






 , Duk L. Na 2,3,4,5, Hee Jin Kim 1,2,3,4,6, Hyemin Jang 7, Jason K. Sa 8, Bae Sung Ko 1,2, Jong Wook Chang 1,2,9,*
, Duk L. Na 2,3,4,5, Hee Jin Kim 1,2,3,4,6, Hyemin Jang 7, Jason K. Sa 8, Bae Sung Ko 1,2, Jong Wook Chang 1,2,9,*
1 Department of Health Sciences and Technology, Samsung Advanced Institute for Health Sciences and Technology (SAIHST), Sungkyunkwan University, 06355 Seoul, Republic of Korea
2 Cell and Gene Therapy Institute (CGTI), Research Institute for Future Medicine, Samsung Medical Center, 06351 Seoul, Republic of Korea
3 Alzheimer’s Disease Convergence Research Center, Samsung Medical Center, 06351 Seoul, Republic of Korea
4 Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, 06351 Seoul, Republic of Korea
5 Happymind Clinic, 06061 Seoul, Republic of Korea
6 Department of Digital Health, SAIHST, Sungkyunkwan University, 06355 Seoul, Republic of Korea
7 Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, 03080 Seoul, Republic of Korea
8 Department of Biomedical Informatics, Korea University College of Medicine, 02841 Seoul, Republic of Korea
9 Cell & Gene Therapy Research Institute, ENCell Co., Ltd., 06072 Seoul, Republic of Korea
Abstract
Several preclinical studies have reported elevated levels of tau following the induction of chronic cerebral hypoperfusion (CCH) in Alzheimer’s disease mouse models. The objective of this study was to first induce CCH in a mouse model of tauopathy over an extended period of up to 6 months and to subsequently investigate the effects of CCH on tau accumulation and alterations in the transcriptome.
Three-month-old P301S tauopathy mice were randomly allocated to either a Sham or CCH group. The common carotid arteries (CCAs) of the CCH group were bilaterally implanted using 0.75-mm inner diameter ameroid constrictors. Prior to surgery, Doppler ultrasound imaging was acquired, with follow-up imaging at 1, 3, and 6 months postoperatively. Brain tissue samples were obtained, and hemispheres were dissected and divided for separate analysis.
No significant differences in phosphorylated and total tau protein levels were found in either Sham or CCH left cortical hemispheres or hippocampal lysates. Immunohistochemical staining of phosphorylated tau in the right hemisphere revealed similar findings. Compared with the Sham group, transcriptomic deconvolution revealed a significant reduction of memory B cells in the CCH group (p = 0.029).
To clarify the effects of chronic hypoperfusion on tau pathology, more than one surgical method of hypoperfusion should be used in future studies.
Keywords
- tau
- chronic cerebral hypoperfusion
- tauopathy
- ameroid constrictor
- common carotid artery
In humans, the contribution of chronic perfusion to the accumulation of phosphorylated tau (pTau) protein remains uncertain [1, 2]. However, induction of progressive chronic cerebral hypoperfusion (CCH) has been reported to increase hippocampal pTau levels in animal models [3, 4]. Neurofibrillary tangles, mostly consisting of pTau, are a major hallmark of Alzheimer’s disease (AD) and related neurodegenerative diseases or tauopathies [5, 6]. The PS19 (P301S) transgenic mouse model is a representative tauopathy mouse model. This model is reported to express the human mutant tau protein at a level fivefold higher than that of endogenous tau protein [7]. A literature review revealed that limited research has been conducted on this particular tauopathy mouse model to assess the direct effects of chronic cerebral hypoperfusion on the progression of tau pathology.
The induction of hypoperfusion in mouse models is commonly achieved via stenosis of the common carotid artery (CCA) [8, 9] for both acute and chronic time periods. The surgical methods of hyperfusion range from simple ligation techniques using silk/nylon sutures [3, 10, 11], uni- or bilateral microcoils [12, 13, 14], to uni- or bilateral ameroid constrictors [15, 16, 17, 18, 19]. The severity of common carotid artery occlusion and the heterogeneity of the pathology vary greatly depending on the method of hypoperfusion and whether one or both arteries are occluded. In contrast to microcoils, where a severe decline in cerebral blood flow is observed following implantation [20, 21], hypoperfusion is gradually induced when ameroid constrictors are implanted due to their inner material, which is composed of hygroscopic casein [20]. This material expands progressively on contact with water [22, 23]. Generally, the endpoints for hypoperfusion models in rodents (mice and rats), classified as either gradual or chronic, do not exceed 28 days [24, 25, 26, 27]. It remains uncertain as to whether such a duration is sufficient for the induction of CCH for addressing the underlying issues of chronic cerebral hypoperfusion and the subsequent cascade of events.
In this study, 3-month-old P301S tauopathy mice were progressively subjected to CCH for up to 6 months by bilateral implantation of ameroid constrictors. The aim was to induce a gradual and chronic reduction in blood supply to the brain over 6 months. The objective was to determine whether hypoperfusion alone could affect the accumulation of the tau protein and alter transcriptome levels in a mouse model of tauopathy. It was hypothesized that the 6-month period of chronic hypoperfusion would result in a notable elevation in pTau levels and a rapid acceleration of disease progression, which would in turn give rise to major alterations in transcriptomic profiles.
The Institutional Animal Care and Use Committee of the Research Institute for Future Medicine (RIFM) at the Samsung Medical Center approved this animal study on June 29, 2021 (approval number: 20210629002). The Institute is committed to adhering to the guidelines set forth by the Institute for Laboratory Animal Resources (ILAR) and is a facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All experiments were conducted in accordance with the ARRIVE guidelines 2.0. Genotyping was conducted using genomic DNA extracted from the tail snips of littermates born by mating PS19 (P301S) transgenic and B6C3 wild type mice originally procured from the Jackson Laboratory (Bar Harbor, ME, USA). This transgenic mouse model expresses a mutant human microtubule-associated protein tau. The expression of the human tau protein is approximately fivefold higher than that of the endogenous mouse tau protein [7]. Experiments were performed using transgenic mice that were 3–4 months old (n = 32). Mice were surgically implanted with ameroid constrictors (MC-0.75-TI, inner diameter: 0.75 mm, Research Instruments SW, Escondido, CA, USA) at an early age to allow for the gradual occlusion of the arteries to be observed over an extended period of time. Four separate PS19 mice and four separate B6C3 mice were used as positive and negative controls, respectively, for the tau enzyme-linked immunosorbent assay (ELISA). Mice were fed ad libitum and maintained on a 12-hour light/12-hour dark cycle.
PS19 mice were placed in the supine position and surgical procedures were performed under 2% isoflurane (1015, Hana Pharmaceuticals, Seoul, Republic of Korea) as general anesthesia. CCH was induced according to the methodology described in a previously reported procedure [10]. After removal of the fur and povidone iodine sterilization, a midline incision was made over the thyroid bone. The carotid arteries were exposed by retracting the omohyoid and sternomastoid muscles and by meticulously dissecting surrounding fascia and sheath, including the vagus nerve. A 6-0 silk thread (Ethicon, Cincinnati, OH, USA) was used to wrap the ameroid constrictors (inner diameter: 0.75 mm; Research Instruments SW, Escondido, CA, USA) around each CCA. An opening slit on one side of the ameroid constrictor allowed it to be wrapped around the vessel. In the Sham group, the same procedure was performed without the use of the ameroid constrictors after exposure of both CCAs. After surgery, the midline incision was closed with a 5-0 silk suture (Ethicon, Cincinnati, OH, USA). Prior to inducing CCH for 6 months, a preliminary study was performed to determine the optimal duration of CCH induction in PS19 mice. PS19 mice were sacrificed (Sham: n = 3, CCH: n = 5) at either 1 or 3 months (Fig. 1). PS19 mice were subjected to CCH for up to 6 months. Following this period, the mice were deeply anesthetized with 1.5–2.0 isoflurane (1015, Hana Pharmaceuticals, Seoul, Republic of Korea) and sacrificed via transcardial perfusion (Sham: n = 6, CCH: n = 10) at 9–10 months of age (Fig. 1A).
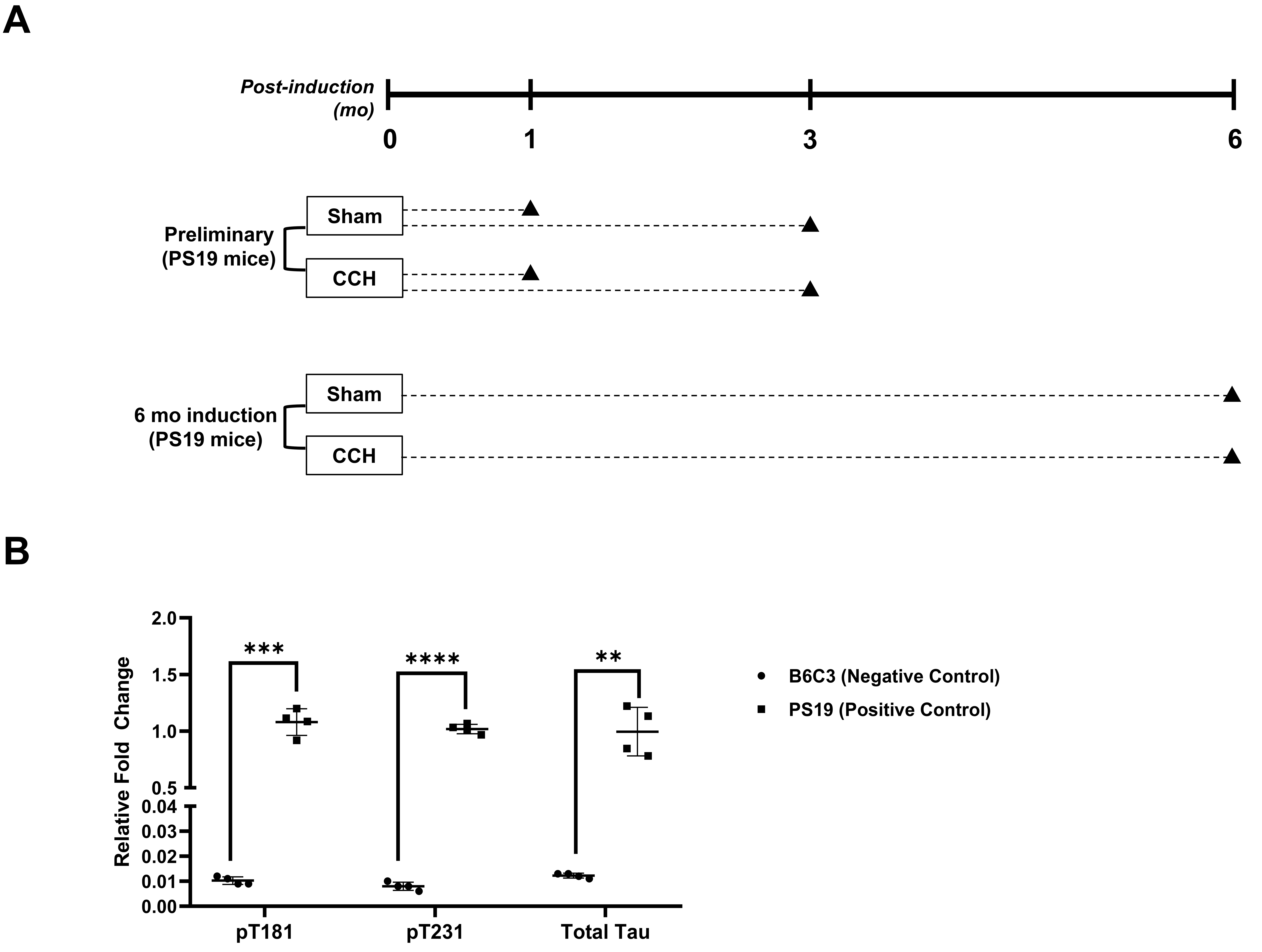 Fig. 1.
Fig. 1.
Schematic diagram illustrating overall study design. (A) In the
preliminary experiments, PS19 mice were allocated to one of two experimental
groups: the Sham group or the CCH group. Mice were subsequently sacrificed either
1 or 3 months following surgery. Subsequently, PS19 mice were subject to a
6-month period of chronic cerebral hypoperfusion and subsequently sacrificed. (B)
An initial experiment performed using positive (PS19) and negative (B6C3)
controls (each group: n = 4) validated the experimental procedures for
pT231, pT181, and total tau ELISA. Statistical significance was defined at
***p
Peak systolic velocity (PSV; mm/s) was measured for both the Sham and CCH groups before surgery (pre) and at 1, 3, and 6 months post-surgery. PSV was measured for both the left and right CCAs. Mice from the Sham and CCH groups were anesthetized with 1.5–2.0% isoflurane (1015, Hana Pharmaceuticals, Seoul, Republic of Korea) during the acquisition of the ultrasound image. During image acquisition mice were restrained in a supine position on a pad equipped with a heater, temperature sensor, and electrocardiogram electrodes (serial number: 2100-0286, Visual Sonics Inc., Toronto, ON, Canada). Ultrasound images were acquired using a Vevo® 2100 Imaging System and an MS-550D (22–55 MHz) transducer probe (serial number: 2100-0286, Visual Sonics Inc., Toronto, ON, Canada). Image acquisition was by pulsed wave Doppler and Color Doppler modes. PSV was calculated using Vevo® 2100 1.6.0 software (serial number: 2100-0286, Visual Sonics Inc., Toronto, ON, Canada).
To assess changes in motor coordination, rotarod tests were conducted [28] prior to animal sacrifice. Over the course of 3 consecutive days, three trials were conducted each day in which mice were placed on a revolving cylindrical rod. Prior to each trial, the mice underwent a 1-minute training session on the rotating rod at a speed of 4 revolutions per minute (RPM). In the actual trial, the speed of the revolving rod was increased from 4 to 40 rpm, and the trial lasted for a total of 4 minutes. The latency to fall was recorded during the aforementioned time period. Additionally, the mice were permitted a 10-minute recovery interval between each trial.
To validate the ELISA procedures, ELISA (pT181, KHO0631; pT231, KHB8051, Invitrogen, Carlsbad, CA, USA) was first performed to quantify pTau (pT181, pT231) and total tau protein levels using homogenized brain samples from PS19 (positive control; n = 4) and B6C3 (negative control, n = 4) mice (Fig. 1B).
After a period of 1, 3, or 6 months of inducing CCH, PS19 mice were sacrificed and samples of brain tissue were harvested. On the day of sacrifice mice were euthanized by cardiac perfusion (only PBS) under deep anesthesia (2% isoflurane, 1015, Hana Pharmaceuticals, Seoul, Republic of Korea). The hippocampus was excised from the cortex of the left hemisphere of the Sham and CCH groups and the resulting tissue samples were stored separately in a freezer (TFF900S-UULTF, Thermo Fisher Scientific, Waltham, MA, USA) at –80 °C. For the samples harvested after 1 and 3 months, pTau (pT181, pT231) and total tau protein levels were quantified via ELISA. For the samples harvested at post 6 months, half of the Sham (n = 3) and CCH (n = 5) samples (both hippocampus and cortex) were used for quantification of pTau (pT181, pT231) and total tau protein levels via ELISA. Phospho-tau 217 (pT217, 59672, Cell Signaling Technology; Danvers, MA, USA) protein levels were also quantified via ELISA. The manufacturer’s instructions were followed in the use of each ELISA kit, with each sample tested in duplicate wells. For each of the tau ELISAs, each sample was measured three times and results were averaged.
Whole-transcriptome sequencing was conducted on the remaining samples (Sham: n = 3, CCH: n = 5). The total RNA concentration was extracted and quantified using the Quant-IT RiboGreen assay. To evaluate the quality of total RNA, samples were analyzed using a TapeStation RNA screen tape (5067-5576, Agilent Technologies, Santa Clara, CA, USA). Only high-quality RNA (RNA integrity number greater than 7) was used for library construction. An independent preparation of the library was conducted for each sample using the Illumina TrueSeq Stranded mRNA Sample Prep Kit (20020595, Illumina, San Diego, CA, USA). The poly-A-containing mRNA molecules were purified using poly-T-attached magnetic beads (61002, Thermo Fisher Scientific, Waltham, MA, USA), followed by fragmentation and complementary DNA (cDNA) synthesis using SuperScript II reverse transcriptase (18064014, Thermo Fisher Scientific, Waltham, MA, USA) and random primers. Subsequently, the products underwent further purification and enrichment through PCR and the libraries were quantified using the KAPA library quantification kit (07960140001, Roche, Pleasanton, CA, USA) for Illumina sequencing platforms. Alignment of the whole-transcriptome sequencing reads to the reference human genome (GRCh37, hg19) was performed using the STAR 2.7.11b alignment tool (https://github.com/alexdobin/STAR). Gene expression levels were quantified and determined based on fragments per kilobase of transcript per million mapped reads. These values were subsequently log2-transformed and used in differentially expressed gene (DEG) analysis using the DEseq2 1.46.0 software (http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html). Gene set enrichment analysis was performed to acquire pathway-level activities. The mRNA expression levels of genes associated with AD were evaluated and the gene expression of immune cell types was also identified via immune deconvolution analysis using CIBERSORTx software (http://cibersortx.stanford.edu).
To validate mRNA sequencing results, the gene expressions of Glutathione s-transferase pi 1 (Gstp1) and Fibroblast growth factor receptor 1 (Fgfr1) were validated by carrying out real-time quantitative polymerase chain reaction (RT-qPCR). With the remaining RNA samples following mRNA sequencing, cDNA was synthesized using the AccuPower® RocketScript™ Cycle RT PreMix kit (K-2241, Bioneer, Daejeon, Republic of Korea). RT-qPCR was subsequently performed by mixing the respective cDNA samples (1 µg per sample) with the SYBR green master mix (4367659, Applied Biosystems, Foster City, CA, USA). All samples were subjected to a 40-cycle amplification at 95 °C for initial holding (10 min), 95 °C for denaturation (15 sec), and 57 °C for annealing (1 min).
All cDNA samples were assayed in triplicate. Results were normalized against Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) levels and quantified using the 2-ΔΔCt method proposed by Livak and Schmittgen [29]. The primer sequences were: Fgfr1, forward, GCCTCACATTCAGTGGCTGAAG; reverse, AGCACCTCCATTTCCTTGTCGG (Origene, Rockville, MD, USA), Gstp1, forward, TGGAAGGAGGAGGTGGTTACCA; reverse, GGTAAAGGGTGAGGTCTCCATC (Origene, Rockville, MD, USA), and Gapdh, forward, CAGTATGACTCCACTCACGG; reverse, GTGAAGACACCAGTAGACTCC.
The right hemispheres of the Sham and CCH groups were fixed in 4% paraformaldehyde (Biosesang, Seongnam, Republic of Korea) then paraffin blocked. Serial sections measuring 4 µm were made for immunohistochemical (IHC) staining. Previously reported methods were used to deparaffinize the slides and perform heat antigen retrieval [30]. The phospho-tau (pT231) primary antibody (1:400, MN1040, ThermoFisher Scientific, Waltham, MA, USA) was used for IHC and visualized using 3,3′-diaminobenzidine (DAB, K3468, Dako, Santa Clara, CA, USA). CD68 macrophage expression (1:200, ab125212, Abcam, Cambridge, UK) was observed by immunofluorescence using Alexa Fluor 594 (A-11012, Thermo Fisher Scientific, Waltham, MA, USA). Upon completion of IHC, a Scanscope AT scanner (Leica Biosystems, Wetzlar, Germany) was employed to scan the slides, then pTau expression in the cortex, hippocampus, and thalamus of the Sham and CCH groups was quantified using the Vectra® Automated Imaging System (PerkinElmer Applied Biosystems, Waltham, MA, USA). Two adjacent stained and imaged paraffin slides were used. Phenochart (Vectra version 2.6.0) software (PerkinElmer Applied Biosystems, Waltham, MA, USA) was used to delineate rectangular regions of interest (ROIs) for each brain region. Subsequently, the DAB optical density threshold was calibrated to quantify pTau expression for the aforementioned ROIs.
To evaluate the histologic changes caused by CCH, a hematoxylin and eosin (H&E) stain was first performed [10]. Luxol fast blue (IW-3005, IHC World, Woodstock, MD, USA) was used for myelin staining and to specifically determine white matter changes in the corpus callosum following CCH. The Scanscope AT scanner was used for the scanning of the H&E and Luxol fast blue-stained slides. The corpus callosum area for both the Sham and CCH groups was assessed using the Otsu threshold algorithm [31] in the ImageJ analysis software (https://imagej.nih.gov/ij/, National Institutes of Health (NIH), Bethesda, MD, USA).
GraphPad Prism 8.0 software (GraphPad Software, Inc., San Diego, CA, USA) was
used for the statistical analysis. Values are given as mean
A preliminary study was conducted to ascertain the optimal time point for
induction of CCH in PS19 mice. Sham and CCH mice were sacrificed at either 1 or 3
months (Fig. 1A). Prior to assessing the 1- and 3-month samples from the Sham and
CCH groups, the positive and negative control samples were examined using ELISA.
Statistical analysis revealed significant differences in pT181 (p
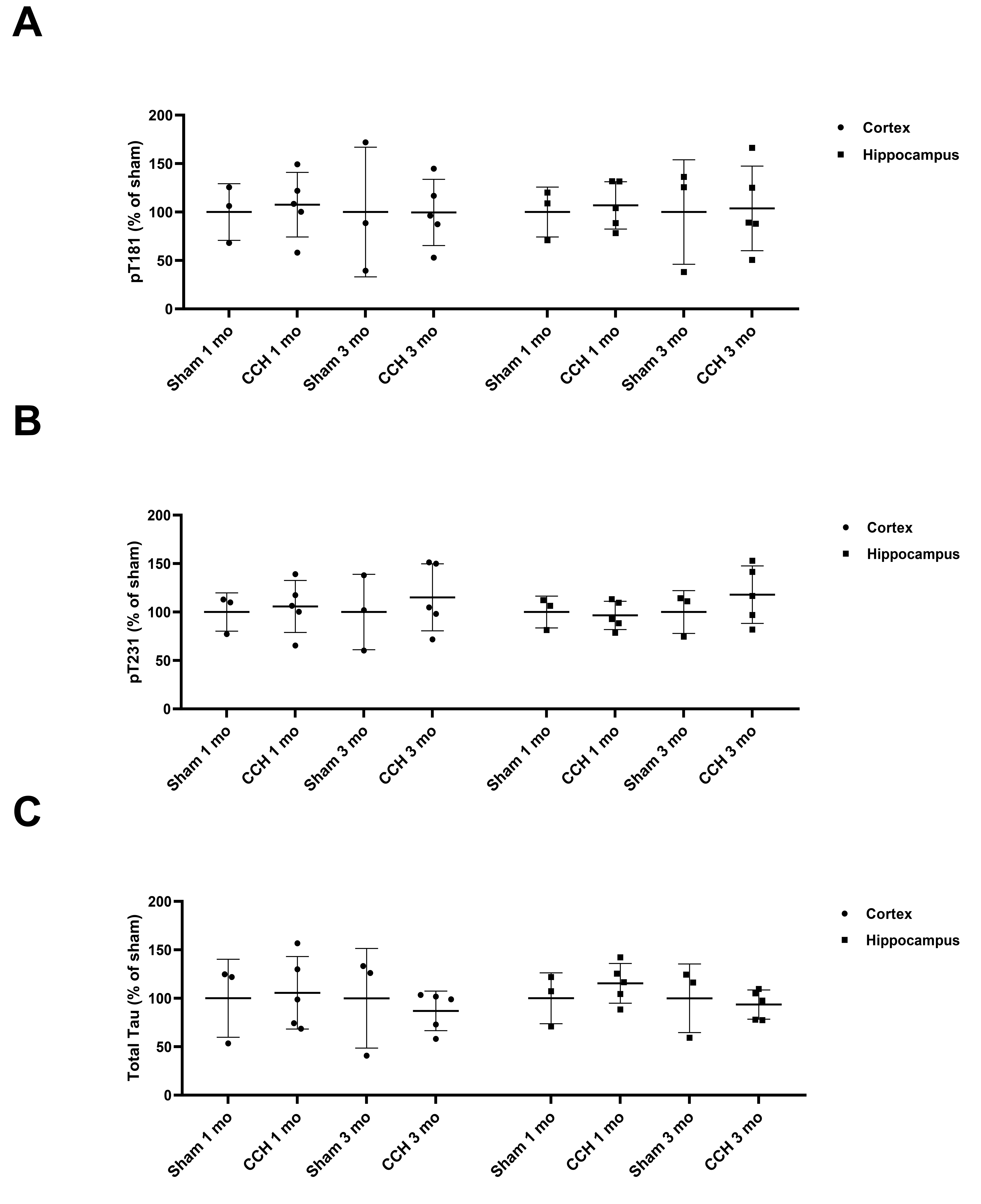 Fig. 2.
Fig. 2.
Pathological alterations of the tau protein in PS19 mice induced
with chronic cerebral hypoperfusion for up to 1 or 3 months. PS19 mice were
sacrificed 6 months after sham or CCH surgery. Phosphorylated tau (A) pT181, (B)
pT231, and (C) total tau levels were determined via ELISA using brain homogenates
from the right cortex and hippocampus of the Sham (n = 3) and CCH
(n = 5) groups at 1 and 3 months (Mann-Whitney U test). Values are shown
as a percentage of Sham, mean
A comprehensive analysis of the mice in the CCH group 3 months after CCH induction revealed that two of the five mice examined exhibited histopathological alterations such as loss of hippocampal neurons and early signs of infarction (foamy macrophages), with alterations in hippocampal structure after H&E staining noted in one mouse. The presence of lipid-laden macrophages or foamy macrophages was identified in the corpus callosum and cortex of the CCH-induced PS19 mouse. To ascertain the presence of macrophages, IHC was employed. Based on IHC staining, CD68-positive cells were observed to be strongly visible at the sites where foamy macrophages were identified by H&E staining (Supplementary Fig. 1).
The outcomes observed following induction at 1 and 3 months indicate a gradual accumulation of tau. To ascertain the chronic effects of hypoperfusion on tau phosphorylation, it was necessary to extend the duration of CCH induction for a period exceeding 3 months. Consequently, the final endpoint for the remaining set of experiments was set to occur 6 months post-induction.
For the Sham group, eight mice were observed over the course of the study (6 months). Unexpectedly, two mice died before the final endpoint of the study was reached. A total of five (out of 15) CCH mice died within 3 months of the surgical procedure. In comparison with the Sham group, a consistent decline in peak systolic velocity (mm/s) was observed in the CCH group across all time points (1, 3, and 6 months post-surgery) in both the left (1 month: p = 0.0001, 3 months: p = 0.000046, 6 months: p = 0.00016) and right (1 month: p = 0.000099, 3 months: p = 0.000046, 6 months: p = 0.00016) CCAs (Fig. 3A,B).
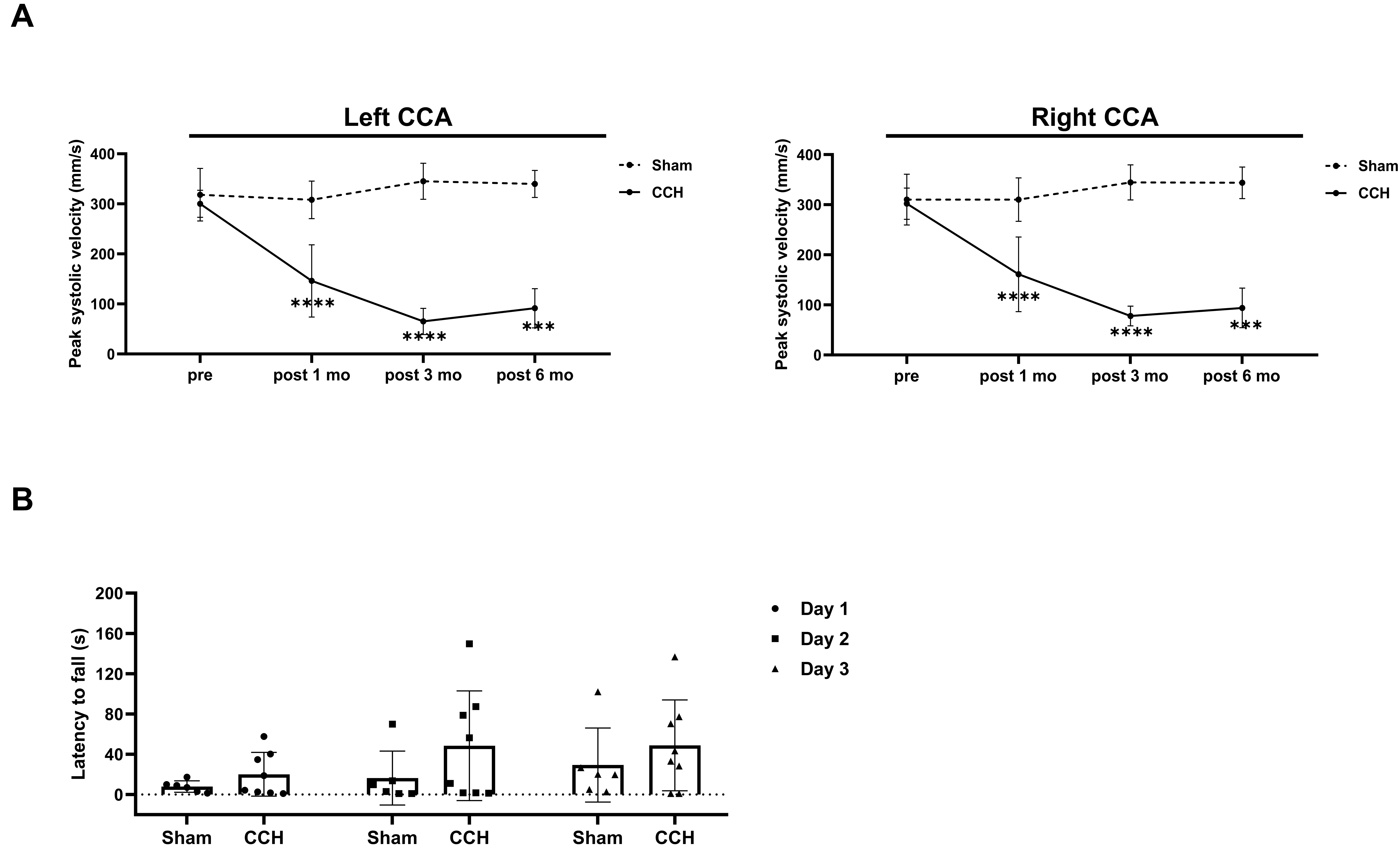 Fig. 3.
Fig. 3.
Induction of chronic cerebral hypoperfusion up to 6 months. (A)
At various time points (pre-, 1, 3, and 6 months) following surgery, the peak
systolic velocity (PSV; mm/s) of both the left and right CCAs of both the Sham
and CCH groups was examined. A statistically significant and consistent decline
in PSV was observed over time for both bilateral PSVs. Statistical significance
is defined as ****p
The rotarod test was repeated to examine changes in motor function. The latency to falls from the revolving rod was measured for each experimental animal. For all three test days, the CCH group demonstrated a tendency to remain on the rotarod for longer durations than the Sham group, although the difference was not statistically significant (Fig. 3B).
Using the separate cortical and hippocampal lysates of both the Sham and CCH groups, the pTau (pT181, pT231, pT217) and total tau protein levels were quantitated using ELISA. A non-significant increasing trend in pT181 (Fig. 4A) and pT231 (Fig. 4B) levels was observed for both the cortical (pT181, pT231) and hippocampal (pT181, pT231) lysates of the CCH group in comparison with those of the Sham group. No significant difference in total tau levels was observed for the cortical or hippocampal lysates of the CCH group (Fig. 4C), although there was a non-significant trend toward elevated levels of pT217 in the cortex and hippocampus of the CCH group compared with the Sham group (Fig. 4D).
 Fig. 4.
Fig. 4.
Quantitative analysis of phosphorylated tau and total tau
following induction of chronic cerebral hypoperfusion up to 6 months. PS19 mice
were sacrificed 6 months after Sham or CCH surgery. ELISA was used to measure
phosphorylated and total tau: (A) pT181, (B) pT231, (C) total tau, and (D) pT217,
in the right hemisphere (cortex and hippocampus separately) brain homogenate from
the Sham (n = 3) and CCH (n = 5) groups. All four tau ELISAs
showed no significant differences between the groups (Mann-Whitney U test).
Values are shown as a percentage of Sham, mean
Following ELISA, accumulation of pTau (pT231) was histologically evaluated across various regions in the PS19 mouse parenchyma (cortex, hippocampus, and thalamus). Significant elevation of pTau accumulation was not seen in the CCH group across all three brain regions in comparison with the Sham group, although there was a discernible tendency towards moderately elevated cortical pT231 levels, with a slight non-significant attenuation in the thalamus of the CCH group relative to the Sham group (Fig. 5). The progression of the disease was evaluated based on changes in either the white matter tract or the corpus callosum, which were assessed using Luxol fast blue. However, no significant differences were observed between the Sham and CCH groups in the corpus callosum (Supplementary Fig. 2).
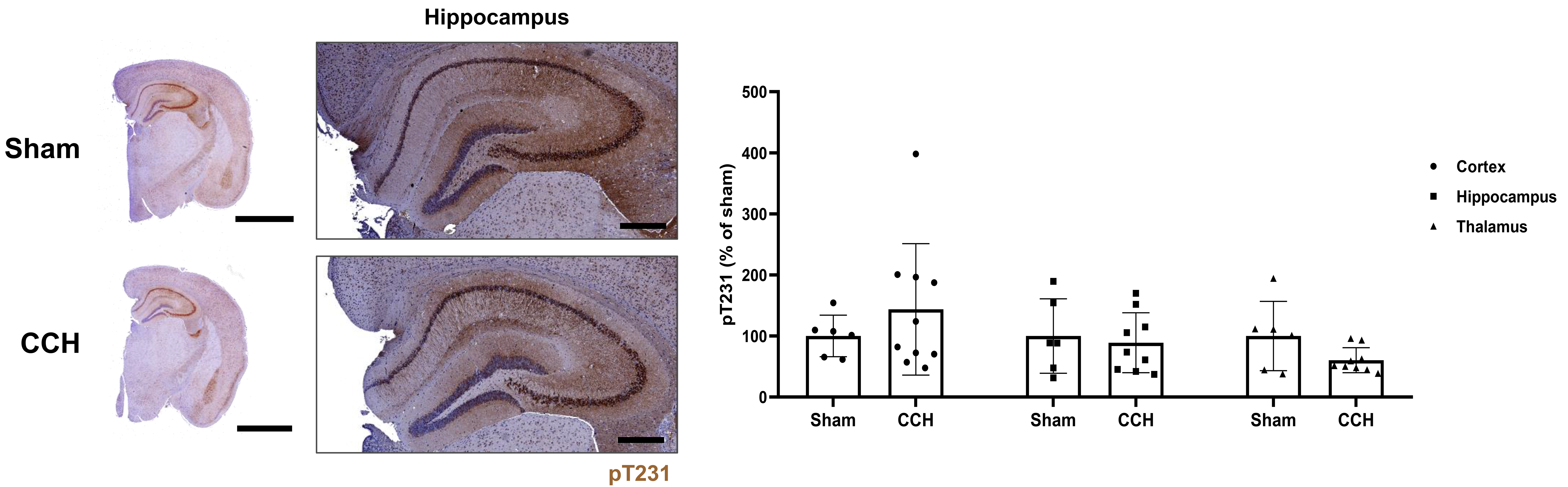 Fig. 5.
Fig. 5.
Detection of alterations in phosphorylated tau in the cortex,
hippocampus, and thalamus. Representative 3,3-diaminobenzidine
immunohistochemical stained images showing the expression of phosphorylated tau
(pT231, dark brown) for both the Sham and CCH groups. Nuclei were counterstained
with hematoxylin. pT231 expression was quantified for both groups in the cortex,
hippocampus, and thalamus (% of Sham, Mann-Whitney U test). Scale bar: right
hemisphere: 2 mm, hippocampus: 400 µm. Values are shown as a
percentage of Sham, mean
To identify genes that are uniquely enriched in the CCH and Sham groups based on anatomical locations, genome-wide DEG analysis was initially performed. Notably, functional analysis using the Gene Ontology database revealed that the Sham group exhibited an upregulation of genes related to the immune system and synaptic activity, whereas the CCH group displayed an upregulation of genes related to cell cycle and DNA repair mechanisms (Fig. 6A). Moreover, the Fgfr1 pathway was collectively upregulated in the CCH group, while the immune system was upregulated in the Sham group (Fig. 6B). Both the cortex and hippocampus of the CCH group exhibited higher Gstp1 mRNA expression levels compared with the matched Sham group for both brain regions (Fig. 6C). The immune deconvolution analysis of the hippocampal samples indicated a statistically significant reduction (p = 0.029; solid red arrow) in the abundance of B memory cells in the CCH group in comparison with the Sham group (Fig. 6D). To validate the mRNA sequencing results, RT-qPCR was conducted using the respectively remaining RNA samples. A noticeable, but non-significant, increase in Fgfr1 levels was apparent in both the cortex and hippocampus of the CCH group when compared with the Sham group (Fig. 6E). No statistically significant difference was observed in Gstp1 expression between the two groups (Fig. 6F).
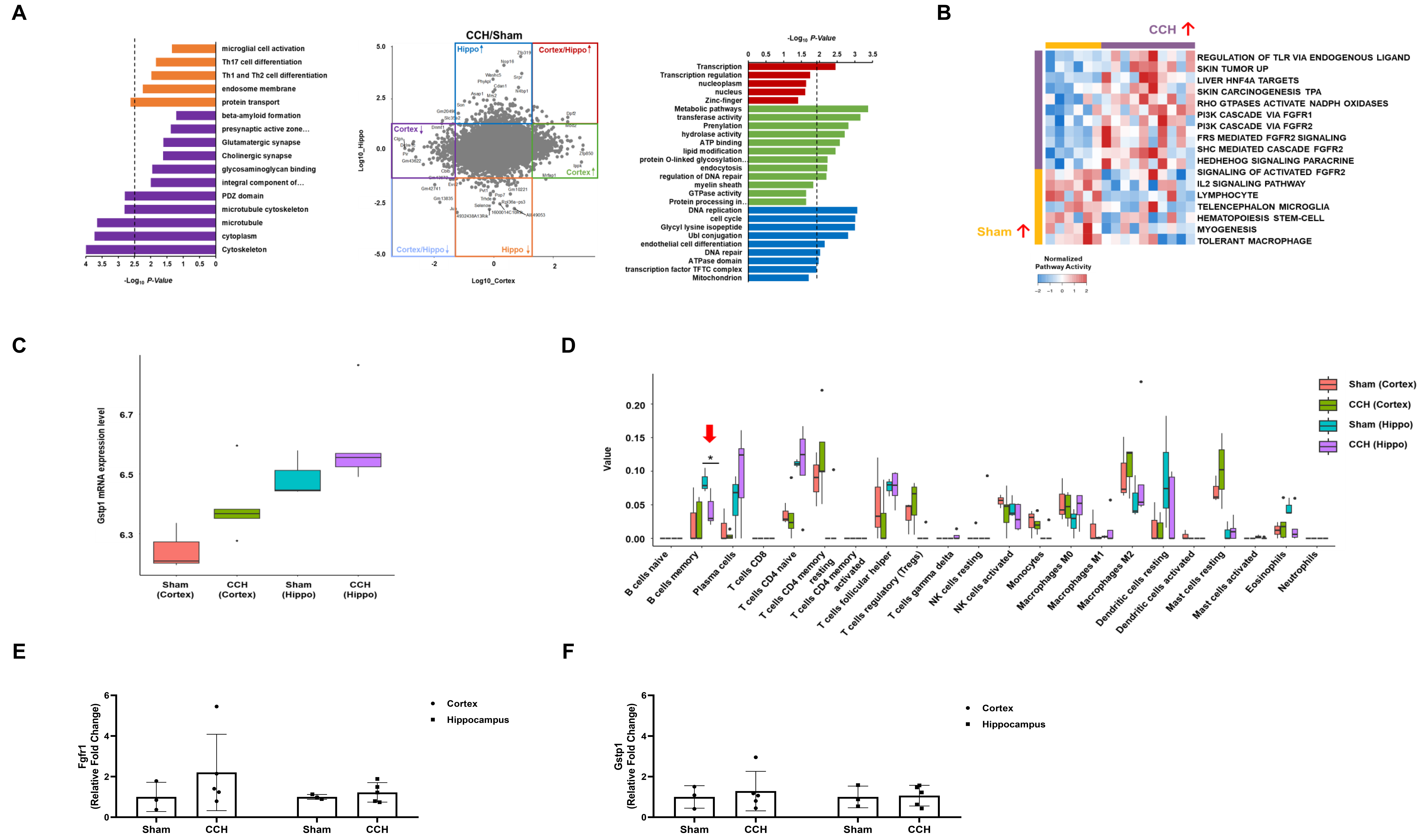 Fig. 6.
Fig. 6.
Whole-transcriptome sequencing analysis of hippocampal and
cortical lysates and validation of results using RT-qPCR. At the 6-month
sacrifice point, the Sham and CCH group brain tissues were harvested and the
cortex and hippocampus (Hippo) from the left hemisphere were homogenized for
transcriptome sequencing analysis. (A) DEG and Gene Ontology analyses. The colors
represent genes or pathways significantly enriched in the corresponding groups
based on the experimental models. Blue: CCH-Hippo; Red: CCH-Cortex & Hippo;
Green: CCH-Cortex; Orange: Sham-Hippo; Light Blue: Sham-Cortex & Hippo; Purple:
Sham-Cortex. (B) DEG and single sample gene set enrichment analyses. (C)
Gstp1 mRNA expression. The box plot encompasses the first and third
quartiles, with the median indicated by a line extending from the box. The
whiskers extend to 1.5 times the interquartile range. (D) Immune deconvolution
analyses using CIBERSORTx. (E) qPCR validation of Fgfr1 mRNA expression.
(F) qPCR validation of Gstp1 mRNA expression. Values are expressed as
mean
In this study, chronic cerebral hypoperfusion was initially induced in a tauopathy mouse model by bilateral implantation of ameroid constrictors. Subsequently, the impact of hypoperfusion on pTau levels and transcriptomic profiles was assessed. Successful induction of hypoperfusion in mice was demonstrated by the reduced peak systolic velocity observed over time using Doppler ultrasound. To the best of the authors knowledge, this is the first time that cerebral hypoperfusion using ameroid constrictors in a PS19 mouse model has been induced chronically for up to 6 months. A review of the existing literature revealed that bilateral implantation of ameroid constrictors is a common procedure in APP23 mice [32], a common transgenic mouse model for AD, but not for tauopathy. Most groups induced CCH in APP23 mice by bilateral implantation of ameroid constrictors with an inner diameter of 0.75 mm for up to 8 months [33, 34, 35]. While the majority of groups did not examine the impact of CCH on tau phosphorylation in APP23, several groups have noted that the number of taupositive cells exhibiting phosphorylation increased significantly in the cortex and thalamus of these mice when CCH was induced up to 8 months [36, 37].
Although a time-dependent decrease in peak systolic velocity was observed, neither motor function nor subtle pathological changes were noted in the CCH-induced PS19 mice. These findings suggest that, although chronic, the overall induction of CCH was relatively mild. This may account for the relatively minor alterations in tau pathology observed using ELISA and IHC, as well as the upregulation of the Fgfr1 signaling pathway as revealed by mRNA sequencing analysis. As statistically significant differences were not noted between the Sham and CCH groups for pT231, pT181, and total tau, an additional tau ELISA was conducted using pT217, which has recently been identified as a more specific and sensitive biomarker for tau pathology and AD progression compared with pT181 [38, 39]. However, although increasing trends were observed, statistical significance was not reached in the CCH group. Here, PS19 mice that had been induced with CCH and subjected to histological staining did not display any significant indications of ischemic injury or neuronal death for up to 6 months. Nevertheless, the sustained reduction of blood flow to the brain appears to have initiated the Fgfr1 pathway, which is renowned for its pivotal role in neuronal cell proliferation and survival [40], as a protective response to the hypoperfusion. This might explain why an upregulation of genes related to cell cycle and DNA repair mechanisms was evident in the CCH group, with the DEG and kyoto encyclopedia of genes and genomes (KEGG) analyses showing significant differences. The potential for Fgfr1 expression to mediate neuroprotection against AD neuropathology has also been a subject of investigation [41, 42]. It is possible that cell survival pathways were initiated by adaptive responses to cope with reduced oxygen and nutrient supply brought about by cases of mild or moderate chronic cerebral hypoperfusion. The generation of mild hypoxia via mild CCH may suppress the immune system, which might explain the reduced expression of memory B cells observed in the CCH-induced PS19 mice.
This study had several limitations. First, the impact of CCH on the regulation of the immune environment was not fully examined. For instance, an assessment of changes in immune cytokine expression was not carried out. However, we used transcriptomic analysis to evaluate immune-related signaling pathways and the distribution of immune cells, which may parallel cytokine expression patterns. Second, while the duration of CCH induction is a critical determinant of CCH severity, an equally, if not more, important factor may be the method of surgical induction. Despite being implanted for up to 6 months in this study, ameroid constrictors with an inner diameter of 0.75 mm may not have been sufficient to induce severe CCH. However, unilateral, permanent ligation of the right common artery for up to 6 weeks in an APPswe/PS1 transgenic mouse model also induced mild chronic cerebral hypoperfusion [43]. Therefore, further research that utilizes multiple methods of hypoperfusion to induce hypoperfusion across multiple time points in more than one transgenic mouse model of tauopathy is required. Third, there was inconsistency in sample sizes between the Sham and CCH groups. Based on previous studies and the extended sacrifice time point, it was anticipated that a higher mortality rate would be observed. However, the sample sizes were maintained at levels sufficient for statistical analysis and meaningful conclusions. Lastly, the PS19 mouse model may not have been a suitable model to observe chronic exacerbation of tau pathology, given the potential for aggressive accumulation of tau pathology during the early stages of development in this model.
The present study aimed to demonstrate the effects of chronic cerebral hypoperfusion, induced for a period of up to 6 months via the bilateral implantation of ameroid constrictors, on the regulation of pTau and transcriptome levels. Chronic cerebral hypoperfusion in PS19 mice, even when extended to 6 months, induced modest tau pathology and significant inflammatory responses without marked changes in motor function or white matter integrity. It can be postulated that prolonged exposure to mild hypoxia due to CCH may have activated neuronal survival pathways and dysregulated immune responses, as evidenced by a significant decrease in memory B cells in CCH-induced PS19 mice. These findings collectively indicate that hypoperfusion may not be the sole or even primary driving force underlying tau deposition. A complex interplay between hypoperfusion, tau pathology, and immune response appears to exist, suggesting a need for further investigation to fully elucidate these mechanisms.
AD, Alzheimer’s disease; CCH, Chronic cerebral hypoperfusion; CCA, Common carotid artery; pTau, Phosphorylated tau; Fgfr1, Fibroblast growth factor receptor 1; Gstp1, Glutathione S-transferase p1; Gapdh, Glyceraldehyde 3-phosphate dehydrogenase; H&E, Hematoxylin and eosin; IHC, immunohistochemistry.
The authors declared that they have not used Artificial Intelligence in this study. The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
NKL contributed to the study design, performed the experiments, analyzed and interpreted the data, and wrote the manuscript. NKL, DLN, and JWC contributed to data analysis and interpretation. NKL, DLN, and JWC contributed to the study conceptualization and design. HJK and HJ contributed to the study design and revision of the manuscript. BSK helped perform the experiments. JKS helped analyze the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The project, titled Inducing chronic cerebral hypoperfusion in a Tau Alzheimer’s disease mouse model, was conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) of the Research Institute for Future Medicine at Samsung Medical Center (approval number: 20210629002). This committee reviewed and approved the study protocol on June 29, 2021.
Not applicable.
This research was funded by the Ministry of Education, Republic of Korea (grant number: RS-2021-NR065630, the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI)), funded by the Ministry of Health & Welfare, Korea (grant number: HR14C008), Future Medicine 2030 Project of the Samsung Medical Center (grant number: SMO1250091).
Jong Wook Chang is additionally affiliated with ENCell Co., Ltd., which was not involved in the study design, data collection, analysis, interpretation, the writing of this article, or the decision to submit it for publication. The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN26108.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.