





 , Lili Wang 2, Aoyu Hu 1, Shanshan Zhang 2,*
, Lili Wang 2, Aoyu Hu 1, Shanshan Zhang 2,*
1 The Second School of Clinical Medicine, Zhejiang Chinese Medical University, 310053 Hangzhou, Zhejiang, China
2 Department of Acupuncture, The Second Affiliated Hospital of Zhejiang Chinese Medical University, 310005 Hangzhou, Zhejiang, China
Abstract
The blood-brain barrier (BBB) consists of endothelial cells enmeshed by brain microvessels, surrounding basement membrane, pericytes and astrocyte pedicles. It serves as a natural barrier between the blood and brain tissue and both its structural and functional integrity play a crucial role in protecting the central nervous system (CNS) from harmful substances and maintaining the internal stability of the brain. As an important component of the BBB and a hub in the neurovascular unit that links neurons and the cerebral microvasculature, astrocytes play a key role in providing structural support and dynamic regulation of the BBB. In this review, we describe both the physiological structure and mechanistic functions of the BBB and astrocytes, and explores the role of astrocytes in the development, stabilization, destruction and repair of the BBB. Finally, we outlines the therapeutic potential of targeting these mechanisms for CNS disorders associated with BBB disruption.
Keywords
- astrocyte
- BBB
- nervous system
- glial cells
The blood brain barrier (BBB) is formed by the walls of the brain’s capillaries and neuroglia. It constitutes one of the internal barriers involved in the immune processes of the body [1, 2]. The structural core of the BBB is its unique endothelial cells (ECs) [3], which possess continuous, non-fenestrated tight junctions (TJs) composed of specific junctional proteins, as well as an intact basement membrane (BM), pericytes and extracellular matrix (ECM) [4]. Together, these structures play an important role in both protecting the central nervous system from harmful substances and maintaining the stable state of the intracerebral environment. In addition to the TJ itself, transporter proteins play a critical role in protection. These proteins are expressed in the endothelial cells of the central nervous system (CNS) and allow various nutrients to enter the brain [1]. ECs are characterised by a low transcytosis rate, with a low rate of vesicle-mediated transcellular translocation. This helps maintain the integrity of the BBB and keeps the brain microenvironment stable.
Astrocytes are neural cells derived from the ectoderm and neuroepithelium [5], representing the most abundant type of glial cell in the CNS. They are integral components of the neurovascular unit, playing a crucial role in the maintenance of brain homeostasis. Astrocytes display a spongiform morphology, with a diameter typically ranging from 40–60 µm [5]. Their primary structural features include the cell body, cellular processes and expression of the glial fibrillary acidic protein (GFAP). The intricate nature of their morphological architecture underpins their functional diversity, allowing them to perform a wide array of physiological roles within the CNS [6].
The most prominent structural characteristic of astrocytes is their processes, which are typically arranged in a radial pattern. These processes form tight junctions with neuronal synapses and the ECs of blood vessels [5]. Through these processes, astrocytes regulate signaling between neurons and glial cells, thereby maintaining proper function throughout CNS development and aging. Additionally, astrocytes play a pivotal role in the formation and functional maintenance of the BBB through their interactions with ECs, directly regulating the dynamic stability of the BBB [7, 8, 9, 10, 11, 12]. Another key structural feature of astrocytes is GFAP, a well-established marker and structural protein [13]. As a major component of the astrocytic cytoskeleton, GFAP provides mechanical support, preserves structural integrity and facilitates stable interactions with other cells, including neurons and endothelial cells. Upon astrocyte damage, GFAP is released into the cerebrospinal fluid and blood, serving as an indicator of brain injury and neuroinflammation [14]. This release further disrupts the integrity of the BBB, exacerbating neuronal damage and inflammatory responses [15, 16].
These structural and functional characteristics underscore the pivotal role of astrocytes in maintaining the stability and function of the BBB. Through their interactions with ECs via their processes, astrocytes not only provide essential structural support for BBB formation but also regulate its permeability, ensuring that harmful substances are effectively prevented from entering the brain, thus safeguarding the CNS from potential injury. Previous transplantation study has demonstrated that isolated astrocytes are capable of inducing barrier properties in newly formed blood vessels, thereby promoting BBB formation [13]. Furthermore, by examining the roles of astrocytes in various regions of the CNS, Yang et al. [17] have elucidated their involvement in stabilizing the internal environment of the BBB. These functions are inherently linked to the unique structural features of astrocytes. This article will further explore the underlying mechanisms involved in these processes.
The close apposition of astrocyte end-feet to cerebral blood vessels [1] enables astrocytes to promote the structural properties of the nascent vascular barrier [13]. This support manifests through the induction of EC barrier properties and facilitation of BM formation. Furthermore, the BBB’s selective permeability and metabolic supply are mediated by diverse transport proteins (e.g., GLUT1, excitatory amino acid transporters (EAAT), P-glycoprotein (P-gp) expressed on ECs) [1]. GLUT1 facilitates unidirectional glucose transport from the blood into the brain, providing energy essential for BBB homeostasis. EAAT removes excess glutamate from the brain, mitigating neurotoxicity and preserving BBB integrity. P-gp promotes efflux and restricts influx, protecting the BBB from toxic substances. Astrocytes structurally support the BBB by regulating the expression of these transporter proteins. The following sections detail the specific roles of astrocytes in maintaining BBB integrity. The structural support of astrocytes for BBB is listed in Fig. 1.
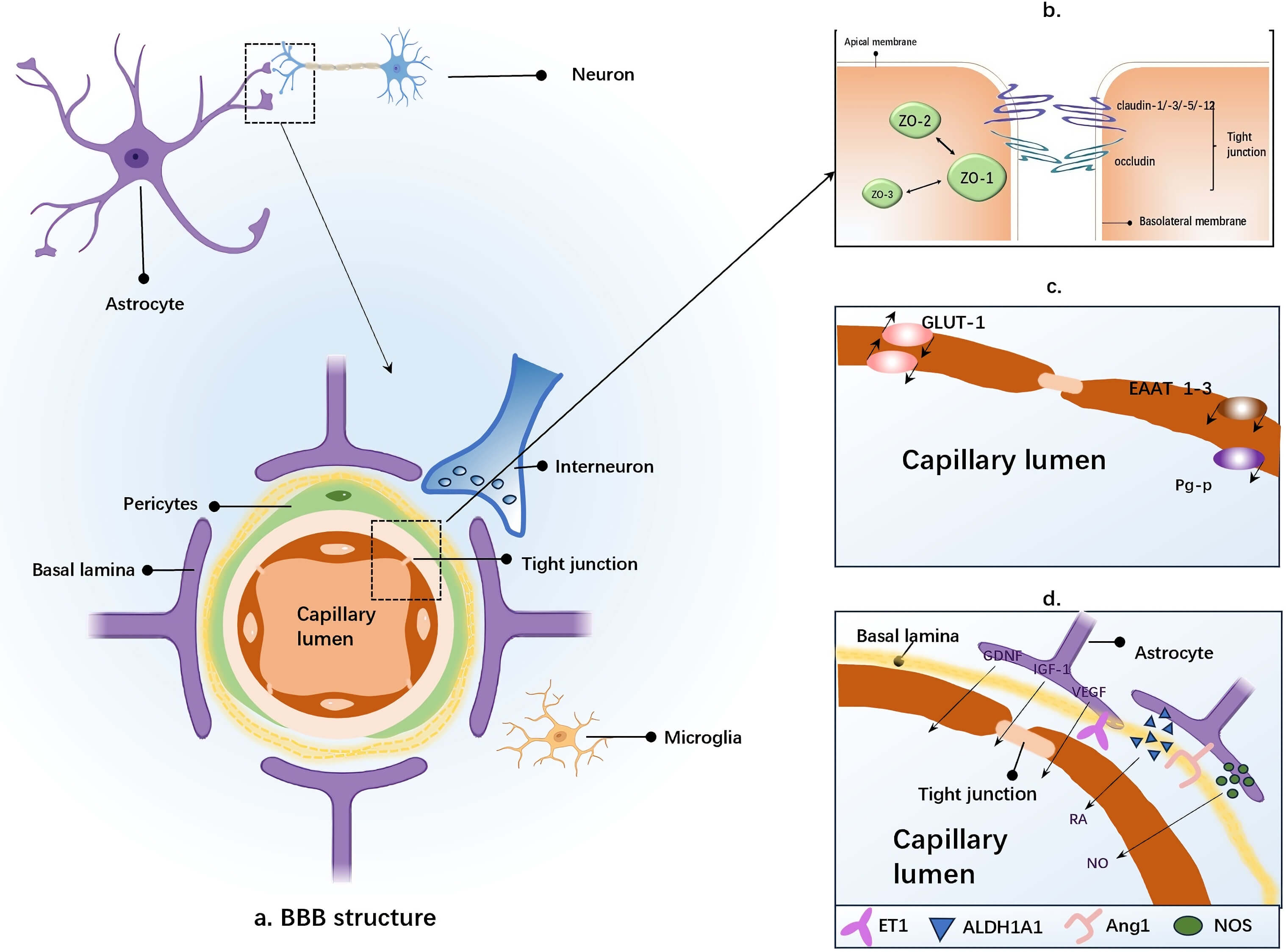 Fig. 1.
Fig. 1.
Cellular Components of the BBB. (a) The structural core of the BBB is formed by capillary endothelial cells, which are encased by the basement membrane and the perivascular end-feet of astrocytes. Astrocytes establish cellular connections with neurons, while pericytes and microglia are also depicted. (b) Schematic representation of tight junction protein structure: The tight junction complex is composed of transmembrane proteins (such as claudins, occludin, and junctional adhesion molecules), actin filaments, and cytoplasmic scaffolding proteins, including zonula occludens (ZO). (c) Characteristics of brain endothelial cells observed in cell culture: These cells express various transporters and receptors, such as EAAT 1–3, GLUT1, and P-gp, which contribute to the structural support and barrier functions of the BBB. (d) Induction of BBB properties by factors secreted by astrocytes (e.g., Ang-1, ET-1, GDNF, RA, VEGF, IGF-1, NO): These factors, regulated by the brain microenvironment and astrocytes, exert distinct effects on BBB properties under different physiological or pathological conditions. ZO, zonula occludens; EAAT, excitatory amino acid transporters; GLUT1, glucose transporter 1; P-gp, P-glycoprotein; BBB, blood-brain barrier; Ang-1, Angiopoietin-1; ET-1, Endothelin-1; GDNF, glial cell derived neurotrophic factor; RA, retinoic acid; VEGF, vascular endothelial growth factor; IGF-1, insulin-like growth factor-1; NO, nitric oxide. The figure was created using Microsoft PowerPoint (2016, Microsoft Corporation, Redmond, WA, USA).
ECs constitute the core cellular component of the BBB, forming a low-permeability barrier that controls substance transport between blood and brain tissue. Astrocytes, positioned adjacent to ECs, secrete signaling molecules that induce and maintain EC barrier properties. Park et al. [18] demonstrated that optimal BBB development requires direct contact between ECs and astrocytes, indicating an astrocytic contribution to EC differentiation and barrier function.
Adjacent ECs are interconnected by TJ and adherens junction proteins, both regulated by astrocytes. TJ structures comprise transmembrane proteins (occludin, claudins, junctional adhesion molecules), actin filaments and cytoplasmic scaffolding proteins (zona occludens (ZO)) [19]. TJ formation, mediated by these proteins, is pivotal for regulating BBB permeability [17]. TJ protein expression is 1.5-2-fold higher in EC-astrocyte co-cultures compared to EC monocultures [18], reflecting the essential supportive role of astrocytes in TJ assembly. Otani et al. [19] found that introducing cultured astrocytes near normally leaky vessels increased TJ protein expression and EC tightening, resulting in reduced leakage. This suggests astrocytes influence both TJ formation and expression.
The BBB basement membrane formation (BBB-BM), a specialized ECM [20], consists primarily of type IV collagen, laminin, nidogen (often referred to as entactin) and perlecan [1, 21]. These are proteins synthesized mainly by ECs, pericytes and astrocytes. Astrocytes ensheath capillaries, arterioles and venules and secrete trophic factors that maintain EC barrier properties [22]. Astrocyte-derived apolipoprotein E (ApoE) binds the LRP1 receptor on pericytes, inhibiting cyclophilin A-mediated activation of matrix metalloproteinase-9 (MMP-9), thereby participating in BBB-BM stabilization [23]. Concurrently, astrocytes strengthen the BM via polarized end-feet expressing aquaporin-4 (AQP4), which connect to the cerebrovascular BM. Crucially, astrocytes secrete laminin, a major structural BM component [1]. Laminin not only reinforces the BM and stabilizes pericytes [20] but also induces AQP4 expression [24]. Yao et al. [21] demonstrated that laminin increases AQP4 expression, contributing to BBB-BM integrity. An early study by Xu et al. [20] revealed that astrocyte-secreted laminin deficiency leads to BBB disruption and intracerebral hemorrhage. Collectively, these findings underscore the critical role of astrocyte structure and function in BBB-BM formation.
Encoded by SLC2A1, the glucose transporter 1 (GLUT1) is highly
expressed at the BBB, predominantly localized within the cerebral vasculature and
on astrocyte end-feet [25, 26]. It transports glucose unidirectionally from blood
to brain, supplying energy to neurons and glia and is essential for BBB integrity
[27]. Given that astrocyte end-feet ensheath cerebral vasculature, astrocytes
represent a mandatory pathway for GLUT1-mediated glucose transport
[28, 29, 30]; astrocyte regulation of GLUT1 is thus key to maintaining BBB
integrity and function. Zheng et al. [31] observed vasogenic brain edema
in zebrafish following GLUT1 knockdown; GLUT1 function is
highly conserved in humans. Pearson et al. [32] noted that
GLUT1 deficiency impairs glucose transport across the BBB, indirectly
compromising BBB stability. These findings highlight GLUT1’s importance
in BBB homeostasis. Astrocytes regulate GLUT1 through two primary
mechanisms: (i) Releasing factors (e.g., Wnt ligands, transforming growth
factor-beta (TGF-
EAAT is a high-affinity, sodium-dependent transporter responsible for
the majority of CNS glutamate uptake, compensating for the lack of extracellular
catabolic enzymes [35]. Excess glutamate exerts neurotoxic effects, including
neuronal death and BBB disruption. EAAT clears glutamate from the brain
against a concentration gradient, maintaining amino acid homeostasis [36, 37].
Astrocyte-derived EAAT1 and EAAT2 are primarily responsible for
synaptic glutamate clearance at the BBB [38, 39, 40, 41], with EAAT2 alone
accounting for ~80% of uptake [42, 43]. This clearance prevents
excitotoxicity, neuroinflammation and cerebral edema, thereby directly and
indirectly maintaining BBB integrity [44]. Astrocytes coordinate
EAAT-mediated glutamate metabolism [45]. Increasing EAAT2
transcription and membrane localization via secreted TGF-
P-gp, a member of the ATP-binding cassette (ABC) transporter superfamily, is localized to the luminal plasma membrane of BBB ECs during telencephalic development, serving as an early marker of BBB differentiation [49]. P-gp maintains CNS homeostasis and protects the BBB from blood-borne toxins by actively effluxing diverse substrates and restricting compound uptake into ECs [50, 51, 52, 53]. Baello et al. [54] co-cultured astrocytes with ECs and observed a significant increase in P-gp levels, particularly in neonatal astrocytes. This suggests astrocytes enhance P-gp expression, thereby safeguarding efflux capacity, protecting against harmful substances and preventing BBB damage.
BBB ECs exhibit a low rate of vesicle-mediated transcytosis, a process involving endocytic uptake of material into vesicles followed by exocytic release on the opposite side. This process functions synergistically with pericytes and astrocytes [55]. At the BBB, transcytosis selectively transports macromolecules (e.g., proteins, drugs) into the brain parenchyma [1], while lysosomes degrade or return substances to the peripheral circulation, preventing toxin accumulation and sealing the BBB [55, 56]. Mfsd2a, a lipid transporter highly expressed in CNS ECs, suppresses transcytosis and is associated with low BBB permeability. Evidence indicates astrocyte regulation of Mfsd2a, suggesting astrocytes modulate BBB permeability by influencing Mfsd2a-mediated transcytosis through co-regulatory mechanisms [55].
Beyond their role in inducing and maintaining long-term barrier properties,
astrocytes secrete signaling factors that bidirectionally regulate EC
permeability [57]. Protective factors include Ang-1, GDNF,
retinoic acid (RA), and insulin-like growth factor-1 (IGF-1) [57, 58, 59, 60, 61, 62, 63],
which shield ECs from apoptosis and promote functional recovery. Some
EC-disrupting factors, such as astrocyte-derived ET, cause endothelial cell
apoptosis and down-regulation of TJ-related proteins, leading to BBB disruption.
Other specific factors have different mechanisms for regulating BBB permeability
in different states. These include nitric oxide (NO) and vascular endothelial
growth factor (VEGF), among others. Additionally, signaling pathways activated by
astrocytes regulate BBB function. The Hh (Hedgehog),
Wnt/
| Category | Signaling factor | Abbreviation | Effect on BBB permeability |
| EC protection | Angiopoietin-1 | Ang-1 | Maintains BBB integrity and low permeability by stabilizing vasculature, enhancing TJs, and inhibiting inflammation. |
| Glial-derived neurotrophic factor | GDNF | Enhances barrier function via GDNF receptor activation, supporting neuronal survival and regulating endothelial function. | |
| Retinoic acid | RA | Synthesized by astrocytes (ALDH1A1+), maintains barrier integrity, regulates permeability, and promotes repair. | |
| Insulin-like Growth Factor-1 | IGF-1 | Reduces BBB permeability and damage by acting as a neurotrophic factor. | |
| Nitric Oxide | NO | Low concentrations generated by eNOS maintain barrier integrity under homeostasis. | |
| Vascular Endothelial Growth Factor | VEGF | Physiological levels contribute to vascular homeostasis maintenance. | |
| EC Disruption | Nitric Oxide | NO | Overproduction by iNOS during inflammation causes BBB destruction. |
| Vascular Endothelial Growth Factor | VEGF | Secreted by reactive astrocytes in pathology, disrupts BBB integrity. | |
| Endothelin | ET | Astrocyte synthesis and upregulation of ET-1 exacerbate CNS inflammation, leading to BBB destruction. |
eNOS, Endothelial Nitric Oxide Synthase; iNOS, Inducible Nitric Oxide Synthase; CNS, central nervous system.
Primarily derived from astrocytes, Ang-1 stabilizes vascular structure, enhances TJs, inhibits inflammation, maintains BBB integrity and low permeability [64]. Mechanistically, astrocyte-secreted Ang-1 binds the Tie2 receptor on ECs, activating the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway to upregulate TJ proteins (e.g., occludin, claudin-5). Concurrently, it inhibits the RhoA/ROCK pathway, reducing EC contraction and preventing TJ disassembly. Ang-1 also suppresses neuroinflammation via the SNHG14/miR-223-3p/NLRP3 pathway, providing neuroprotection [65].
This astrocyte-secreted neurotrophic factor, enriched in end-feet, is a key mediator of endothelial network formation [66]. GDNF ensures neuronal survival and regulates EC function by activating its receptors on neurons and ECs [63]. Specifically, GDNF binding to the rearranged during enstransfection receptor on ECs activates the PI3K/Akt pathway, resulting in increased TJ protein expression (occludin, claudin-5), enhanced membrane localization and activity of efflux transporters (P-gp, breast cancer resistance protein (BCRP)), and modulation of the Rac1/RhoA balance to stabilize the cytoskeleton. GDNF also acts on pericyte receptors, enhancing vascular coverage and inhibiting leakage [67].
RA, an active vitamin A metabolite, is synthesized via enzymes including ALDH1A1/A2/A3 [68]. The role of RA on the BBB is mainly in maintaining barrier integrity, regulating permeability and participating in pathological repair. Adam et al. [69] observed strong ALDH1A1 expression in mature astrocytes, suggesting that ALDH1A1 is highly enriched in astrocytes. Therefore, it is assumed that astrocytes regulate BBB permeability via the RA signaling pathway. Adam et al. [69] concluded from their analysis of human post-mortem brain tissue and in vitro experiments that human fetal astrocytes promote the formation of the brain endothelial barrier and increase the expression of BBB-specific genes via this pathway. To further determine the expression of BBB-specific proteins induced by RA, the Mizze experimental group performed a Western blot analysis of ZO-1 and VE-calmodulin, which are important for BBB integrity. The results indicated that the levels of these proteins increased in the group treated with RA. In summary, astrocytes participate in RA formation through ALDH1A1 and also induce EC differentiation and promote BBB formation through the RA signaling pathway.
Mainly sourced from astrocytes and peripheral plasma [70], IGF-1 acts as a
neurotrophic factor via the IGF-1 receptor, promoting neurogenesis, neuronal
survival, neuroprotection and repair [71]. Some researchers pointed out that
IGF-1’s anti-apoptotic effects in ECs and its reduction of Evans blue dye
extravasation, suggesting BBB protection [70]. Astrocytes contribute to elevated
IGF-1 levels in the infarcted cortex, partly via mesenchymal stem cell mediation
[70]. Mechanistically, astrocyte-derived IGF-1 activates the MARK/ERK and
PI3K/Akt pathways in ECs, inhibiting GSK-3
NO, a vasodilator involved in neurovascular coupling [72], is synthesized by NO
synthase (NOS). Expression of eNOS and iNOS on astrocyte end-feet implicates
astrocytes in NO-mediated BBB regulation. Physiological NO concentrations,
generated by astrocytic eNOS, maintain barrier integrity via the sGC-cGMP-PKG
pathway, which phosphorylates occludin and ZO-1 to stabilize TJs; eNOS also
directly prevents inflammation and apoptosis [73, 74, 75, 76, 77]. Conversely, under
inflammation, astrocytic iNOS produces excessive NO, generating peroxynitrite
that degrades TJ proteins, activates MMPs to degrade the BM, and triggers
NF-
Predominantly astrocyte-derived [78], VEGF prevents EC apoptosis and protects BBB integrity under physiological conditions, but downregulates TJ proteins, increases permeability and pathologically disrupts the BBB [79, 80, 81, 82]. Here, it is proposed that astrocyte-secreted VEGF bidirectionally regulates BBB function. Physiological VEGF maintains vascular homeostasis by activating the PI3K/Akt pathway in ECs, promoting angiogenesis and survival and transiently enhancing TJ protein expression via VEGFR2 activation [83]. Pathologically, reactive astrocytes secrete VEGF, activating the VEGFR2/PKC pathway to induce TJ protein endocytosis and degradation, increase transcytosis, inhibit P-gp (causing toxin accumulation) and amplify inflammatory infiltration, culminating in BBB damage and brain injury [79, 84]. Hence, astrocyte-derived VEGF acts as a “double-edged sword”: Essential for vascular development/repair at physiological levels, but destructive to the BBB when pathologically overexpressed.
ET, a potent vasoconstrictor mediating CNS inflammatory responses [85], exacerbates BBB damage, as ET blockade significantly reduces leakage [86]. Among ET isoforms (ET-1, ET-2, ET-3), astrocyte-synthesized ET-1 is upregulated and critically involved in cerebral ischemic injury pathogenesis. Cheng et al. [87] found astrocyte-specific ET-1 overexpression increased brain injury susceptibility following MCAO-induced ischemia. Collectively, astrocytes exacerbate CNS inflammation and BBB destruction by upregulating ET-1 expression.
Sonic hedgehog (Shh), secreted by astrocyte end-feet, promotes BBB formation and integrity. Shh binding to the patched-1 receptor on ECs relieves inhibition of smoothened, activating the pathway. This directly regulates transcription of target genes (e.g., Ang-1) via Glioma-associated oncogene (Gli) transcription factors, upregulating BBB barrier function [88].
The continuous activation of the Hh signaling pathway reduces permeability between endothelial cells by increasing the expression of tight junction proteins. This prevents toxins, immune cells, or macromolecules from infiltrating the brain parenchyma from the blood. Among these proteins, the transcription factor Gli-1 increased the expression of ECs after activation of the Hh pathway, while the TJ protein regulator SRY-box transcription factor 18 (SOX18) peaked within two hours of activation. This indicates that Gli-1 can upregulate TJ expression [88]. Thus, the Hh signaling pathway in the CNS endothelium results in increased expression of TJ proteins, which are indispensable for the maintenance and stabilization of the BBB.
The Hh signaling pathway also provides an anti-inflammatory balance within the
CNS by inhibiting the NF-
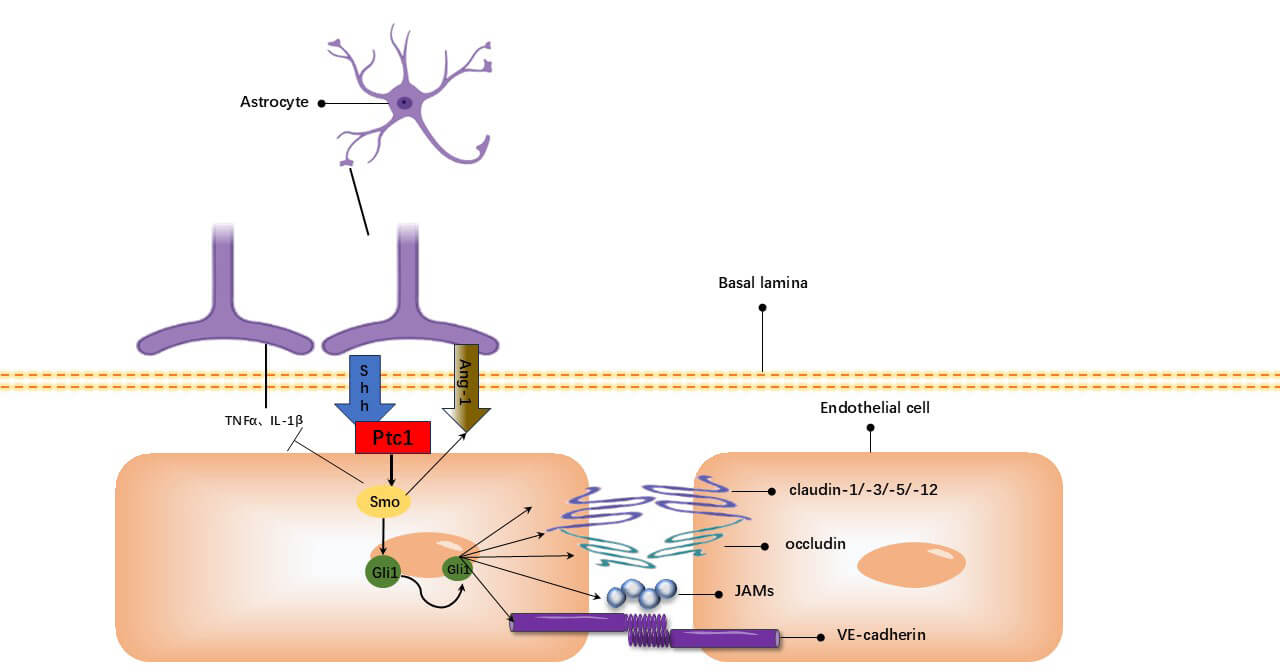 Fig. 2.
Fig. 2.
Hh signaling pathway. Astrocyte-derived Shh binds Ptch-1,
activating Smo. This leads to Gli1-mediated transcriptional upregulation of Ang-1
and TJ proteins (occludin, JAMs, VE-cadherin, claudin-1/-3/-5/-12). The pathway
concurrently inhibits TNF-
Essential for BBB integrity and CNS homeostasis [93], this pathway centrally
regulates BBB EC differentiation and function [94]. It dominates BBB formation
during embryogenesis, inducing cerebral angiogenesis and barrier properties. In
adults, it maintains EC TJs and low permeability, supporting post-injury repair.
Astrocyte-secreted Wnt ligands (e.g., Wnt7a/7b) bind EC receptors (Frizzled,
low-density lipoprotein receptor-related protein 5/6 (LRP5/6)), activating the
pathway via paracrine signaling. This inhibits
The Wnt/
The Wnt/
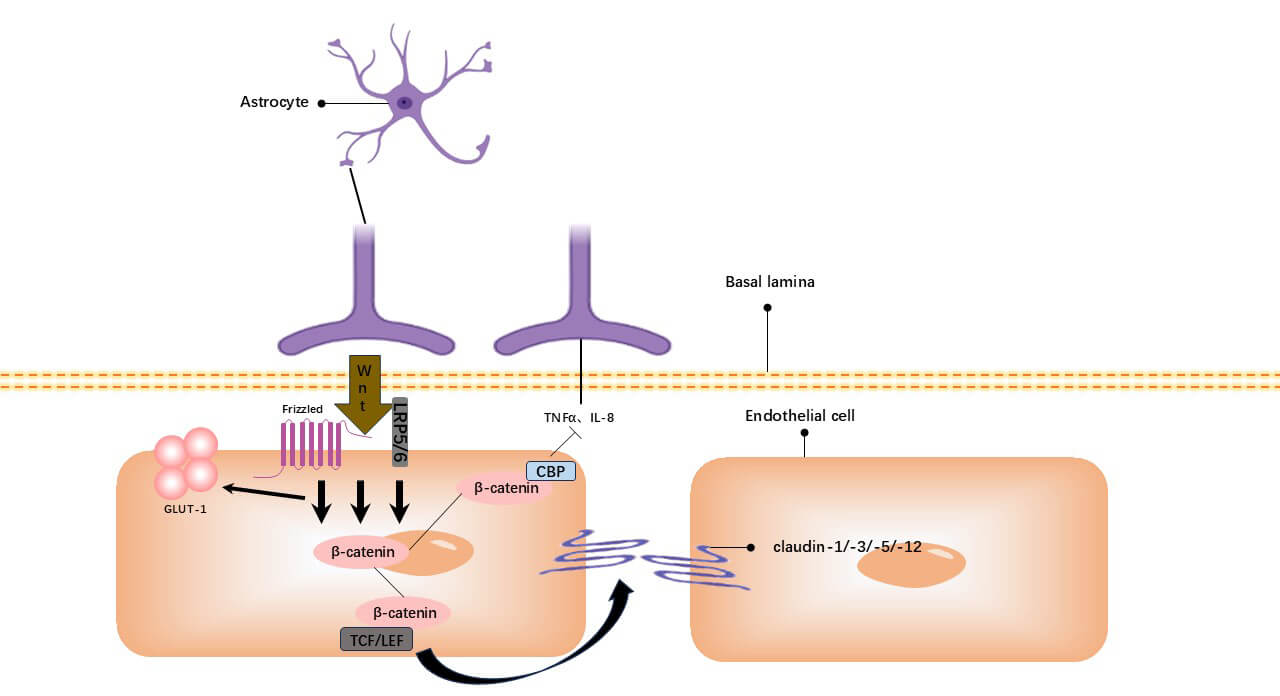 Fig. 3.
Fig. 3.
The Wnt/
The Wnt/
Astrocytes precisely regulate BBB permeability via TGF-
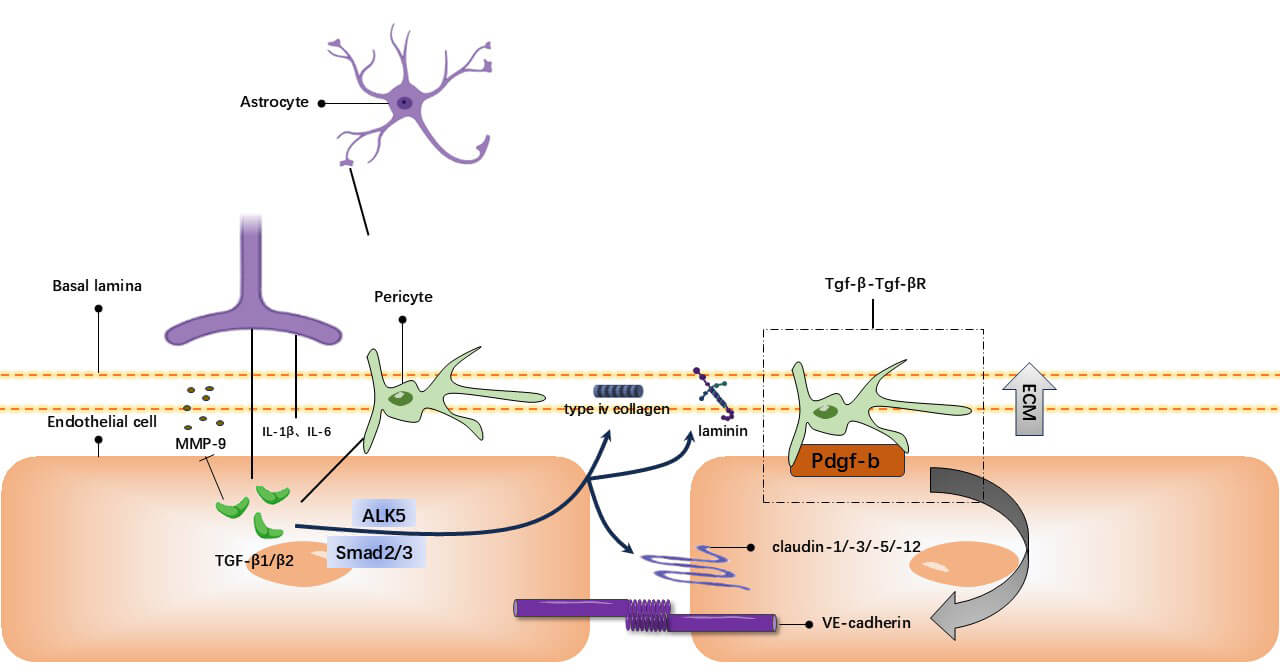 Fig. 4.
Fig. 4.
TGF-
The TGF-
As detailed above, astrocyte involvement in BBB formation, maintenance, disruption and repair is fundamentally governed by their structural specializations, secreted signaling factors and activation of specific pathways. Critically, the pathogenesis of several CNS disorders—including multiple sclerosis (MS), Parkinson’s disease (PD), Alzheimer’s disease (AD), ischemic stroke, and epilepsy—is intimately linked to BBB dysfunction. Below, the aforementioned astrocyte regulatory mechanisms contributing to disease pathogenesis are explored and evaluated for their potential as therapeutic targets.
MS is a group of autoimmune neuroinflammatory disorders characterized by demyelination, axonal damage and ultimately neurodegeneration with progressive disability. Dysregulation of astrocytic synaptic modulation is one of the pathogenic mechanisms underlying MS [100, 101]. As key regulators of neuroinflammation [102], astrocytes become overactivated under chronic inflammatory conditions, leading to glial scar formation, secretion of various chemokines (e.g., CCL2, CXCL10, CXCL12, FASL and TRAIL) and MMP-9, disruption of BBB tight junctions and interaction with immune cells to facilitate their infiltration into the brain parenchyma, thereby inducing neurotoxicity and attacking neuromyelin sheaths [103]. It has been shown that tumor necrosis factor (TNF) produced by astrocytes can directly exert cytotoxic effects by interacting with TNF receptors on neurons, thus mediating neurodegenerative processes [102, 104].
Activation of astrocytic sphingosine 1-phosphate (S1P) signaling also
contributes to MS pathogenesis; as a disorder of sphingolipid metabolism, its
severity closely correlates with the degree of demyelination and axonal damage
[105, 106]. Additionally, lipolactosylceramide activates the
cPLA2-MAVS-NF-
PD is the second most prevalent neurodegenerative disorder, characterized by
motor disturbances (e.g., tremor, rigidity, bradykinesia) [111]. Its pathological
hallmarks include the loss of dopaminergic neurons in the substantia nigra pars
compacta and aggregation of misfolded
AD is one of the most common late-onset neurodegenerative diseases,
characterized by dementia, cognitive decline and memory impairment [121]. Its
pathological features include intraneuronal neurofibrillary tangles and
extracellular amyloid-
In summary, during AD pathogenesis, astrocytic glutamate uptake mechanisms
become dysregulated. Concurrently, activated astrocytes promote inflammatory
responses, driving A
Ischemic stroke, the second leading cause of death globally, is characterized by
ischemic and hypoxic necrosis of brain tissue resulting from disrupted cerebral
blood supply. In this process, astrocyte-mediated inflammatory responses play a
key role. Within two to six hours of ischemic injury onset [137], activated
astrocytes secrete large quantities of inflammatory factors (e.g.,
TNF-
Based on these findings, mitigating post-ischemic brain damage may be achievable by inhibiting astrocyte activity (e.g., via cotton seed oil or analogous agents) or inducing A2 astrocyte polarization. Further in-depth understanding of key signaling pathways regulating A2 astrocyte differentiation and the mechanisms governing A1-to-A2 astrocyte transition (e.g., S100A10) will facilitate exploration of A2 astrocytes’ roles in maintaining BBB homeostasis.
Epilepsy is a chronic neurological disorder caused by highly synchronized
abnormal neuronal discharges in the brain, characterized by recurrent, transient
and stereotypical CNS dysfunction [146]. Following epileptogenesis, reduced
EAAT expression in astrocytes triggers the release of pro-inflammatory
cytokines and chemokines [147]. In adult epileptic rats, inhibition of
IL-1
The BBB is a critical structural and functional component of the CNS, with its unique structural and functional properties serving to restrict the entry of harmful substances into the brain parenchyma. Additionally, it plays a pivotal role in maintaining the homeostasis of the cerebral microenvironment. Astrocytes, the most abundant glial cells in the CNS, are pivotal for maintaining BBB integrity [149]. Astrocytic perivascular endfeet are directly involved in the formation of the neurovascular unit and these endfeet act as critical nodes in cerebral metabolic processes [150]. Such functions, encompassing metabolism, cholesterol synthesis, neurotransmitter uptake and biosynthesis, represent core mechanisms underlying the maintenance of BBB microenvironmental homeostasis [151].
Astrocytes leverage their structural specializations to induce EC differentiation, upregulate TJ protein expression, enhance basement membrane integrity and regulate the BBB in coordination with transporter proteins on ECs. GLUT1, a key glucose transporter, is highly expressed at the BBB, with astrocytes facilitating neuronal glucose supply through GLUT1-mediated uptake. EAAT, the primary glutamate transporters, enable astrocytes to clear glutamate from the synaptic cleft, thereby preventing excitotoxicity, neuroinflammation and cerebral edema, as well as mitigating the initiation and progression of neurodegenerative diseases such as AD, PD and MS. Furthermore, P-gp actively effluxes various substrate molecules and limits compound uptake by ECs, thereby maintaining local microenvironmental homeostasis and shielding the BBB from blood-borne toxins. Moreover, astrocytes can modulate BBB permeability by regulating endothelial proteins, leveraging the BBB’s intrinsic low endocytic activity.
Astrocytes also exert either protective or deleterious effects on the BBB
through the secretion of diverse factors. Secreted factors such as
Ang-1, GDNF, RA, and IGF-1 have been demonstrated to enhance
BBB barrier properties and upregulate TJ expression. Conversely, certain
factors—including VEGF, NO and ET—exert deleterious effects by inducing EC
apoptosis and downregulating TJ-related proteins. These factors interact in a
coordinated manner, enabling astrocytes to dynamically regulate BBB stability.
Beyond secreted factors, signaling pathways are also critical: Hh,
Wnt/
Astrocytes are further involved in the dynamic regulation of the nervous system and contribute significantly to the pathogenesis of neurodegenerative diseases [152]. In this review, multiple disorders have been highlighted and illustrated—including MS, PD, AD, ischemic stroke and epilepsy—as has the fact that astrocytes employ diverse mechanisms to protect, disrupt, or repair the BBB under pathological conditions, such as releasing excessive pro-inflammatory factors, modulating neuronal excitability, altering TJ protein expression and inducing reactive astrocyte polarization. Moving forward, a deeper understanding of the multifaceted roles and underlying mechanisms of astrocytes in BBB regulation may facilitate the design of more targeted therapeutic strategies aimed at BBB modulation, thereby opening new avenues for restoring BBB integrity in neurological disorders.
Although current research has thoroughly explored the role of astrocytes in the BBB, several gaps and unresolved questions remain to be addressed. The following outlines the limitations of current studies and potential directions for future research.
First, research into astrocyte heterogeneity is insufficient. Astrocytes are not a homogeneous cell type; rather, they exhibit functional diversity across various brain regions and physiological states. Current understanding of astrocytic heterogeneity is limited, particularly concerning the influence of distinct astrocytic subtypes on BBB stability and function. Future investigations should aim to explore the distribution and functional differences of astrocytic subtypes across brain regions, thereby elucidating their specific roles within the context of BBB regulation.
Second, the long-term effects of astrocyte functional regulation on BBB integrity are not yet fully understood. While existing studies have established the pivotal role of astrocytes in BBB formation and maintenance, there remains a significant gap in the understanding of their sustained influence, particularly in how they continuously regulate the BBB following neurotrauma or inflammatory events. For instance, while astrocytes can modulate BBB stability by secreting various signaling factors, their long-term impact on the BBB during chronic pathological conditions and the sustainability of these regulatory actions remain unclear. Moreover, the roles of astrocytes in different pathological states—whether protective or detrimental—are often time-dependent. In neurodegenerative diseases such as AD and PD, the long-term response of astrocytes may either exacerbate or alleviate BBB damage. Therefore, future research should prioritize elucidating the mechanisms underlying the long-term regulation of astrocytic function, with particular emphasis on their role in chronic diseases. Such studies will be crucial for the development of long-term therapeutic strategies aimed at stabilizing the BBB.
Finally, a gap persists between basic research and clinical application. Although numerous fundamental studies have provided valuable insights into the role of astrocytes in BBB regulation, translating those findings into effective clinical therapies remains challenging. Many studies are based on animal models or cell cultures, which do not fully capture the complexity of human diseases. Future research must place greater emphasis on clinically relevant studies, including clinical trials and human-specific models, to validate the therapeutic potential of astrocyte regulation of the BBB.
Astrocytes play an indispensable role in maintaining the integrity and function of the BBB. In terms of structural adaptation, astrocytes play a role in maintaining BBB permeability by influencing endothelial cell differentiation and regulating the expression of tight junction proteins. Meanwhile, their roles in neurotransmitter uptake, glucose transport, and protection against neurodegenerative diseases highlight their important contributions to brain homeostasis. In addition, the factors secreted by astrocytes can both protect and damage the BBB, reflecting their dual roles in health and disease. Finally, the article mentions the dynamic regulation of BBB by astrocytes under pathological conditions, highlighting their potential as therapeutic intervention targets for neurological diseases. However, after summarizing, we found that there are still some research gaps, especially in the heterogeneity of astrocytes, long-term regulatory mechanisms, and the insufficient translation of basic research into clinical applications. Future research should focus on these areas to improve strategies for stabilizing the BBB and enhancing the treatment of neurodegenerative diseases.
Conceptualization, SSZ and LLW; review of the literature, DF and AYH; writing—original draft preparation, DF; writing—review and editing, SSZ and LLW. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
Funding for this research was provided by the National Natural Science Foundation of China (NO. 82405544) and Zhejiang Provincial Natural Science Foundation of China under Grant (NO. LQ23H270009).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.