Alzheimer’s disease (AD) is characterized by the buildup of amyloid-
(A) outside neuronal cells in the form of senile plaques and
intracellular neurofibrillary tau tangles. In addition, it is accompanied by
synaptic loss, neuroinflammation, cognitive decline, and neuronal death. In a
survey done in the United States, the long-term care costs for people with
dementia are anticipated to reach 360 billion dollars in 2024 and almost 1
trillion dollars by 2050 [1]. However, no cure or treatment is available either
to stop or reverse the disease progression. To develop effective therapy, it is
necessary to understand the pathogenesis of AD.
Misfolded proteins and protein degradation systems have contributed
significantly to the understanding of AD. For instance, the ubiquitin variant
Ubiquitin-B+1 frameshift mutant (UBB+1) affects proteasome degradation and can be seen early in AD pathology. It
has been shown to stimulate amyloid and tau pathology in cellular models [2]. A
dysfunction in proteostasis not only leads to protein misfolding but also
activates the innate immune system. This can be attributed to mitochondrial
stress and the activation of the inflammation pathway triggered by the nucleic
acid-sensing factors. In the case of proteasomal dysfunction, one consequence
could be the leakage of mitochondrial DNA (mtDNA) from the damaged mitochondria
into the cytosol. This leaked mtDNA acts as a danger-associated molecular
pattern, thereby activating the cyclic GMP–AMP synthase (cGAS) and stimulator of
interferon genes (STING) DNA-sensing pathway, to cause neuroinflammation [3, 4, 5]
(Fig. 1).
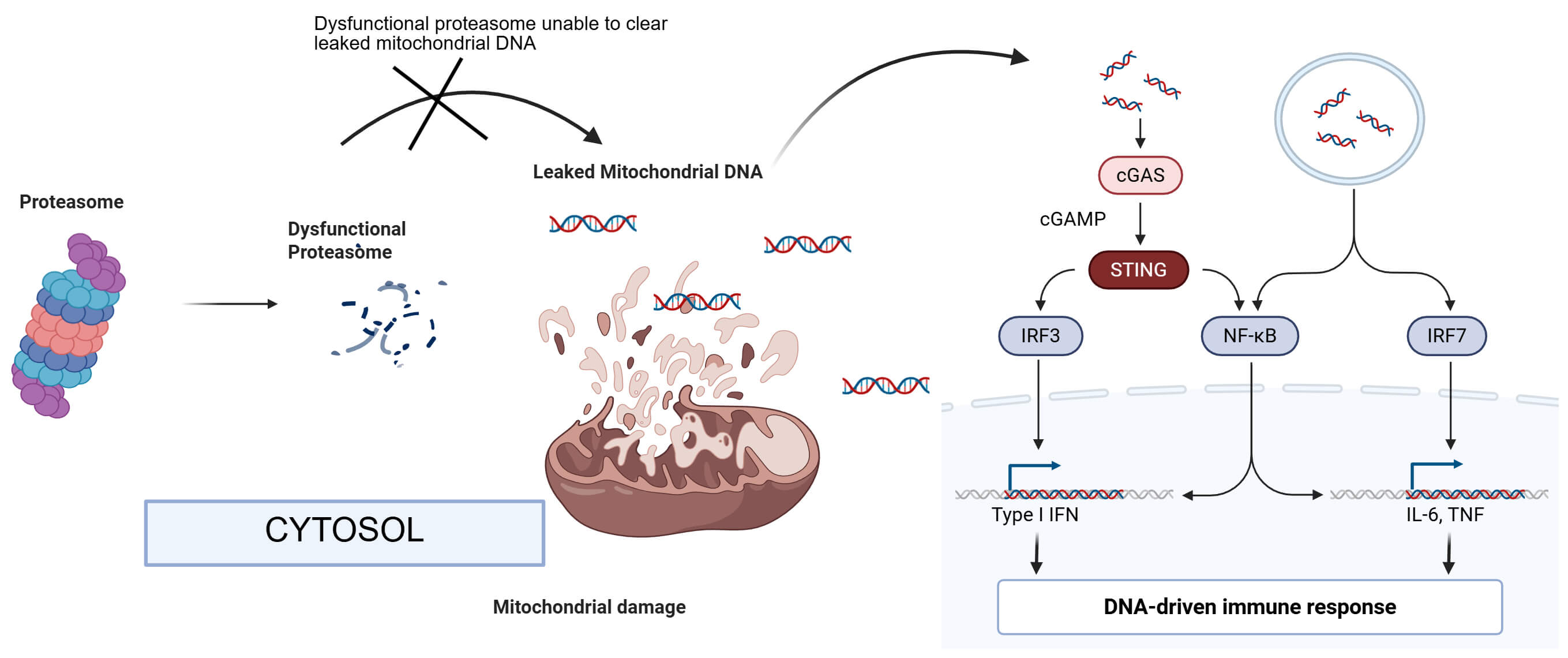 Fig. 1.
Fig. 1.
Activation of the cGAS-STING pathway. Dysfunctional
proteasome induces the alteration of mitochondrial structure and function,
leading to the release of mtDNA into the cytosol, where mtDNA activates
cGAS-STING and the downstream signaling pathways to cause neuroinflammation
responses. cGAS, cyclic GMP–AMP synthase; STING, stimulator of interferon genes;
cGMP, cyclic guanosine monophosphate; IRF3, interferon regulatory factor 3; IRF7, interferon regulatory factor 7. The figure was created in BioRender https://BioRender.com.
1. Proteasome Dysfunction Links to Neurodegeneration and mtDNA Release
The ubiquitin-proteasome system, or ubiquitin-proteasome system (UPS), is vital for clearing abnormal proteins
that could trigger inflammation if accumulated. Neurons are particularly
vulnerable to UPS impairment due to their high reliance on precise protein
homeostasis for function and survival. Findings from the studies of the 5FAD and
tau-P301S mice revealed that the synaptic proteasome function is impaired even in
the early stages, a phase before overt plaque formation, correlating with early
memory deficits [6]. Blocking proteasome function in healthy neurons causes
AD-like effects, such as oxidative stress, synaptic loss, and cognitive decline.
Conversely, boosting UPS activity can reverse these effects [6]. Deletion of a
26S proteasome subunit causes neurodegeneration and Lewy-like inclusions,
accompanied by abnormal mitochondria, linking proteasome failure to mitochondrial
dysfunction and neuronal damage that extends beyond protein aggregation [5].
Increased production of reactive oxygen species (ROS) can damage mitochondrial
lipids and proteins, compromise membrane integrity, and ultimately cause membrane
rupture. This occurs due to abnormal protein aggregation caused by proteasomal
failure, which disrupts redox balance. Although UPS is involved in mitochondrial
quality control, its impairment weakens the removal of damaged mitochondrial
proteins, leading to oxidative stress that eventually causes mitochondrial
membrane collapse. This collapse can then leak mitochondrial DNA into the
cytosol. Also, it has been shown in a neuron-specific proteasome knockout mouse
that the cGAS-STING pathway was activated, as evidenced by increased protein
levels of cGAS and STING, and pro-inflammatory factors, such as STAT1,
NF-B, IL-1, TNF-, and IL-6, as well as signs of
neurodegeneration, including decreased brain weight and necroptosis markers
(phosphorylated mixed lineage kinase domain-like protein (MLKL) and receptor-interacting protein kinases (RIP) kinases). These results link proteasomal dysfunction
to immune responses and cell death in the brain [4].
2. mtDNA Release and cGAS-STING Pathway Activation in AD
The cGAS-STING pathway drives chronic inflammation in aging and
neurodegeneration [7]. cGAS detects double-stranded DNA in the cytosol and
produces cyclic GMP, activating STING on the endoplasmic reticulum. STING then
promotes the transcription of type I interferons and inflammatory genes, mainly
protecting against infections. In AD, cytosolic mtDNA acts as a trigger [4, 8, 9]. Proteasome-deficient mouse brains exhibit increased pathway activity,
linking proteostasis issues to immune responses [4, 5, 8]. In the 5FAD mouse
model, cytosolic DNA triggers microglial cGAS production, whereas the deletion of
cGAS reduces amyloid plaques and improves cognition [8]. Blocking STING with H151
reduces amyloid and inflammation in these mice [10]. While the main effector
cells are microglia, neuronal activation can also occur under stressful
conditions, particularly with aging or tau pathology [11]. Activation of
cGAS-STING in microglia primarily enhances neuroinflammation by increasing the
release of inflammatory mediators, including IL-1, TNF-, and
IL-6, and alters microglial homeostasis, including phagocytosis. However, in
neurons, cGAS-STING activation affects the cell intrinsically, leading to
synaptic dysfunction and cell senescence, which directly contributes to
neurodegeneration.
3. Therapeutic Implications
Recognizing that proteasomal dysfunction can trigger the cGAS-STING cascade in
AD provides a new avenue for treatment. One approach is enhancing proteostasis at
its root. In one study, activation of the proteasome through the cAMP/protein kinase A (PKA)
signaling pathway, as with rolipram, restored protein turnover and rescued
A-induced synaptic failure [6]. Although proteasome-activating compounds
are still being developed, other methods, such as activating proteasome subunits
or enhancing autophagy, could also prevent the cascade [12]. Additionally,
enhancing the removal of damaged mitochondria (mitophagy) is another strategy.
Removal of defective mitochondria limits mtDNA leakage. Treating aged mice with
mitophagy boosters, such as urolithin A, lowers cytosolic mtDNA, attenuates
cGAS-STING activation, and supports neuronal health [9]. Similarly, the use of
metabolic approaches to replenish intracellular NAD+ has been shown to inhibit
the inflammation triggered by the cGAS-STING pathway. Supplementing NAD+ in a
transgenic AD mouse model reduced the inflammation as well as the cellular
senescence triggered by the cGAS-STING pathway, thereby improving neuronal
resilience and function [13]. Again, inhibiting the cGAS-STING pathway itself is
another approach [14, 15]. The STING inhibitor H-151 is protective in AD models,
and the drugs that inhibit cGAS are also being explored in inflammatory diseases
[8, 10, 14]. However, these therapeutic strategies face several challenges and
limitations. The broad suppression of the STING pathway by STING inhibitors such
as H-151 can weaken antiviral defenses or cause off-target effects, raising
safety concerns with long-term use. Also, approaches used to enhance proteasome
activity or mitophagy are hard to confine to neurons, since the pathway is active
in many tissues, and systemic activation can disrupt normal proteostasis
elsewhere. These approaches also differ from traditional therapies targeting
amyloid or tau, as they focus on upstream processes such as improving
proteostasis, innate immune signaling, and mitochondrial function, rather than
directly clearing misfolded proteins and protein aggregates. Since impairments in
proteostasis can worsen A and tau pathology, and cGAS-STING inhibition
can lower amyloid levels in AD models, adapting combination therapy is a
promising treatment strategy.
In conclusion, the discovery that proteasomal dysfunction activates the
mtDNA-cGAS-STING pathway in the brain changes our understanding of AD, linking
protein aggregation and neuroinflammation. This reveals targets for therapy, such
as enhancing mitophagy. Exploring these areas is vital, as restoring proteostasis
and controlling innate immunity could break the cycle of protein buildup and
inflammation, offering hope for new treatment strategies against AD and dementia.
The role of the cGAS-STING pathway in neuroinflammation, compared with other
innate immune drivers, such as those mediated by toll-like receptor (TLR) or NOD-like receptor pyrin domain containing 3 (NLRP3), has not been
established, and such comparisons may represent an area of interest for potential
future research.
Author Contributions
AD and HW made substantial contributions to the work. AD drafted the manuscript and HW reviewed and edited it. Both AD and HW were involved in searching literature and revised the work to ensure the accuracy of the cited work. HW was also responsible for acquisition of funding. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This research was funded by National Institutes of Health (NIH) grant R01AG072510.
Conflict of Interest
The authors declare no conflict of interest. Hongmin Wang is serving as one of the Editorial Board members of this journal. We declare that Hongmin Wang had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Bettina Platt.



