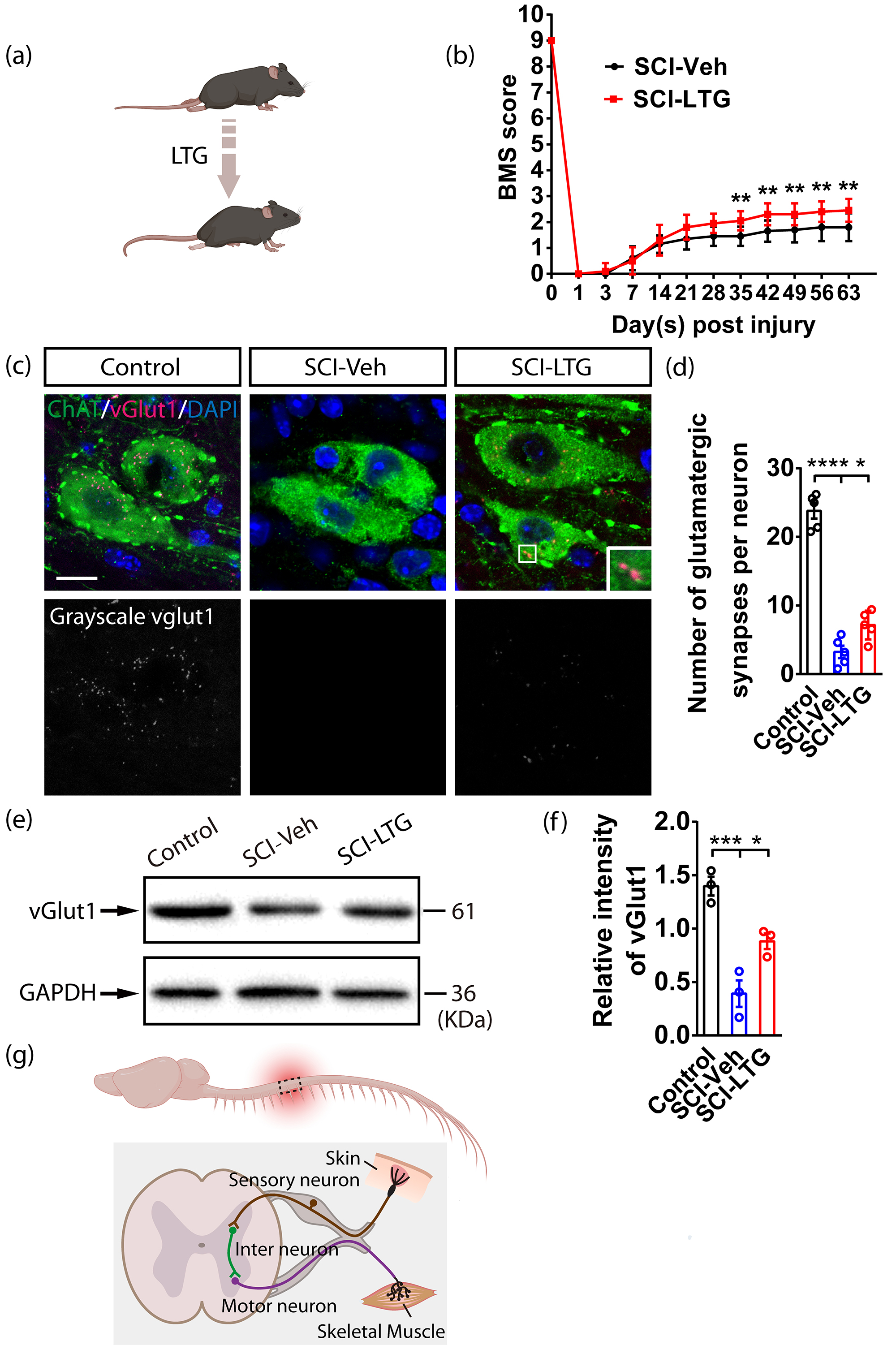 Fig. 2.
Fig. 2.
Enhanced BMS scores and synapse survival in
LTG-treated mice following SCI. (a) Diagram showing motor function recovery in
mice following LTG treatment. (b) The timeline displays changes in BMS scores in
SCI-Veh and SCI-LTG groups. Data are presented as the mean
Subsequently, we investigated the basis for the improved motor function in LTG-treated mice. Given the enhanced neuronal survival observed in the LTG group (Fig. 1), we hypothesized that synapse survival was also increased, which could partly explain the improved BMS scores. To verify this, we performed co-immunostaining of ChAT and vGlut1 at 9 weeks after SCI. This confirmed the presence of a higher number of glutamatergic synapses in the SCI-LTG group compared with the SCI-Veh group (Fig. 2c,d). Western blot analysis of vGlut1 further supported this finding, indicating more glutamatergic synapses in the LTG-treated group (Fig. 2e,f) (The original western blot data for Fig. 2e are provided in Supplementary Fig. 1). These results suggest that the improvement in BMS scores observed in LTG-treated mice after SCI may be due to enhanced survival of spinal synapses and neurons (Fig. 2g).
We next conducted a series of analyses to elucidate the neuroprotective mechanism of LTG. First, TUNEL staining was performed to detect apoptotic neurons in both the SCI-Veh and SCI-LTG groups. The results showed a reduction in apoptotic neurons in the SCI-LTG group compared to the SCI-Veh group 4 weeks after SCI (Fig. 3a–c). Following this, we conducted western blot analysis of key apoptosis-related proteins, including Bcl-2, Bax, and cleaved caspase-3, 7 days post-injury (Fig. 3d,e) (The original western blot data for Fig. 3e are provided in Supplementary Fig. 2). The SCI-LTG group showed significantly increased Bcl-2 expression levels in comparison to the SCI-Veh group (Fig. 3f), along with reduced expression levels of Bax and cleaved caspase-3 (Fig. 3g,h). These results indicate that LTG effectively suppresses neuronal apoptosis, which may underlie its neuroprotective effects.
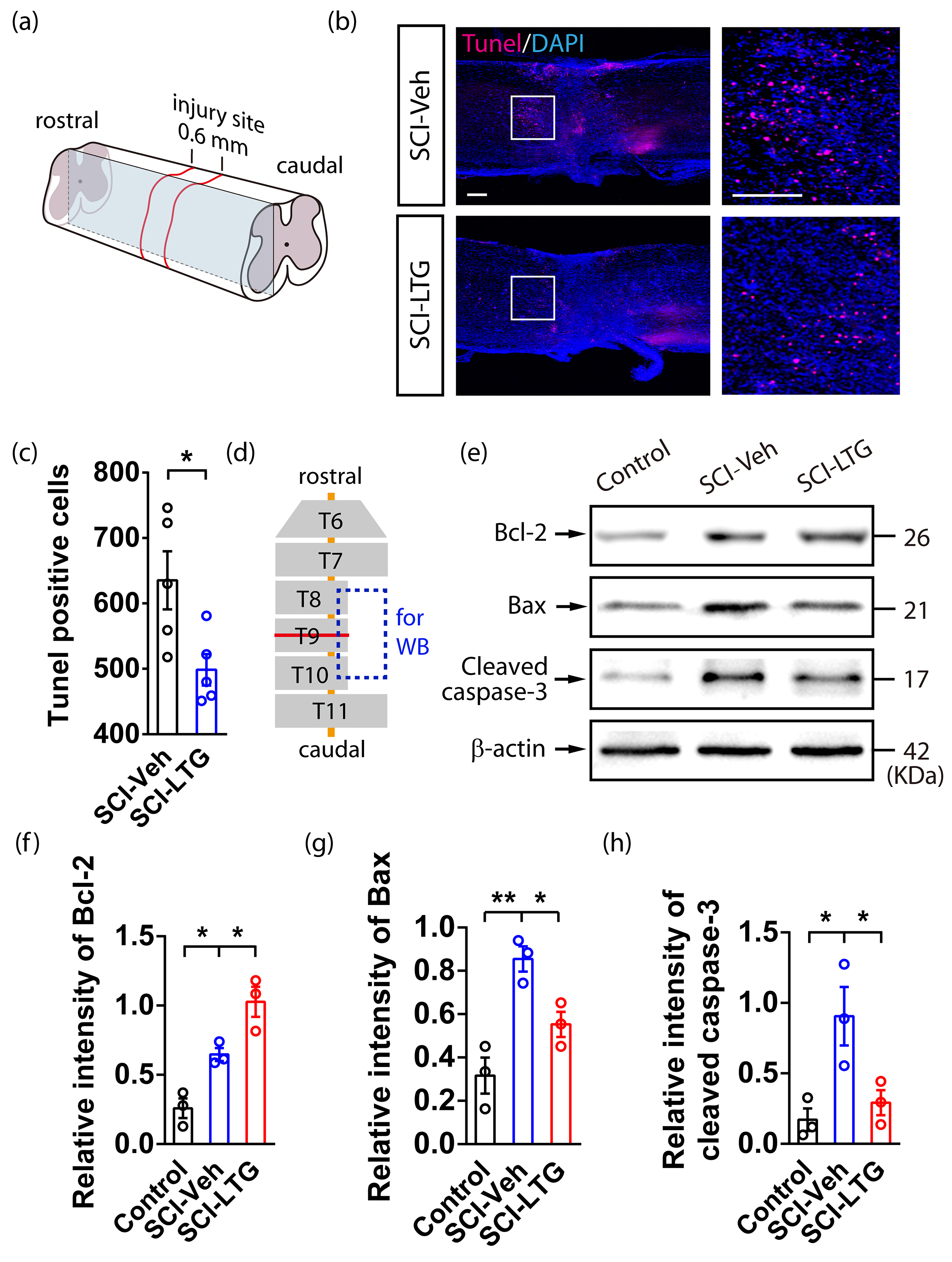 Fig. 3.
Fig. 3.
LTG may reduce neuronal apoptosis in mice after SCI. (a)
Diagram illustrating TUNEL staining around the injury site in the mouse spinal
cord following SCI. (b,c) TUNEL assay (b) and quantitative analysis (c)
of apoptotic neurons in the SCI-Veh and SCI-LTG groups 4 weeks post-injury. The
number of TUNEL+ neurons was counted, and data are presented as the mean
To further investigate how LTG suppresses apoptosis and promotes neuronal survival after SCI, we examined whether these effects are linked to the activation of autophagy. We first assessed LC3-II expression as an indicator of autophagosome formation. At 7 days after SCI, a significant increase in LC3-II levels was observed in the SCI-LTG group compared with the SCI-Veh group, indicating enhanced autophagosome formation (Fig. 4a,b) (The original western blot data for Fig. 4a are provided in Supplementary Fig. 3). To determine whether this increase was due to elevated autophagosome production or impaired autolysosome degradation, we examined the expression of p62. This marker accumulates when autolysosome degradation is inhibited. p62 expression was significantly lower in the SCI-LTG group compared to the SCI-Veh group at 7 days post-SCI, suggesting that LTG treatment promotes both autophagosome formation and autolysosome degradation (Fig. 4c). Immunostaining further corroborated these findings, showing reduced p62 and elevated LC3-II levels in the SCI-LTG group compared to the SCI-Veh group (Fig. 4d–f).
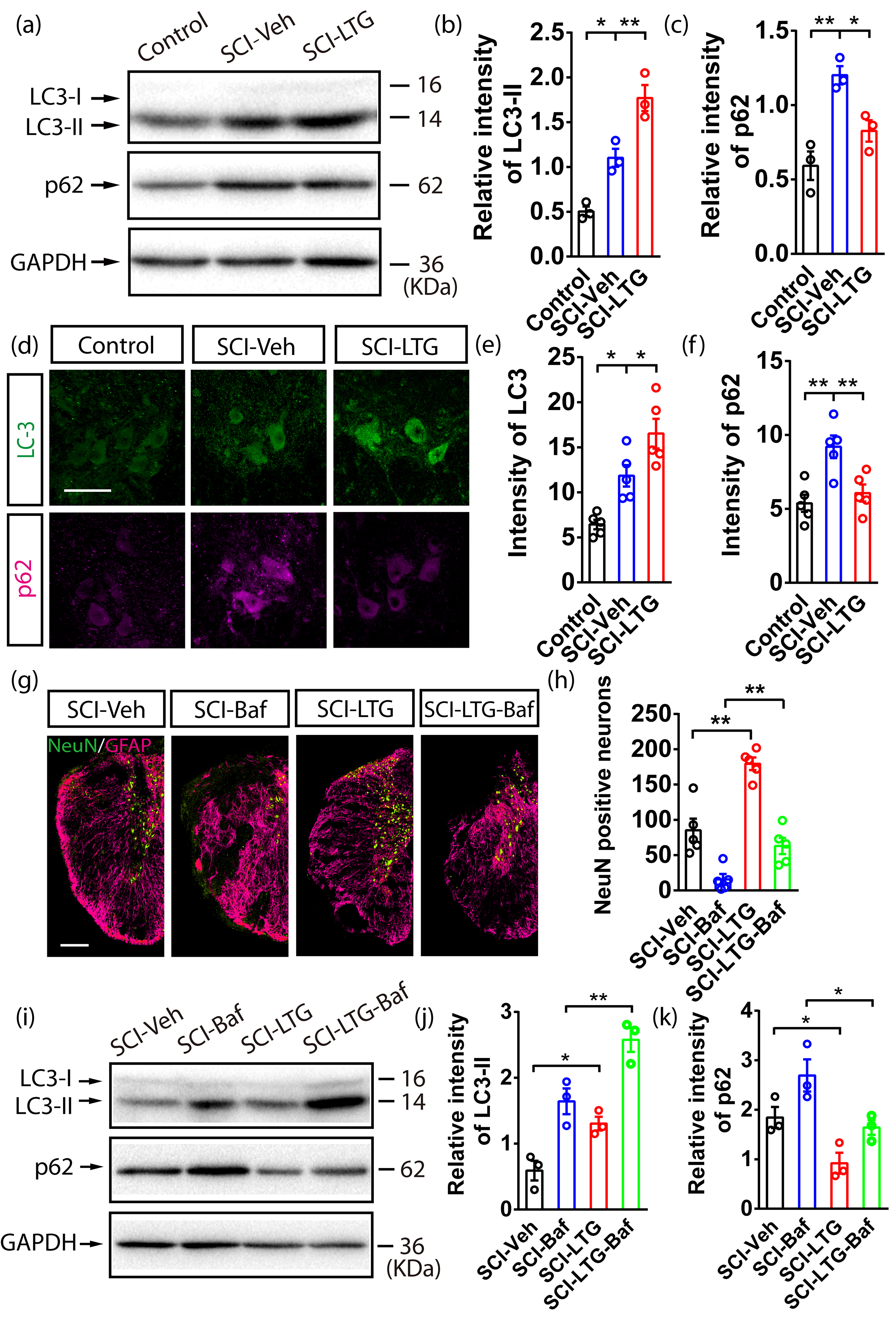 Fig. 4.
Fig. 4.
Enhanced autophagy flux by LTG attenuates neuronal death in
SCI. (a–c) Western blot (a) and quantification of LC3-II (b) and p62 (c)
expression at the lesion site in Control, SCI-Veh, and SCI-LTG groups 7 days
after SCI. Data are presented as the mean
To further elucidate the role of autophagic flux in neuronal survival, we employed Baf, a specific inhibitor of autolysosome degradation [25]. Co-immunostaining for NeuN and GFAP at 4 weeks post-SCI revealed a significant reduction in surviving spinal neurons in the SCI-Baf and SCI-LTG-Baf groups compared with the SCI-Veh and SCI-LTG groups that did not receive Baf treatment. Notably, among the Baf-treated groups, the SCI-LTG-Baf group exhibited a higher neuronal survival rate than the SCI-Baf group (Fig. 4g,h). These findings suggest that Baf treatment decreases the number of surviving neurons by blocking lysosome degradation, thereby exacerbating neuronal death after SCI. LTG mitigates this effect by promoting autophagy flux, which is essential for the survival of spinal neurons.
To assess autophagy-lysosome changes after Baf treatment, p62 and LC3-II were measured at the lesion area 7 days post-SCI. Both the SCI-Baf and SCI-LTG-Baf groups showed increased p62 and LC3-II levels, indicating that Baf effectively suppressed autolysosome degradation in the autophagy-lysosome pathway. In addition, the SCI-LTG-Baf group exhibited greater autolysosome degradation than the SCI-Baf group, suggesting that LTG partially alleviates Baf-induced suppression of autolysosome degradation (Fig. 4i–k) (The original western blot data for Fig. 4i are provided in Supplementary Fig. 4). Elevated autolysosome degradation in the SCI-LTG-Baf group is reflected by the reduced p62 levels, which suggests enhanced autophagic flux. This may explain the reduced neuron loss in LTG-treated mice after SCI and Baf treatment.
In summary, these results demonstrate that autophagy flux is essential for LTG to promote the survival of spinal neurons after SCI by enhancing autophagosome formation and autolysosome degradation.
Currently, no well-established neuroprotective treatments exist for SCI. This is partly due to the complexity of the injury mechanisms, as demonstrated in animal experiments and clinical studies [4, 5, 6]. SCI involves both direct mechanical tissue damage (primary injury) and biochemical changes that cause delayed or progressive cell death (secondary injury) [5]. The primary injury results in cell stress and plasma membrane rupture, leading to ionic imbalance, excitatory amino acid release, and the formation of oxidative species at the injury site (Fig. 5). These events trigger secondary changes that spread the injury to neighboring intact cells over time. While the primary injury is instantaneous and irreversible, secondary injury occurs gradually and involves specific biochemical, cellular, and molecular changes, thus presenting a therapeutic opportunity.
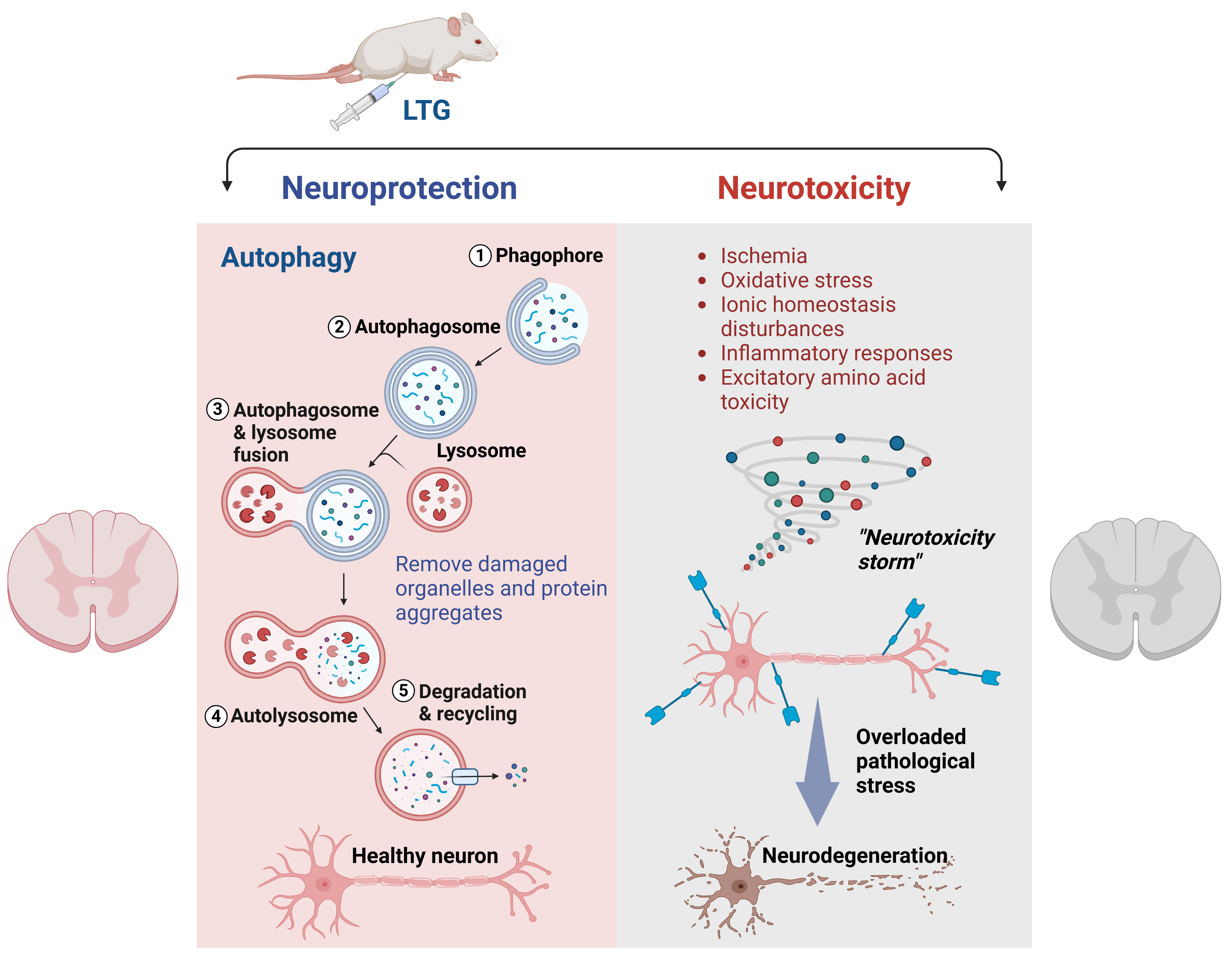 Fig. 5.
Fig. 5.
LTG-mediated neuroprotection via activation of autophagy in SCI. During the secondary injury phase following SCI, neurotoxic stress arises from ischemia, ionic homeostasis disturbances, oxidative stress, inflammatory responses, and excitatory amino acid toxicity. Together, these factors create a “neurotoxicity storm”, leading to pathological stress and subsequent neurodegeneration. LTG can activate autophagy to facilitate the removal of damaged protein and organelle aggregates, thus maintaining neuronal health and sustaining overall cellular homeostasis. Fig. 5 was created using BioRender (https://biorender.com/). The license for BioRender (license number: WL27WGJWOG) is provided.
One significant pathological change during the secondary phase is the excessive release of excitatory neurotransmitters, particularly glutamate. Glutamate binds to its receptors, opens corresponding ion channels, and persists in injured tissue for days after SCI [26]. This dysfunction triggers excitotoxicity, calcium overload, and a cascade of cell death processes, further aggravating the injury [27]. Pharmacological research has shown that LTG inhibits the extracellular accumulation of glutamate, as well as action potential discharges caused by glutamate. While previous research has primarily focused on the anti-convulsant effects of LTG and its role in regulating excitatory neurotransmitter release, our study has drawn attention to a less explored function by demonstrating its ability to activate autophagy. This perspective addresses a critical gap in the current literature, as the autophagy-related effects of LTG remain largely unexplored. Our findings revealed that LTG enhances autophagy flux and provides robust neuroprotection after SCI (Fig. 5). LTG promotes the recovery of motor function, preserves neuronal and synaptic integrity, and reduces the size of the lesion site, thus offering a detailed and comprehensive understanding of its therapeutic potential in acute SCI. Our results are consistent with previous findings that LTG improves neurofunctional recovery and mitigates proteasome inhibition-induced apoptosis, as demonstrated in studies of cell lines and in rat models of hypoxic-ischemic encephalopathy, cerebral ischemia, and subarachnoid hemorrhage [28, 29].
Autophagy is a catabolic process that clears toxic proteins and damaged organelles through the autophago-lysosomal pathway [30]. It acts as an initial survival mechanism, markedly induced during ischemia events and neurotrauma. Following SCI, enhanced autophagy promotes the survival of spinal neurons during ischemia/reperfusion [31]. Basal autophagy suppression in neural cells has been shown to cause neurodegenerative disease in mice. Recent studies suggest that LTG suppresses abnormal spike activity, prevents loss of spines and synaptophysin immunoreactivity, and reduces neuron death in AD mice, likely due to the activation of autophagy [18, 19]. Unlike neurodegenerative diseases, SCI involves acute and rapidly progressing pathological changes such as excitotoxicity, oxidative stress, and inflammation. The present study used an acute injury model to explore the effects of LTG on autophagy in the context of SCI. Our results showed that LTG activates autophagy during the secondary injury phase and attenuates disruption of the autophagy-lysosome pathway, which is crucial for cellular homeostasis.
Autophagy flux, which encompasses the formation of autophagosomes to the degradation of their cargo by lysosomes, plays a significant role in cellular homeostasis. Our data suggest suppressed autolysosome degradation and disruption of the autophagy-lysosome pathway after SCI, but LTG can correct this disruption. To assess whether the neuroprotective effect of LTG involves enhanced autophagy flux, we blocked it using Baf, an autophagy-lysosome pathway inhibitor. Our results showed that blocking lysosome degradation worsens spinal neuron injury, highlighting the importance of an unobstructed autophagy-lysosome pathway for the neuroprotective effect of LTG. Interestingly, some spinal neurons survived Baf treatment, suggesting that autophagy is not the only neuroprotective mechanism involved and warranting further investigation.
Research indicates that early autophagy activation is protective, while late or delayed activation can be detrimental [32]. In LTG-treated mice, enhanced autophagy flux immediately after SCI removes damaged organelles and aggregates, thereby promoting neuron survival. Additionally, scar formation at the lesion site involves neuroimmunology, gliocytes, and other factors. It has been reported that autophagy is induced in both neurons and astrocytes after cerebral ischemia injury in a rat model of cerebral stroke [33]. Our study found a beneficial effect of LTG on the lesion site area. However, the exact autophagic mechanisms and changes in astrocytes remain to be investigated and should provide deeper insight into the role of LTG in SCI recovery.
It is well-documented that Bcl-2 functions as an anti-apoptotic protein, Bax promotes apoptosis, and cleaved caspase-3 can inhibit the anti-apoptotic function of Bcl-2. Intricate interactions between these proteins regulate the initiation and progression of apoptosis [34, 35, 36]. Although apoptosis and autophagy are distinct processes, they are interconnected through shared regulatory networks involving key molecules such as p53, Bcl-2, and serine/threonine kinases [37, 38]. For example, in response to cellular stress such as injury or starvation, c-Jun N-terminal kinase 1 (JNK1) phosphorylates Bcl-2. This disrupts its interaction with Bcl-2-interacting protein (Beclin-1), which promotes autophagy in cell lines. Simultaneously, phosphorylated Bcl-2 binds to Bax, stabilizing mitochondrial membrane integrity and reinforcing its anti-apoptotic role [39, 40]. Parallel studies in rats have shown that resveratrol enhances autophagy-related protein expression while inhibiting apoptosis through activation of the adenosine 5′-monophosphate-activated protein kinase (AMPK) pathway [40, 41]. In the current research, LTG was found to activate autophagy and reduce neuronal apoptosis after SCI, although the exact molecular mechanisms remain to be elucidated. Moreover, it is still unclear whether autophagy activates apoptosis or vice versa, presenting a “chicken or the egg” dilemma. Future research should explore whether autophagy is activated early in acute injury to support survival, with apoptosis becoming dominant later due to prolonged stress. It will be essential to understand how LTG influences these pathways, and to identify its specific mechanisms and targets. Time-series experiments may provide additional clarity on these interactions.
This study provides important insights, but several limitations need to be considered. First, while LTG is shown to activate autophagy and provide neuroprotection, the molecular mechanisms and specific targets involved are not yet fully understood. Further studies are necessary to clarify the exact components and pathways involved. Second, although our focus was on the autophagy-lysosome pathway, autophagy is a complex process that also interacts with other mechanisms, such as apoptosis, oxidative stress, and inflammation. These interactions were not explored in depth, and understanding them is essential for refining LTG-based therapies. Finally, the use of preclinical animal models limits the ability to directly apply findings to human SCI. Species-specific differences in injury response may affect how these results translate to humans. Future studies should include larger and more clinically relevant cohorts to better assess the potential and safety of LTG in human SCI.
Our findings demonstrate that LTG restores the autophagy-lysosomal process in injured spinal neurons by enhancing autophagic flux, which encompasses autophagosome formation and autolysosome degradation. This process reduces neuronal loss, protects synapses, and improves functional outcomes.
The dataset supporting the conclusions of this article is included within the article. Further inquiries can be directed to the corresponding author.
MTZ supervised the research. MTZ, LC and HRG designed and performed the experiments, MTZ, LC, HRG and TL interpreted the data and wrote the manuscript. HRG, LC, and TL analyzed the data. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal experimental protocols employed were meticulously reviewed and approved by the Ethical Committee of Anhui University of Chinese Medicine (Approval No. AHUCM-mouse-2024224) in accordance with the ARRIVE guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
We sincerely thank Dr. Yaobo Liu from the Institute of Neuroscience at Soochow University for his insightful comments on this project, as well as all members of the Liu laboratory for their valuable discussions and support. We are also grateful to Jianzhe Liu from Huangshan Bright Semiconductor Co. Ltd. for his generous support, which has been instrumental in advancing this work.
This study was supported by the Anhui Province Scientific Research Planning Project (No.2023AH050851), National Natural Science Foundation of China (No.81904095, No.82101483).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN37357.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.