





 , Wen-xuan Cao 2, Ming-rui Liu 1, Hao Wu 1, Li-yuan Cui 3, Ying-jiao Liu 1,*
, Wen-xuan Cao 2, Ming-rui Liu 1, Hao Wu 1, Li-yuan Cui 3, Ying-jiao Liu 1,*
1 College of Pharmacy, Jiangxi University of Chinese Medicine, 330004 Nanchang, Jiangxi, China
2 Key Laboratory of Marine Drugs, Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, 266003 Qingdao, Shandong, China
3 State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica & Neuroscience Center, Chinese Academy of Medical Sciences and Peking Union Medical College, 100050 Beijing, China
Abstract
The onset and progression of central nervous system (CNS) disorders are frequently associated with aberrant neuronal death. In addition to the classical forms of cell death such as apoptosis and necrosis, neurons can also undergo alternative modes of death, including ferroptosis, cuproptosis, and ammonia-induced cell death, all of which may involve the participation of astrocytes. Neuronal death is an irreversible process and plays a central role in the pathogenesis of numerous CNS diseases. We found that astrocytes exhibit the capacity to regenerate into neurons, a characteristic that may pave the way for novel therapeutic approaches in the treatment of neurological disorders. Astrocytes represent optimal starting cells for reprogramming techniques due to their anatomical proximity to neurons and their shared origin from common progenitor cells—radial glial cells. Reprogramming techniques encompass the conversion of astrocytes into pluripotent neurospheres or their direct in vivo reprogramming into functional neurons, aiming to replace damaged or lost neurons through processes such as transdifferentiation and dedifferentiation. This article examines the interplay between astrocytes and neuronal survival and degeneration in CNS disorders, as well as two reprogramming strategies for converting astrocytes into neurons, with the aim of establishing a scientific foundation for neuronal repair in the treatment of CNS diseases.
Keywords
- ferroptosis
- cuproptosis
- ammonia-induced death
- astrocytes
- cell reprogramming
- neurorestoration
- neurons
- de-differentiation
- trans-differentiation
- transcription factors
The brain comprises two main cell types: neurons, which are responsible for transmitting information, and glial cells, which provide structural and functional support to neurons. Among these glial cells, astrocytes represent the predominant cell type, comprising approximately 20% to 40% of the glial cell population [1, 2]. Astrocytes can be categorized based on their cellular morphology and anatomical localization: protoplasmic astrocytes are predominantly found in the gray matter, fibrous astrocytes are primarily located in the white matter, and specialized astrocytes include subtypes such as radial astrocytes, Müller cells in the retina, and Bergmann glia in the cerebellum [3]. Astrocytes represent a diverse and heterogeneous population of glial cells [4], exhibiting region-specific variations. Hippocampal astrocytes exhibit a greater degree of interaction with neurons compared to those originating from the striatum [5]. Furthermore, protoplasmic astrocytes in the gray matter demonstrate distinct differences from fibrous astrocytes in the white matter with respect to glutamate and energy metabolism, and astrocytes in the gray matter display a higher propensity for transdifferentiation into neurons than their counterparts in the white matter [4].
Astrocytes are referred to as “support cells” due to their capacity to provide structural and functional support to neighboring neurons and other cellular components of the nervous system [6]. Moreover, astrocytes maintain a functionally integrated and structurally interconnected relationship with neurons. First, astrocytes serve as essential cellular components participating in information processing within the nervous system. In addition to forming both the neuronal network and the astrocyte syncytium network, astrocytes also engage in bidirectional communication with neurons via tripartite synapses [7]. Astrocytes are in a prime position to facilitate and maintain synaptic connections. At tripartite synapses, astrocytes are capable of responding to neuronal activity in a feedback-regulated manner and modulating synaptic activity through bidirectional communication with synaptic neuronal elements, including the detection and response to neurotransmitters released from synapses, thereby regulating synaptic signaling [8]. Second, astrocytes have the capacity to influence neuronal function and regulate synaptic plasticity. Astrocytes are capable of expressing receptors for a wide range of neurotransmitters, including glutamate and gamma-aminobutyric acid (GABA), as well as transporter proteins on their cell surface. These molecular components enable astrocytes to detect and respond to changes in the surrounding chemical environment [9]. At the synapse, astrocytes contribute to the regulation of extracellular ion concentrations—such as sodium and calcium—as well as neurotransmitter levels, thereby playing a critical role in modulating synapse formation, neuronal development, and functional activity. It also contributes to the stability and plasticity of neural circuits through the release of various secreted factors, such as thrombospondin-1 (TSP-1), glypican-4/6 (Gpc4/6), and hevin, which promote both synapse formation and maintenance [10]. Finally, astrocytes play a crucial role in supporting neuronal structural integrity and energy metabolism. They are capable of guiding neuronal migration, promoting the growth of neuronal dendrites and axons, and maintaining a robust oxidative metabolic capacity [11]. Astrocytes metabolize glucose into lactate through the glycolytic pathway and subsequently release substantial amounts of lactate into the extracellular space, where it is taken up by neurons to fulfill their high energy demands and sustain neurotransmitter activity [12]. This process is formally recognized as the astrocyte-neuron lactate shuttle (ANLS). In addition, astrocytes generate a substantial number of lipid droplets (LDs) to facilitate the uptake of fatty acids (FAs) released by neurons and synthesize various antioxidant molecules [13], thereby protecting neurons from FA-induced toxicity.
Neurological disorders such as Parkinson’s disease, stroke, spinal cord injury,
and Alzheimer’s disease result from the loss or dysfunction of neurons and glial
cells in various brain regions. Furthermore, the depletion or pathological
alterations of astrocytes can also contribute to neuronal death and metabolic
disturbances within the central nervous system. Cerebral ischemia can induce the
formation of two distinct phenotypes of reactive astrocytes: neurotoxic A1
astrocytes and neuroprotective A2 astrocytes [14]. Type A1 reactive astrocytes
induce neuronal damage, ultimately resulting in neuronal death [15]. Astrocytes
are induced to produce € that release toxicity induced neuronal
death mediated by interleukin-1
Ferroptosis is an iron-dependent form of cell death, and iron accumulation and lipid peroxidation are the main drivers of ferroptosis and are also strongly associated with oxidative stress and inflammatory responses [20, 21].
Neurotoxic type A1 astrocytes have been demonstrated to induce neuronal ferroptosis in a rat model of epilepsy through the enhancement of lipid peroxidation, thereby promoting ferroptosis. The chemokine C-X-C motif chemokine ligand 10 (CXCL10), secreted by type A1 astrocytes, interacts with C-X-C chemokine receptor 3 (CXCR3) on neuronal cells and promotes cellular ferroptosis through modulation of the cystine/glutamate antiporter (STAT3/SLC7A11) signaling pathway [22, 23]. Zhang et al. [24] demonstrated that a transmembrane protein, TMEM164, may function as a potential therapeutic agent for modulating neurotoxic astrocytes. Overexpression of TMEM164 was shown to suppress A1 phenotypic expression, preserve normal astrocytic functions, and attenuate neuronal death mediated by neurotoxic reactive astrocytes.
During ischemic stroke, glutamate concentrations increase and the Xc-cysteine/glutamate transport system (the Xc-system) becomes inhibited, leading to reduced synthesis of glutathione (GSH). Reduced levels of GSH contribute to decreased expression of glutathione peroxidase 4 (GPX4), which subsequently disrupts the balance of oxidized glutathione (GSSH) [25, 26], leading to impaired redox homeostasis. Furthermore, this deficiency results in the intracellular accumulation of toxic phospholipid hydroperoxides (PLOOH) due to their reduced degradation capacity, thereby accelerating the process of ferroptosis [27, 28]. Type A2 astrocytes are capable of scavenging excess extracellular glutamate through mechanisms including the upregulation of excitatory amino acid transporter proteins (EAATs), nuclear factor erythroid 2-related factor 2 (Nrf2), and antioxidant genes. These processes contribute to the reduction of neuronal excitotoxicity and the attenuation of ferroptosis in neurons [29, 30, 31, 32, 33, 34]. In contrast, neurotoxic A1 phenotype astrocytes promote neuronal ferroptosis by accelerating the ferroptotic process through multiple mechanisms, including the secretion of CXCL10, which enhances STAT3 phosphorylation, inhibits the Xc-system, and facilitates intracellular lipid peroxidation (Fig. 1) [22].
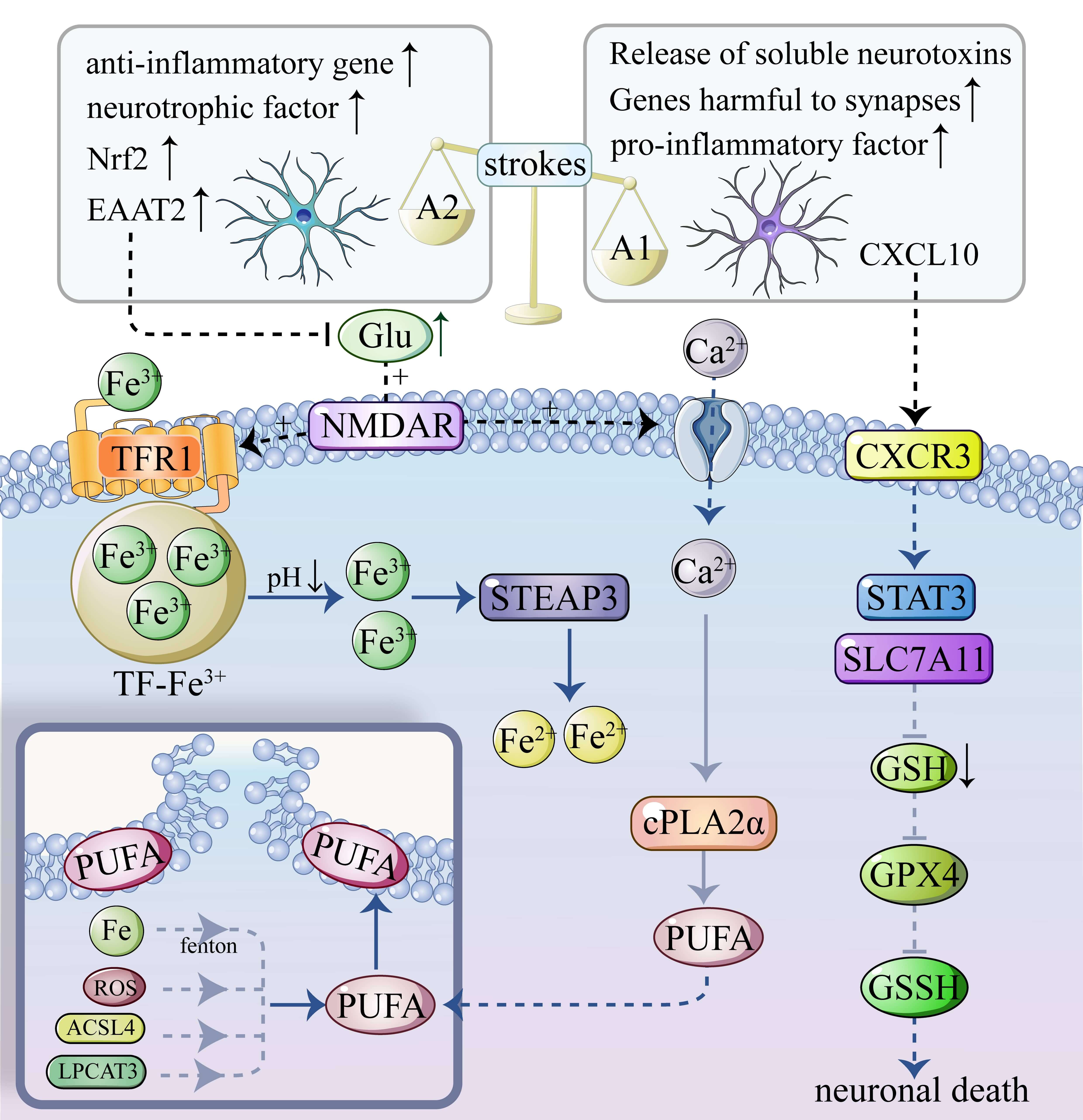 Fig. 1.
Fig. 1.
Ferroptosis in astrocytes and neurons. Fe3+ enters the cell through transferrin receptor 1 (TFR1) and binds to the transferrin (TF). Within the acidic environment of the endosome, Fe3+ is dissociated from TF and subsequently reduced to Fe2+, leading to intracellular iron accumulation. Iron promotes the biosynthesis of polyunsaturated fatty acids (PUFAs), leading to membrane lipid peroxidation, cell membrane disruption, and ultimately accelerated cell death. Following brain injury, elevated glutamate levels further enhance PUFA synthesis. Moreover, stroke not only increases the proportion of neurotoxic A1 astrocytes but also triggers neuroinflammatory responses and neuronal lipid peroxidation, thereby contributing to neuronal death. Type A2 astrocytes can inhibit the elevation of glutamate concentration, a mechanism mediated through the induction of anti-inflammatory factors, neurotrophic factors, Nrf2 activation, and upregulation of EAAT2. Neurotoxic A1 phenotype astrocytes promote the release of soluble neurotoxins and pro-inflammatory factors, as well as the upregulation of synapse-damaging genes. CXCL10 secreted by these cells inhibits the Xc– system, leading to reduced GSH synthesis, decreased GPX4 activity, and disruption of glutathione homeostasis, ultimately resulting in impaired redox balance. The figure is drawn by Adobe Illustrator 2022 (Adobe Inc, Located in San Jose, CA, USA). CXCL10, chemokine C-X-C motif chemokine ligand 10; GSH, glutathione; GPX4, glutathione peroxidase 4; EAAT2, excitatory amino acid transporter protein 2; Nrf2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lyso-phosphatidylcholine acyltransferase-3; STEAP3, six-transmembrane epithelial antigen of prostate 3; STAT3, transcription 3; SLC7A11, cystine/glutamate antiporter solute carrier family 7 member 11.
Disruption of copper ion homeostasis in vivo leads to cytotoxic effects and promotes cell death through multiple molecular mechanisms, including the accumulation of reactive oxygen species (ROS), induction of neuroinflammation, proteasome inhibition, and mitochondrial dysfunction. This newly discovered mode of regulatory cell death has been termed “cuproptosis”. Astrocytes play a critical role in maintaining copper homeostasis within the brain and are capable of mediating copper uptake, storage, and efflux. Astrocytes mediate the uptake of accumulated exogenous copper through the copper transporter protein receptor 1 (CTR1), and the internalized copper subsequently binds to intracellular glutathione and metallothioneins (MT) to maintain cerebral copper homeostasis [35]. A positive correlation has been reported [36] between serum copper levels and the prevalence of stroke in the general adult population, suggesting a potential association with impaired astrocytic function following cerebral ischemia. Astrocyte death disrupts copper homeostasis within the brain, resulting in the release of toxic copper ions into the extracellular fluid and subsequent neuronal cell death [37]. The accumulation of Cu2+ disrupts cellular redox homeostasis and leads to the overproduction of ROS. The presence of cytoplasmic oxidoreductases facilitates the interconversion between Cu+ and Cu2+, which catalyzes the formation of highly toxic hydroxyl radicals through the Fenton reaction, ultimately resulting in neuronal cell death (Fig. 2, Ref. [29]) [38].
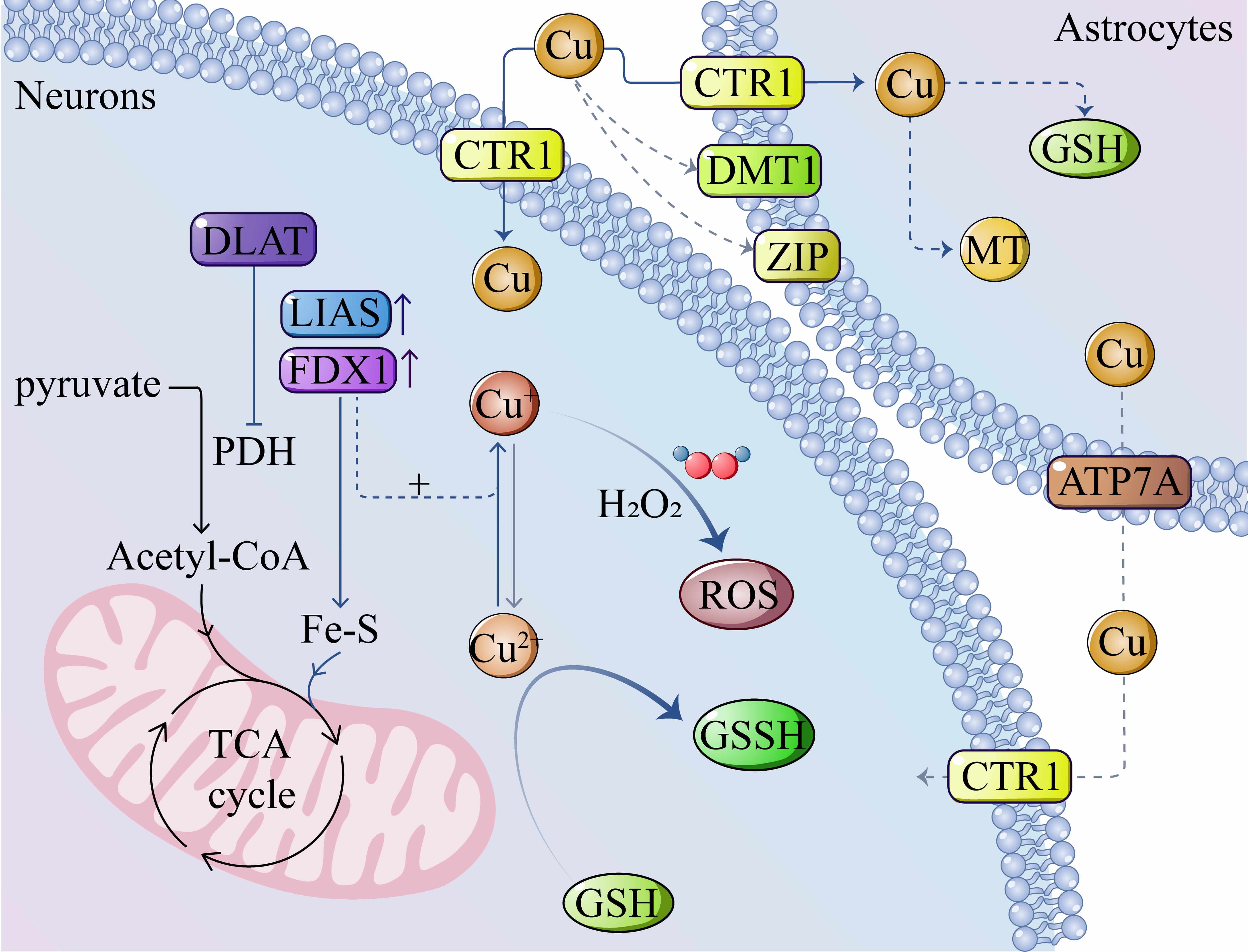 Fig. 2.
Fig. 2.
Mechanisms of cuproptosis in neurons. Copper is transported into the cell through CTR1 and undergoes redox interconversion between Cu+ and Cu2+ mediated by cytoplasmic oxidoreductases. The generated Cu+ reacts with hydrogen peroxide (H2O2), thereby accelerating the production of ROS. Simultaneously, Cu2+ oxidizes GSH to its disulfide form GSSH, thereby reducing intracellular GSH levels and disrupting ROS homeostasis. Ferredoxin 1 (FDX1) mediates the reduction of Cu2+ to the more toxic Cu+, thereby inhibiting Fe-S cluster protein synthesis, promoting mitochondrial protein aggregation, and causing Fe-S cluster loss, which ultimately blocks the tricarboxylic acid (TCA) cycle. Astrocytes take up copper through transporters such as CTR1, divalent metal transporter 1 (DMT1), and the Zrt/Irt-like protein (ZIP), and excess copper binds to GSH to form complexes. Upon binding to sulfhydryl-containing mitochondrial enzymes in the tricarboxylic acid (TCA) cycle, such as dihydrolipoamide acetyltransferase (DLAT), copper inhibits pyruvate dehydrogenase (PDH) activity, thereby disrupting the TCA cycle. Furthermore, the expression of lipoyl synthase (LIAS) and FDX1 reduces Cu2+ to the more toxic Cu+ species, which impairs Fe-S cluster protein synthesis, promotes mitochondrial protein aggregation, and induces Fe-S cluster loss [29], ultimately leading to complete blockade of the TCA cycle. The figure was drawn by Adobe Illustrator 2022. CTR1, copper transporter protein receptor 1; GSSH, oxidized glutathione; MT, metallothionein.
In ischemic stroke, glutamate-induced excitotoxicity plays a central role in neuronal death, while ammonia—a metabolic byproduct of glutamine [39]—exerts cytotoxic effects. Gaseous ammonia readily crosses the blood-brain barrier (BBB) and triggers a range of CNS pathologies, including astrocyte swelling, cerebral edema, neuroinflammatory responses, and other neurological disorders. It also exerts detrimental effects on neuronal cells, including protein denaturation, oxidative stress, and induction of apoptosis. The underlying mechanisms involve modulation of intracellular pH, induction of mitochondrial dysfunction, and enhanced production of ROS and reactive nitrogen species (RNS), which collectively contribute to its neurotoxic effects [39]. Following ischemic stroke, glutamate concentrations markedly increase, while ammonia—generated from glutamine—accumulates in both astrocytes and neurons. This pathological cascade induces oxidative stress, impairs TCA cycle activity in neuronal and glial cells, and ultimately leads to mitochondrial dysfunction [40, 41].
Most extrahepatic organs lack a complete urea cycle, and glutamine is a
temporary storage form of waste nitrogen. Astrocytes constitute the primary
cellular target of ammonia toxicity, as intracellular accumulation of ammonia
triggers excessive glutamine synthesis. This pathophysiological process results
in astrocytic swelling and activation of apoptotic signaling pathways [42],
ultimately leading to secondary neuronal dysfunction. Angelova et al.
[43] demonstrated that even low concentrations of ammonia can induce neuronal
death, either directly or indirectly, while high concentrations of ammonia were
shown to impair phagocytic activity in glial cells and promote apoptosis via
activation of the NF-
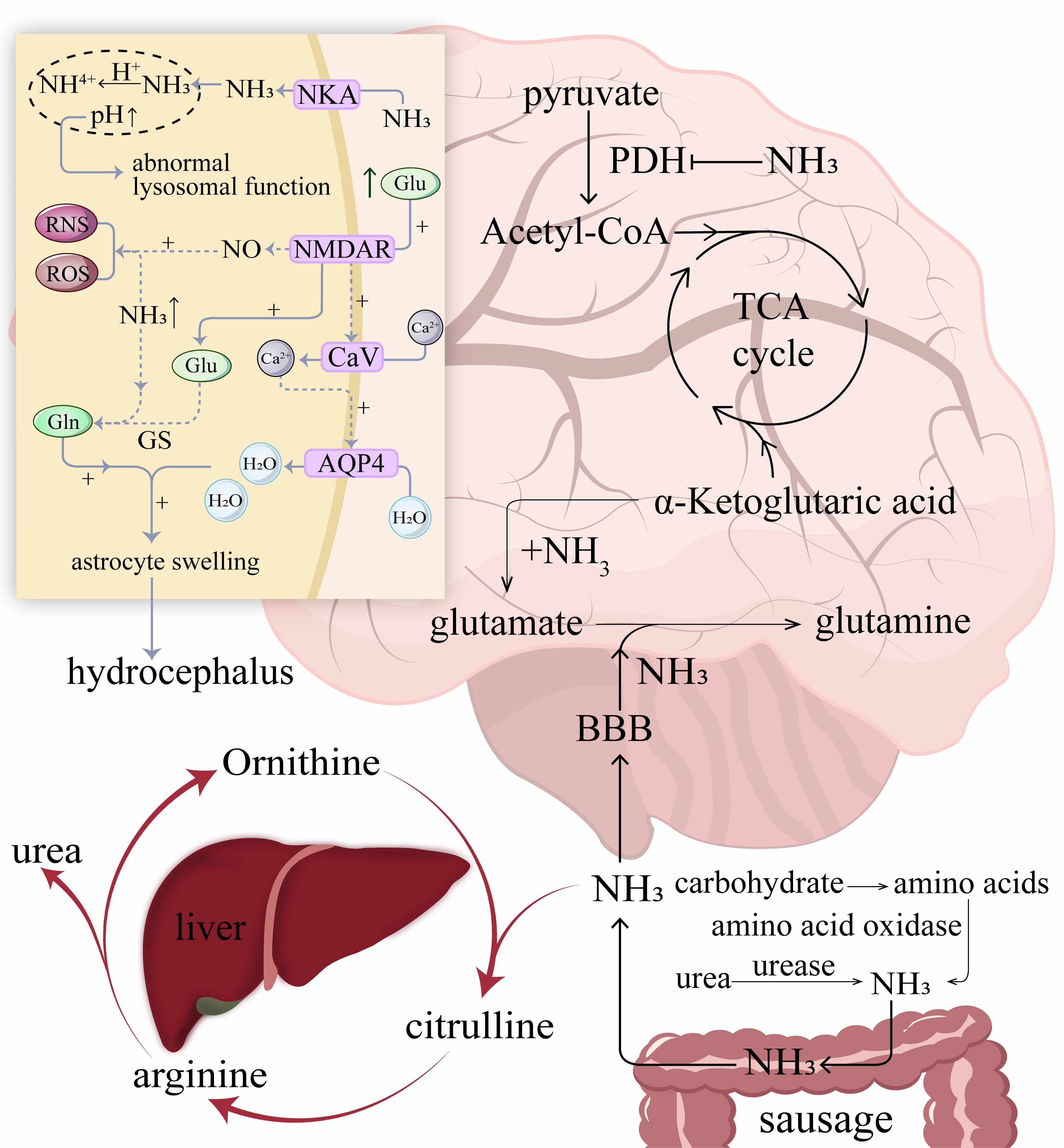 Fig. 3.
Fig. 3.
Mechanisms of ammonia-induced death in neurons. Elevated
concentrations of ammonia (NH3) penetrate the BBB and enter neural cells,
particularly astrocytes, where it inhibits pyruvate dehydrogenase activity and
disrupts the cerebral TCA cycle. Concurrently, ammonia reacts with
In the CNS neural stem cells (NSCs) generate intermediate progenitor cells (IPCs) through continuous self-renewal and asymmetric division [47]. However, the differentiation of neurons is restricted to the embryonic developmental stage. The capacity to generate new neurons has recently been identified in specific regions of the adult brain [48, 49]. Following stimulation, cells become activated and gain self-renewal capabilities, acquiring NSC-like properties and the ability to differentiate into all major cell types of the central nervous system. This phenomenon is referred to as the stem cell response. The cellular microenvironment in which stem cells reside is termed the neurogenic niche, comprising multiple cell populations capable of modulating the specific microenvironment of NSCs [50]. If a cell is transplanted to this site, it may acquire stem cell properties. The neurogenic niches identified in the human brain include the subventricular zone (SVZ) of the lateral ventricles, the subgranular zone (SGZ) of the dentate gyrus, and the striatum [51], all of which are characterized by the abundant presence of astrocytes and NSCs. NSCs exhibit heterogeneity in their differentiation potential. For instance, SVZ NSCs typically differentiate into olfactory bulb interneurons and callosal oligodendrocytes, while SGZ NSCs give rise to dentate granule neurons and astrocytes. Notably, when NSCs from both regions were cultured in vitro under high-concentration growth factor conditions, they demonstrated the capacity to generate cells across all three neural lineages [52]. This suggests that the differentiation potential of neural stem cells is constrained by the neurogenic niche in vivo. With the exception of certain neurogenic niches, most regions of the human brain lack the intrinsic environmental conditions necessary for neurogenesis.
Signaling pathways such as Notch and Sonic Hedgehog (SHH) play critical roles in the differentiation and development of NSCs. SHH signaling has been shown to regulate neurogenesis in both SVZ and SGZ NSCs, promoting increased neurogenesis and cell proliferation in these regions through SHH overexpression [53]. The Notch signaling pathway is a critical regulatory system that determines cell fate by modulating processes such as differentiation, proliferation, and apoptosis. Its activation promotes astrogenesis in neural progenitor cells. Namihira and Nakashima [54] demonstrated that neurons are capable of activating Notch signaling in NSCs. Activation of this pathway induces demethylation of the astrocyte-specific glial fibrillary acidic protein (GFAP) promoter, thereby enabling NSCs to differentiate into astrocytes. Furthermore, Notch signaling enhances the self-renewal and proliferative capacity of NSCs. Following stroke, diminished Notch1 signaling in astrocytes activates a potential neurogenic program within astrocytes located in the striatum and medial cortex [50]. Moreover, the overexpression of specific transcription factors, including NeuroG2, NeuroD1, and Tbr-1/2, has been demonstrated to play a crucial role in neurogenesis [55], offering valuable insights for neuronal replacement strategies.
CNS neurons exhibit limited capacity for regeneration following injury, typically undergoing cellular senescence or apoptosis. Consequently, the brain is unable to restore normal function through neuronal self-renewal [56]. Neurogenesis derived from adult neural stem cells is spatially confined to the SVZ of the lateral ventricular wall and the SGZ of the hippocampal dentate gyrus [57]. Astrocytes, progeny of NSCs, and mature neurons constitute the principal cellular components of the neurogenic niche [58]. Neural progenitor cells exhibiting high proliferative capacity have been shown to express GFAP, a specific marker for astrocytes [59]. Furthermore, ultrastructural analysis of these cells under an electron microscope reveals characteristic features of astrocytes, including bundled intermediate filaments and intercellular gap junctions [60]. It is therefore reasonable to hypothesize that a lineage crossover exists between these cells and astrocytes, which establishes a prerequisite for the neuronal differentiation of astrocytes. A cell type widely present in the nervous system is the radial glial cell (RGC) [61]. Notably, radial glial cells also function as neuronal precursors, thereby suggesting the potential for astrocyte-to-neuron conversion [62].
Astrocytes have been demonstrated to undergo reprogramming into neuronal phenotypes (Fig. 4). Interleukin-6 (IL-6) and leukemia inhibitory factor (LIF), secreted by microglia, are capable of inducing astrocyte dedifferentiation [63]. Laywell et al. [64] demonstrated that astrocytes residing in the neurogenic niche, specifically the SVZ, possess the capacity to generate neurospheres in the adult brain and undergo neuronal differentiation. Although the microenvironment of the neurogenic niche has the capacity to induce astrocyte-to-neuron conversion, signals originating solely from this niche are insufficient to sustain astrocytic de-differentiation [65]. Whether this potential is constrained by the microenvironment of the neurogenic niche in which these cells reside, or instead arises from fundamental differences between them and parenchymal astrocytes, remains to be fully elucidated through further investigation. Furthermore, studies have demonstrated [66, 67] that disparities exist in the efficiency of astrocyte reprogramming between gray and white matter regions of the human brain. Under NeuroD1 induction, neurons derived from astrocytes were readily detectable in gray matter, whereas their detection in white matter proved to be significantly more challenging. In the experiments conducted by Liu et al. [68], it was observed that Dlx2 overexpression effectively reprogrammed astrocytes into neurons within the gray matter striatum, whereas the white matter striatum exhibited only partial reprogramming, which was concurrently associated with neuroinflammatory responses. Further investigation into the influence of specific molecules or signaling pathways present in the microenvironment of both white and gray matter—either on astrocyte reprogramming or on the survival of “regenerated” neurons—may offer valuable insights into identifying the optimal environmental factors that support neuronal “regeneration”.
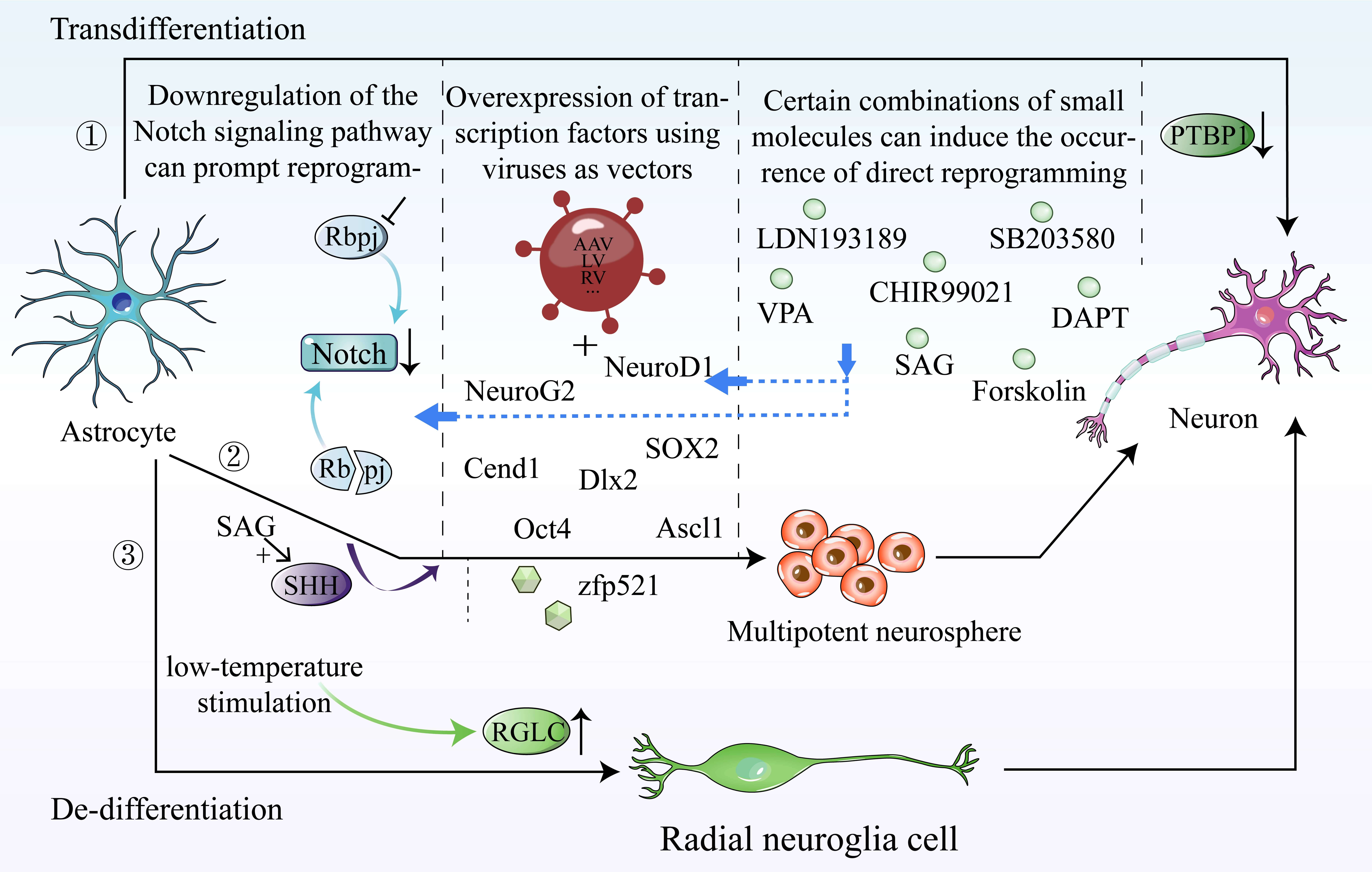 Fig. 4.
Fig. 4.
Astrocytes can be converted into neurons through two distinct reprogramming pathways: direct and indirect reprogramming. Direct reprogramming (line ①), also referred to as transdifferentiation, involves mechanisms such as inhibition of Notch signaling (by inhibiting or deleting the transcription factor Rbpj (immunoglobulin kappa J region)), viral vector-mediated overexpression of transcription factors, and induction by small molecules (the underlying mechanism entails the promotion of transcription factor overexpression and the suppression of the Notch signaling pathway). In contrast, indirect reprogramming (line ②) entails an intermediate step in which astrocytes are first transformed into pluripotent neural progenitor spheres. This pathway can also be mediated by downregulation of Notch signaling, overexpression of key transcription factors, and activation of Sonic hedgehog (SHH) signaling. Line ③ demonstrates how astrocytes can be gently guided to dedifferentiate into radial glial cells through freeze-thaw stimulation. The figure was drawn by Adobe Illustrator 2022. SAG, Smoothened agonist; SHH, Sonic Hedgehog; SOX2, Sry-box; RGLC; DAPT, N-[N-(3,5-difluorophenacetyl)-L-alanyl]- S-phenylglycine t-butyl ester; VPA, valproic acid; Oct4, also known as POU5F1; Ascl1, achaete-scute homolog-like 1; zfp521, Zinc Finger Protein 521; Cend1, neuronal differentiation factor; Dlx2, distal-less homeobox 2; PTBP1, polypyrimidine tract-binding protein 1.
Cell reprogramming techniques are now widely utilized for neural regeneration in the central nervous system [69]. Fibroblasts have been demonstrated to undergo direct reprogramming into neurons in vitro [70, 71, 72]. However, the transdifferentiation of fibroblasts into neurons involves a complex cross-lineage transformation, which significantly elevates the technical challenges associated with this process [73]. Whereas astrocytes are closely associated with neurons and share a common progenitor, the radial glial cells are widely distributed throughout the brain [74, 75]. Moreover, reactive astrocytes exhibit rapid proliferation following brain injury and demonstrate neural stem cell-like potential [76]. Astrocytes therefore represent promising candidates for cellular reprogramming; however, this capability has so far been observed only in specific regions of the brain. Astrocytes can be reprogrammed through both dedifferentiation and transdifferentiation. Dedifferentiation refers to the process in which reactive astrocytes first undergo dedifferentiation to generate self-renewing, pluripotent neurospheres [77], or are induced in vitro to differentiate into radial glial-like progenitor cells, which subsequently mature into functional neurons [78]. Transdifferentiation, also termed direct reprogramming, describes the process by which astrocytes bypass the intermediate pluripotent state and directly convert into neurons [79].
Astrocytes possess intrinsic neural stem cell-like properties; however, they lack an appropriate in vivo microenvironment to activate this potential under physiological conditions [80]. Astrocyte-to-neuron conversion predominantly occurs during the process of brain wound healing [81]. The reinitiation of astrocyte proliferation following brain injury, along with the capacity of distinct astrocyte subpopulations to generate neurospheres, indicates that certain signaling molecules present in the post-injury brain environment may play a role in activating neural stem cell potential. Numerous studies have demonstrated [82, 83, 84] that astrocytes exhibit substantial proliferative capacity and are capable of forming neurospheres in vitro following mechanical puncture or ischemic injuries. As a result, they are increasingly regarded as undergoing an injury-induced dedifferentiation process. During this process, astrocytes initially de-differentiate into neuronal progenitor-like cells before differentiating into neurons (Fig. 5) [85]. However, under in vivo conditions, their differentiation remains confined to the glial lineage and does not extend to neuronal conversion [86].
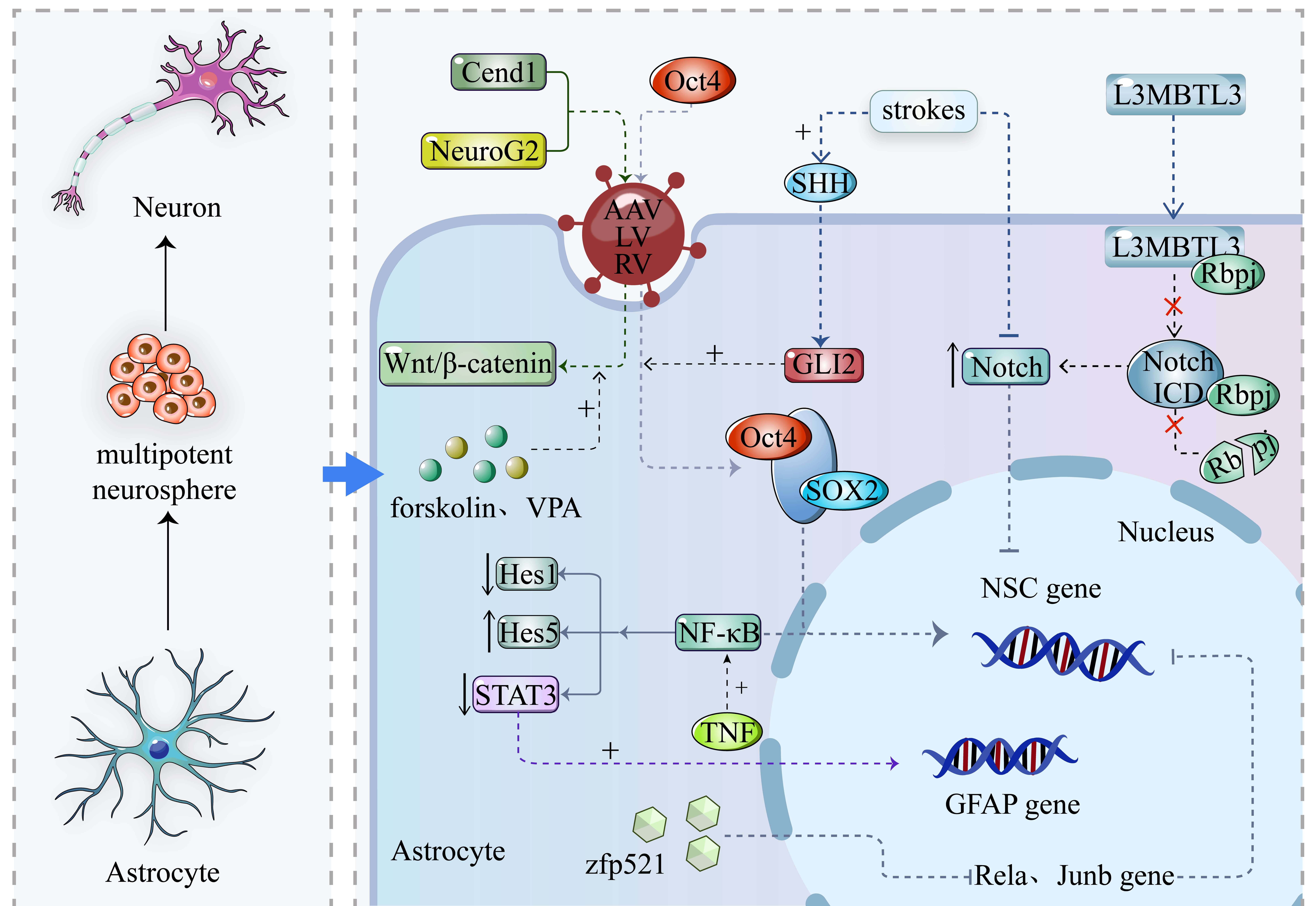 Fig. 5.
Fig. 5.
Mechanisms of astrocyte dedifferentiation into neurospheres.
Viral vectors mediate overexpression of NeuroG2 and Oct4 in astrocytes, whereby
NeuroG2 activates the
Astrocytes exhibit robust upregulation of SHH signaling following brain injury [87]. SHH is a critical signaling protein that serves as a key regulator of neural development, mediates injury-induced hippocampal neurogenesis, promotes the proliferation and differentiation of stem cells, and contributes to the maintenance of the neurogenic niche of neural stem cells [88, 89]. SHH serves as a key inductive signal that activates stem cell-like responses in reactive astrocytes [90, 91]. Sirko et al. [92] demonstrated that SHH signaling can directly target astrocytes and induce them to exhibit neural progenitor cell potential. However, upregulation of SHH signaling alone is insufficient to induce astrocytes to undergo dedifferentiation. Recently, Yang et al. [93] demonstrated that the efficiency of astrocyte reprogramming into neural stem cells can be significantly enhanced through the combined application of ectopic expression of a single transcription factor, Oct4 (also known as POU5F1), and SHH signaling activation. These findings suggest that SHH signaling may serve as an auxiliary cue to improve the efficiency of astrocyte-based cellular reprogramming. Aravantinou-Fatorou et al. [94] demonstrated that astrocytes are capable of forming pluripotent neurospheres under conditions of NeuroG2 and Cend1 dual transduction, exhibiting neural stem cell (NSC)-like proliferative and differentiation capacities. Additional transcription factors, including zinc-finger nuclear protein (Zfp521) [95, 96], SOX2 (Sry-box), and NANOG, have also been shown to induce the dedifferentiation of astrocytes into neural stem cells [97].
The initiation of neurogenic responses in astrocytes is modulated by Notch signaling. Activation of Notch signaling suppresses the differentiation of adult neural progenitor cells, while its inactivation enhances early neuronal differentiation [98]. Shimada et al. [99] demonstrated that reactive astrocytes derived from the post-stroke environment are capable of undergoing dedifferentiation into Rad-NSCs (astrocyte-derived neural stem cell spheroids), and that the knockdown of Presenilin 1 (PS1) and the Notch 1 signaling pathway can regulate the proliferation and self-renewal capacity of neurospheres in vitro [100]. Notch signaling in astrocytes is downregulated following brain injury; however, only a limited number of astrocytes undergo dedifferentiation. The Notch signaling pathway primarily transduces signals via the transcription factor Rbpj [101], which interacts with the Notch intracellular domain (Notch ICD) to activate downstream target genes [102]. Xu et al. [103] identified a novel Rbpj-interacting factor, L3MBTL3, which inhibits the Notch signaling pathway by preventing the binding of Notch ICD to Rbpj through competitive interaction between L3MBTL3 and Notch ICD. Notch signaling can also be blocked by specific deletion of the transcription factor Rbpj [51], thereby promoting enhanced neurogenesis in striatal astrocytes.
Certain signaling molecules and growth factors generated following brain injury
have been shown to activate neural stem cell potential, synergistically interact
with astrocyte dedifferentiation responses, and enhance neurogenesis [47].
Examples include glial cell-derived neurotrophic factors (GDNF, BDNF), vascular
endothelial growth factor (VEGF), and fibroblast growth factor 2 (FGF-2) [104].
Activation of the NF-
The transplantation of cells that have been reprogrammed in vitro and subsequently differentiated into CNS lineages still faces significant challenges in terms of overcoming immune rejection and achieving functional integration. The transdifferentiation approach enables direct in vivo reprogramming into neurons without the need for exogenous cell transplantation, thereby minimizing immune rejection associated with such procedures [108, 109]. A widely employed reprogramming strategy involves the delivery of neuronal transcription factors to astrocytes via viral vectors to induce their ectopic expression. Several types of viral vectors have been utilized for this purpose, including lentiviruses (LVs), retroviruses (RVs), and adeno-associated viruses (AAVs) [110, 111].
Transcription factors (TFs) serve as key regulatory molecules that govern the fate determination of neural cells [112]. They represent critical regulatory factors that modulate the expression of multiple genes within neurons and orchestrate complex processes of neuronal growth and regeneration. TFs exhibit differential expression patterns across various neuronal subtypes [113, 114, 115]. For example, NeuroG2 is predominantly expressed in glutamatergic neuronal progenitors, whereas Ascl1 shows preferential expression in GABAergic interneuronal progenitors [112], contributing critically to the regulation of neuronal diversity. A substantial body of research has established that the overexpression of either single or combined transcription factors can directly reprogram astrocytes into neurons, bypassing the pluripotent neurosphere phase. Furthermore, distinct neuronal subtypes can be generated through the application of specific transcription factors or their combinations [116], as summarized in Table 1 (Ref. [68, 94, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145]).
| Transcription factors | Site of occurrence | Type of neuron induced | References |
| NeuroG2 | cortex, midbrain, spinal cord | glutamatergic/dopaminergic neurons | [117, 118, 119, 120, 121] |
| NeuroD1 | dorsal horn, striatum | glutamatergic neuron | [68, 122, 123, 124, 125, 126, 127] |
| Paired box protein 6 (Pax6) | striatum | / | [128, 129] |
| Mash1 | cerebral cortex | / | [118, 121, 130] |
| Brain-2 (Brn2) | / | glutamatergic/gamma-aminobutyric acid-ergic (GABAergic) neurons | [131, 132] |
| Achaete-scute homolog-like 1 (Ascl1) | dorsal midbrain | glutamatergic/GABAergic neurons | [118, 119, 133, 134] |
| Distal-less homeobox 2 (Dlx2) | cortex, striatum | GABAergic neurons | [68, 117, 121] |
| SOX2 | spinal cord | glutamatergic neuron | [135, 136, 137, 138] |
| Cend1 | / | GABAergic neurons | [139] |
| Zinc Finger Protein 521 (Zfp521) | spinal cord | / | [140] |
| NeuroG2+Mash1 | cortex | glutamatergic neuron | [121, 141] |
| Neuro G2+Cend1 | cortex | glutamatergic/GABAergic/dopaminergic neurons | [94, 139, 142] |
| NeuroG2+Ascl1 | cerebral cortex | glutamatergic/GABAergic neurons | [119, 141] |
| NeuroG2+ Islet-1 (ISl1) | spinal cord | motor neuron | [143] |
| NeuroD1+NeuroG2 | spinal cord | / | [144] |
| NeuroD1+Dlx2 | striatum | GABAergic neurons | [122, 145] |
| NeuroD1+Ascl1+LIM homeobox transcription factor 1 alpha (LMX1A)+miR218 (MicroRNAs) | striatum | dopaminergic neurons | [134] |
Recently, a viral vector-free approach has been developed for cell
reprogramming. This strategy utilizes a combination of small-molecule compounds,
as detailed in Table 2 (Ref. [146, 147, 148, 149, 150, 151, 152, 153]). The mechanism underlying small
molecule-mediated cell reprogramming may involve the activation of transcription
factor overexpression, such as NeuroD1/2 and NeuroG2, or the modulation of
specific signaling pathways, including Notch, transforming growth factor beta
(TGF-
| Small molecule combination | Type of neuron induced | Mechanisms involved | References |
| LDN193189, SB431542, TTNPB, Thiazovivin (Tzv), CHIR99021, valproic acid (VPA), DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]- S-phenylglycine t-butyl ester), SAG (Smoothened agonist), purmorphamine (Purmo) | glutamatergic neuron | Transcriptional activation of NeuroD1 and NeuroG2, DNA methylation in the promoter region of the DFAP gene | [146, 148] |
| DAPT, CHIR99021, SB431542, LDN193189 | glutamatergic neuron | Upregulate NeuroD1 and NeuroG2, and regulate the Notch, GSK-3 |
[149] |
| LDN193189, CHIR99021 | GABAergic neurons | Activation of NeuroD1/2, NeuroG2, MytLl transcription | [147] |
| miR-124, ruxolitinib, SB203580, Forskolin | glutamatergic/cholinergic neurons | Inhibition of Hes1 expression | [150] |
| DBcAMP, ISX9 (isoxazole 9), Forskolin, CHIR99021, I-BET151, Y-27632 | glutamatergic/GABAergic neurons | Upregulation of NeuroG2, NeuroD1, Scrt1, Pou4f1 | [151] |
| VPA, Chir99021, Repsox, Forskolin, i-Bet151 (BET inhibitors), ISX-9 | glutamatergic neuron | Decreased expression of GAP and ALDH1L1, up-regulation of neuron-enriched genes such as MAP2 and NEUN | [152] |
| Kenpaullone, Forskolin, Y-27632, purmorphamine | motor neuron-like cells | NeuroG2, NeuroD1, MytLl, HB9, ISL1, and SMN gene expression were upregulated and GFAP expression was downregulated | [153] |
GSK-3
The administration sequence and dosage of effective small molecule combination protocols require systematic investigation in practical research settings, guided by the functional roles of individual molecules during neuronal differentiation and reprogramming processes. Commonly utilized small molecule compounds include ISX9, i-Bet151, and CHIR99021, which are capable of activating neuron-specific genes and promoting neuronal differentiation and maturation. The underlying mechanisms are summarized in Table 3 (Ref. [66, 149, 158, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179]). MicroRNAs (miRs) serve as potent facilitators of small molecule-mediated reprogramming. For instance, miR-124 [180] and miR-365 have been demonstrated to synergize with small molecules in regulating the astrocyte-to-neuron conversion process, thereby enhancing reprogramming efficiency [181, 182] and playing a crucial role in this approach.
| Small molecule | Full name | Mechanism | References |
| Nurr1 | Nuclear receptor-related transcription factor 1 | Regulation of inflammatory genes and inhibition of inflammatory signaling in microglia and astrocytes | [66] |
| VEGF | vascular endothelial growth factor | Enhancement of Pax6 expression in astrocytes through the MAPK/Erk pathway increases proliferation and differentiation of neural precursor cells | [158, 159] |
| VPA | valproic acid | Enhancement of Oct4, SOX2 reprogramming efficiency for neurogenesis and neuronal maturation | [160, 161, 162] |
| Forskolin | / | Inhibits ALK5, reduces lipid peroxidation, promotes neural reprogramming efficiency, modulates neuronal morphology, and promotes axonal regeneration | [163, 164] |
| Bcl-2 | B cell lymphoma-2 | Reduces oxidative stress damage | [165] |
| BNDF | brain-derived neurotrophic factor | Promotes neuronal development and regeneration | [166] |
| ISX9 | Isoxazole 9 | Activation of neuron-specific genes by Ca2+ in-flow activation of CaMK phosphorylation | [167, 168] |
| i-Bet151 | BET inhibitors | Inhibition of astrocyte gene expression | [169, 170, 171] |
| CHIR99021 | / | Selective inhibition of GSK3 |
[149, 167, 172, 173, 174] |
| miR-124 | MicroRNA-124 | Regulation of the AAK1/Notch pathway promotes neural stem cell (NSC) proliferation and differentiation | [175] |
| PTB | polypyrimidine tract-binding protein | Regulates nerve growth and differentiation | [176] |
| DAPT | N-[N-(3,5-difluorophenacetyl)-L-alanyl]- S-phenylglycine t-butyl ester | Targeting |
[177] |
| SB431542 | / | Inhibition of TGF- |
[172, 178] |
| Ginsenoside Rg1 | / | Suppressing Notch/Stat3 signal pathway | [179] |
Notch signaling plays a critical role in neurogenesis and tissue regeneration. Following stroke, Notch signaling activity in astrocytes is significantly diminished, and inhibition of this pathway can induce astrocytes to adopt neurogenic programs [183, 184]. Elevation of NeuroG2 and Ascl1 signaling in astrocytes leads to suppression of Notch signaling, while inhibition of Notch1 signaling through DAPT treatment [185] or lentiviral vector-mediated shRNA delivery has been shown to significantly enhance cell reprogramming efficiency [84]. Furthermore, Hu et al. [186] observed regional variations in Notch signaling expression across the human cerebral cortex, cerebellum, and spinal cord. Notch signaling was found to be expressed at lower levels in the cortex compared to higher levels in the spinal cord. Consequently, cortical astrocytes exhibited a greater propensity for reprogramming than those derived from the spinal cord.
Polypyrimidine tract-binding protein (PTB) is an RNA-binding protein with diverse biological functions, serving as a key splicing regulator that modulates neuronal developmental processes through its involvement in axon formation, synaptogenesis, and apoptosis of neurons [187]. PTBP1 serves as a critical neuronal reprogramming factor. A substantial body of research has demonstrated that the knockdown or deletion of PTBP1 can directly convert fibroblasts, Müller glial cells (MG), astrocytes, and other GFAP-expressing cell types into functional neurons [188, 189, 190, 191]. However, controversy persists concerning the role of PTB deletion or knockdown in neural reprogramming. Several replication studies [192, 193, 194, 195, 196] have produced findings that contradict those previously reported [191]. Specifically, PTBP1 knockdown in astrocytes located in the substantia nigra or striatum did not generate GFPNeuN-positive cells (indicative of astrocyte-derived neurons). The GFPNeuN signals observed in the study by Qian et al. [191] may instead represent endogenous neurons activated by viral vector-mediated gene leakage. Further experimental validation is required to determine whether PTBP1 knockdown can indeed induce direct in situ reprogramming of astrocytes.
Numerous instances of neuronal death or irreversible damage are observed in various central nervous system (CNS) disorders. Astrocytes, which maintain close functional interactions with neurons, have been found to actively participate in the neuronal death process. Following brain injury, astrocytes adopt two distinct reactive phenotypes: A1 and A2. A2 phenotype astrocytes exert neuroprotective effects and mitigate neural damage through the secretion of neurotrophic and anti-inflammatory factors. In contrast, A1 phenotype astrocytes exhibit neurotoxic properties and release neurotoxins as well as pro-inflammatory mediators, which contribute to accelerated neuronal demise. Following traumatic brain injury, the dysregulation of iron, copper, and ammonia homeostasis in the central nervous system triggers a cascade of pathological events, including lipid peroxidation, inflammatory responses, and other related processes, ultimately leading to neuronal apoptosis. Therefore, identifying effective strategies and elucidating the underlying mechanisms to rescue compromised and degenerating neurons represent critical objectives in neuroscience research. Modulating astrocytic polarization by inhibiting the A1 phenotype while enhancing the expression of the A2 phenotype has emerged as a promising therapeutic strategy for managing CNS disorders and mitigating neuroinflammatory processes. Specific protein molecules, including transmembrane protein 164 (TMEM 164), Krüppel-like transcription factor 4 (KLF-4), and Homer scaffolding protein 1 (Homer 1), have demonstrated the capacity to effectively suppress A1 astrocytic activation and facilitate the polarization toward the neuroprotective A2 phenotype [197, 198].
Neuronal regeneration represents a promising therapeutic approach in the treatment of neurological disorders. Among current interventions, stem cell therapy has become a more widely adopted strategy. Commonly utilized stem cell sources include fetal ventral midbrain tissue, embryonic stem cells, neural stem cells, and induced pluripotent stem cells (iPSCs) [199]. However, this therapeutic approach faces several challenges, including immune incompatibility, ethical concerns associated with fetal tissue utilization, and the potential risk of tumor formation [200], all of which impose significant limitations on its clinical application. Current advances in cellular reprogramming involve utilizing astrocytes as the source cells and converting them into neurons through the modulation of transcriptional networks or signaling pathways. This process encompasses both indirect and direct reprogramming strategies. Astrocytes play a pivotal role in regulating neurogenesis, primarily functioning as NSCs or components of the neurogenic niche during this process [201]. In addition to astrocytes, oligodendrocytes [202, 203], and microglia [124, 204] have also been demonstrated to undergo neuronal conversion. Reactive astrocytes are considered an ideal cell source for neuronal regeneration due to their high proliferative potential, close developmental proximity to neuronal lineages, and functional involvement in glial scar formation [150]. Direct reprogramming techniques enable cells to bypass intermediate pluripotent states and be converted directly into specific neuronal subtypes, both in vivo and in vitro [205]. By overcoming the limitations associated with stem cell-based transplantation—such as immunogenic rejection, tumorigenic potential, and ethical concerns—this approach heralds a new era in the treatment of neurological disorders.
A widely utilized strategy in cellular reprogramming involves modulating gene expression through the application of defined combinations of transcription factors [206]. The extent of neuronal integration following transplantation is influenced by multiple factors, including the origin of astrocytes, the specific transcription factor employed, the anatomical site of transplantation, and the developmental stage of the recipient animal [119]. Recently, a promising reprogramming strategy has emerged through the application of chemical reprogramming techniques that utilize small-molecule compounds [148, 207, 208], which represent non-viral and non-integrative methodologies. This approach circumvents the need for viral vectors in transgene delivery and enables the withdrawal of small molecules from the culture environment upon completion of the reprogramming process. For instance, induction with VCRs (valproic acid, CHIR99021, Repsox) facilitates the conversion of somatic cells into neural progenitor cells (NPCs) without requiring exogenous gene expression [209]. Human fibroblasts were successfully converted into neurons through the application of a cocktail of seven small molecules, including VCRF (VCR + trichostatin) as well as SP600125, GO6983, and Y-27632 [167, 210]. Furthermore, small molecules offer several advantages, such as low cost, ease of acquisition and storage, controllability, and favorable cellular permeability [207]. Currently, however, reprogramming with small molecules remains inefficient and involves complex molecular mechanisms that are not yet fully understood. The biological processes in which these molecules participate and mediate in vivo have not been clearly defined, and in vivo small molecule reprogramming continues to pose significant challenges, necessitating further research to achieve more precise and efficient outcomes.
Cell reprogramming technology offers novel perspectives for the “regeneration” of neurons within the brain and holds significant potential for the treatment of neurological disorders such as stroke. In vitro reprogramming can be initiated with relatively homogeneous cell populations, allowing for direct microscopic observation of cell fate transitions. In contrast, in vivo reprogramming takes place within complex neural microenvironments containing diverse cell types. Therefore, it is crucial to employ well-established and rigorous lineage tracing methodologies to monitor the reprogramming process and validate the cellular origin of induced neurons [211]. The potential of neuronal repair in the human brain is immense; however, current cell reprogramming techniques still face several limitations and challenges. First, the efficiency of direct reprogramming still requires significant improvement. Following cerebral ischemia, both astrocytes and neurons undergo damage and cell death, which not only limits the conversion efficiency of astrocytes into neurons but also fails to enhance reprogramming efficiency even when viral vectors encoding the anti-apoptotic cytokine Bcl-2 are employed [212]. Second, the phagocytosis mediated by activated phagocytes during the neuroinflammatory response following cerebral ischemia may lead to the degradation of delivery vectors, thereby preventing them from exerting therapeutic effects. Moreover, the human brain has a limited capacity for viral vector tolerance [213], necessitating strict control over their dosage. Thirdly, the selection of an appropriate viral vector delivery system is a critical consideration in the design of gene delivery strategies [214]. Fourth, under reprogramming stimulation conditions, astrocyte-derived neurons fail to achieve complete functional maturation or establish functional presynaptic outputs [141]. This represents a critical barrier that must be addressed in contemporary neural repair research. Finally, cellular reprogramming involves the reconfiguration of transcriptional networks and is not yet a fully established transgenic technique. The underlying molecular mechanisms require further investigation to refine and optimize the reprogramming model, with the aim of identifying more effective therapeutic strategies for clinical brain injury. Promoting the “regeneration” of brain neurons has the potential to significantly alleviate neurological dysfunction caused by extensive neuronal loss following cerebral ischemia, and may serve as a valuable reference for the clinical treatment of central nervous system disorders.
The dual role of astrocytes in relation to neurons highlights their complex involvement in the nervous system. On one hand, under pathological conditions such as cerebral ischemia, type A1 astrocytes become abnormally activated and release inflammatory mediators and oxidative stress molecules, which can induce neuronal death. Moreover, the excessive proliferation of reactive astrocytes that form glial scars can also impede neural regeneration. On the other hand, astrocytes have the potential to be reprogrammed into neurons, thereby promoting nerve regeneration. By modulating specific transcription factors (such as NeuroD1/2 and NeuroG2), astrocytes can be induced to undergo neuronal conversion. This capability offers a novel therapeutic avenue for neurological disorders. The balance between these dual functions may be influenced by the regulation of microenvironmental signaling pathways, and a comprehensive understanding of the underlying mechanisms holds significant value for advancing nerve repair research.
Conceptualization and Methodology: YJL; Validation and Writing—Original Draft: YJL, CY and WXC; Substantial contributions to the conception or design of the work: YJL, CY, WXC, MRL, HW, LYC; Formal Analysis: YJL, CY, and MRL; Drawings and Tables: YJL, CY, MRL; Writing—Review & Editing: YJL, CY, HW, and LYC. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the National Natural Science Foundation of China (82360782), Jiangxi Province Natural Science Foundation (20232BAB206170), Science and technology Project of Jiangxi Administration of Traditional Chinese Medicine (2021A333), Science and technology Project of Jiangxi Provincial Health Commission (202211412), Jiangxi University of Chinese Medicine Science and Technology Innovation Team Development Program (CXTD22002).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.