








 , Hongxiang Hong 1, Guanhua Xu 1,3, Mingjie Xia 3, Tianyi Wang 1,2, Zheng Zhou 1,2, Jiale Huang 1,2, Qihao Fu 1,2, Zhiming Cui 1,3,*
, Hongxiang Hong 1, Guanhua Xu 1,3, Mingjie Xia 3, Tianyi Wang 1,2, Zheng Zhou 1,2, Jiale Huang 1,2, Qihao Fu 1,2, Zhiming Cui 1,3,*
1 Department of Spinal Surgery, The Second Affiliated Hospital of Nantong University, 226001 Nantong, Jiangsu, China
2 Medical School of Nantong University, 226001 Nantong, Jiangsu, China
3 Research Institute for Spine and Spinal Cord Disease of Nantong University, 226001 Nantong, Jiangsu, China
Abstract
Spinal cord injury (SCI) constitutes a profoundly debilitating neurological disorder precipitating motor and sensory function impairment. Curtailing microglia-driven neuroinflammation alongside oxidative stress proves indispensable for efficacious SCI patient management. Poliumoside (POL), a phenylethanoid glycoside molecule, manifests anti-inflammatory, antioxidant, and neuroprotective capacities. Nevertheless, documentation concerning its SCI therapeutic efficacy remains sparse.
Systemic drug toxicity for two POL dosages (15 mg/kg, 30 mg/kg) was evaluated across multiple organs. An SCI murine model was generated employing Allen’s technique. Mice received random assignment into sham, SCI, and SCI+POL cohorts. Intraperitoneal POL administration ensued for 7 consecutive days post-trauma. Histological staining probed tissue and cellular alterations. Functional recuperation was assessed via the Basso Mouse Scale (BMS), hindlimb flexion scoring, and footprint examination. RNA sequencing (RNA-seq) explored POL’s therapeutic impact within SCI. Immunofluorescence detected the axonal marker neurofilament 200 (NF200), myelin marker myelin basic protein (MBP), and the glial scar indicators ionized calcium-binding adapter molecule 1 and glial fibrillary acidic protein (IBA1, GFAP); Western blot (WB) identified the nerve growth-associated protein 43 (GAP43). WB and immunofluorescence quantified inflammatory and oxidative stress markers. POL’s regulatory function within the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) cascade was scrutinized both in vivo and in vitro.
POL intervention induced no systemic organ toxicity. POL-treated mice exhibited pronounced locomotor function enhancement, diminished neuronal tissue depletion, elevated neuronal survival, and attenuated demyelination. RNA-seq analysis illuminated POL’s SCI therapeutic mechanism linkage to axonal regeneration, the phosphatidylinositol signaling apparatus, and the neuronal framework. POL concurrently attenuated glial scar formation and potentiated axonal and myelin regeneration. Mechanistically, POL suppressed pro-inflammatory cytokines and oxidative stress mediators while activating the PI3K/AKT/mTOR pathway.
POL mitigated murine spinal cord injury-induced neuroinflammation and oxidative stress through PI3K/AKT/mTOR signaling pathway activation. Furthermore, POL treatment contracted the glial scar expanse within the injury epicenter and fostered axonal regeneration coupled with myelin regeneration. Consequently, POL enhances post-SCI motor function and accelerates neural function restoration.
Keywords
- axon
- neuroinflammation
- oxidative stress
- PI3K
- poliumoside
- spinal cord injury
Spinal cord injury (SCI) arises from direct or indirect mechanical trauma, precipitating partial sensation or motor function forfeiture below the lesion site, variable disability levels, or complete paralysis plus diverse complications [1]. Its elevated disability incidence and grave complications impose considerable socioeconomic encumbrance [2]. SCI pathophysiology encompasses primary mechanical trauma and secondary damage. Primary injury stems from the initial traumatic spinal cord assault, inflicting irreversible neural tissue structural compromise. Subsequently, ischemic conditions and edematous manifestations subsequent to hemorrhagic events and vascular thrombosis intensify the primary insult, culminating in progressive tissue deterioration and functional impairment [3]. Secondary SCI injury hallmarks encompass neuroinflammation, oxidative stress, and demyelination, exacerbating tissue destruction while suppressing axonal regrowth within the lesion zone [4]. Neuroinflammation proves instrumental in post-SCI secondary injury, chiefly orchestrated by central nervous system (CNS) microglia [5].
Microglia, the CNS’s resident immune sentinels, crucially uphold neural
equilibrium during physiological states [6]. Post-SCI, however, microglia undergo
swift activation responding to pathological stimuli, adopting a pro-inflammatory
configuration that liberates excessive inflammatory mediators: tumor necrosis
factor-
Axonal and neural regeneration in the CNS represent a complex yet pivotal biological process critical for functional recovery following SCI, but remain severely limited in adult mammals due to multifaceted intrinsic and extrinsic barriers [11]. The regenerative potential of injured axons depends on the reactivation of growth cones, which requires coordinated regulation of cytoskeletal dynamics through Ras homologous (Rho) GTPases and activation of pro-regenerative signaling pathways including phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) and rat sarcoma virus oncogene (Ras)/rapidly accelerated fibrosarcoma oncogene (Raf)/mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK)/extracellular signal-regulated kinase (ERK), collectively enhancing the neuron’s intrinsic growth capacity [12].
Poliumoside (POL, Fig. 1A) is a natural phenylethanoid glycoside compound [13].
Investigations reveal substantial anti-inflammatory properties, antioxidative
capability, anti-apoptotic action, and neuroprotective potency [14, 15], with POL
mitigating oxidative impairment and pulmonary inflammation within
TNF-
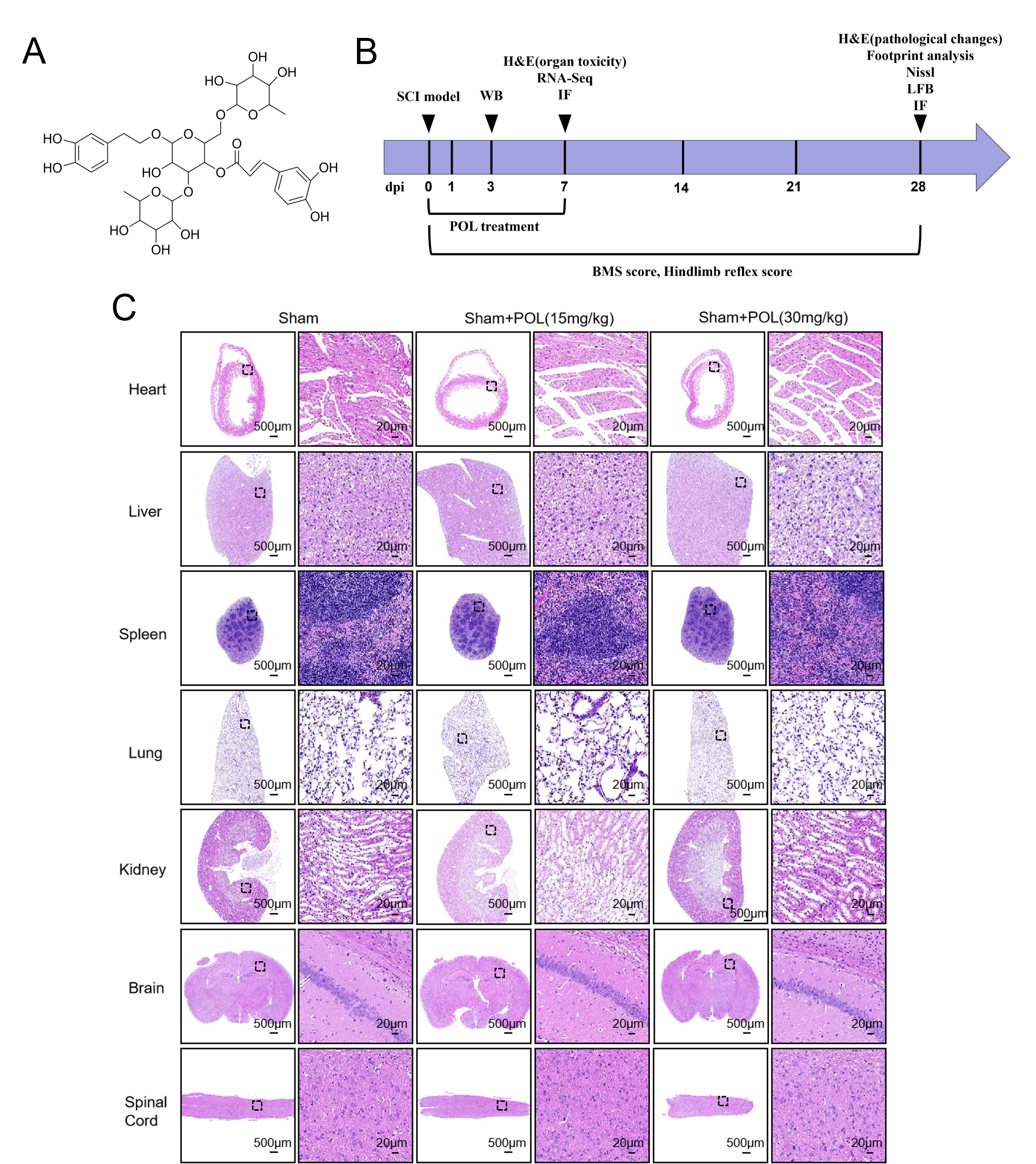 Fig. 1.
Fig. 1.
POL treatment did not induce organ toxicity in SCI mice. (A) POL molecular configuration. (B) Chronological schema for SCI murine interventions and assessments. (C) H&E analysis reveals negligible organic pathology in cardiac, hepatic, splenic, pulmonary, renal, cerebral, or spinal tissues from low/high-dose POL specimens. Scale bars = 500 µm, 20 µm. POL, Poliumoside; SCI, Spinal cord injury; H&E, Hematoxylin-eosin; WB, Western blot; BMS, Basso Mouse Scale; LFB, Luxol Fast blue; dpi, days after injury; IF, Immunofluorescence; RNA-Seq, Ribonucleic Acid Sequencing.
The PI3K/AKT/mTOR signaling cascade engages multifarious regulatory and effector molecules, operating as a principal governor of diverse CNS neurophysiological phenomena encompassing neuronal cellular expansion, viability, and axonal ontogenesis [21]. PI3K/AKT pathway activation proves particularly consequential within SCI’s pathological cascade, since activation delays inflammatory reactions, precludes glial cicatrix formation, and advances functional neurological restitution [22]. Additionally, post-SCI mTOR signaling stimulation exerts multifarious governance across distinct neurobiological processes including axonal reconstitution, neuroinflammatory modulation, and glial scar establishment [23]. Consequently, we probed whether POL intervention modulates the PI3K/AKT/mTOR signaling axis to repress neuroinflammation and oxidative stress while facilitating axonal regeneration after SCI.
Eighty-one female wild-type C57BL/6J mice (8-week-old, 20–30 g) were procured from Nantong University’s Animal Center (Nantong, Jiangsu, China). Specimens were maintained under controlled temperature/humidity with 12-hour photoperiods. Breeding and experimental procedures occurred at Nantong’s Animal Experiment Center under regulatory compliance. All protocols adhered to ARRIVE (2.0) guidelines with ethical approval from Nantong University’s Animal Experiment Ethics Committee (No. S20250318-004). A comprehensive chronological flowchart delineated the experimental design (Fig. 1B).
Anesthesia was induced in the mice via i.p. injection of ketamine (K2753, Sigma-Aldrich, St. Louis, MO, USA) (80 mg/kg). The spinal cord on the T10 vertebrae was struck with a percussion device (68099, RWD Life Science, Shenzhen, China) to produce spinal cord contusion. After impact, the mice still exhibited tail-flick reflexes, and spinal cord hemorrhage was observed in the central region. The mice were placed on a heating pad for recovery. Post-recovery, bilateral hindlimbs showed no ankle joint movement, corresponding to a Basso Mouse Scale (BMS) score of 0, indicating establishment of a successful SCI model. In the sham group, mice underwent laminectomy only, wherein the vertebral laminae were completely removed, followed by sequential suturing of the muscle layer, fascial layer, and skin. For treatment, mice in the SCI group received i.p. injections of POL (HY-N0033, MedChemExpress, Woodbridge, NJ, USA) at doses of 15 mg/kg and 30 mg/kg immediately after successful model establishment and after resuscitation. Subsequent injections were administered every 24 h for 7 consecutive days. Sham and SCI groups received i.p. injection of an equivalent volume of saline daily. To prevent urinary retention, SCI mice underwent daily assisted bladder expression until recovery of spontaneous voiding function.
Motor function recovery in SCI mice was assessed using the BMS, hindlimb reflex score, and footprint analysis. To minimize subjective bias, all evaluations were performed by two independent evaluators, blind to group identification, who were familiar with the experimental protocol but not directly involved in the study. (1) BMS score: hindlimb motor function was assessed using the BMS on days 1, 3, 7, 14, 21, and 28 after SCI (days after injury = dpi) [24]; (2) hindlimb reflex score: At 1, 3, 7, 14, 21, and 28 dpi, mic Subsequentlye were suspended at a height of approximately 30 cm for 14 sec. Hindlimb motor function was assessed using the following scoring criteria: 0, Normal; 1, Incomplete hindlimb extension; 2, Hindlimb clasping; 3, Hindlimb paralysis; (3) Footprint analysis: at 28 dpi, mice were tested for gait analysis using dye-marked limbs (red: forelimbs; blue: hindlimbs) on a paper-lined runway. Quantitative assessment of locomotor recovery included measurements of stride length and stride width from the footprints.
After being deeply anesthetized by intraperitoneal injection of ketamine at a
dose of 80 mg/kg, mice were euthanized by carbon dioxide asphyxiation to ensure
death prior to tissue collection. Subsequently, the abdomen was incised and the
diaphragm was punctured to expose the heart. Cold phosphate-buffered saline (PBS,
ST476, Beyotime, Shanghai, China) and 4% polyformaldehyde (P6148, Sigma-Aldrich) solution were sequentially injected into the heart. After the
perfusion was completed, the damaged central spinal cord segment (1.5 cm in
length) was removed and immediately immersed in 4% polyformaldehyde solution for
preservation. Then, dehydration treatment was carried out and sections were made
using a paraffin sectioning machine (Kedee KD-202A, Hisure, Jinhua, Zhejiang,
China). Thin sections of 5 micrometers were cut longitudinally. For the
immunofluorescence experiment, the preparation process of the tissue samples was
the same as described above. Subsequently, the tissue was stained using the
instructions provided by the Nissl staining kit (G1036, Servicebio, Wuhan, Hubei,
China) and observed under a microscope (DM2500, Leica, Berlin, Germany). The
specific parameters are as follows: Bright field, 4
Hematoxylin-eosin (H&E) staining was executed employing an H&E staining kit
(G1005, Servicebio). Paraffin-embedded spinal cord sections underwent xylene
dewaxing (5 min), graduated ethanol rehydration, hematoxylin immersion (5 min),
eosin counterstaining (2 min), and acid alcohol differentiation (10 sec) prior to
dehydration and mounting. Section evaluation utilized a DM2500 microscope (Leica)
under bright-field illumination (4
Luxol Fast blue (LFB) staining was performed using the LFB staining kit (G1092,
Servicebio). For LFB myelin staining, sections were incubated in 0.1% Luxol Fast
Blue for 2 h, differentiated in lithium carbonate for 10 sec until clear
gray-white matter contrast was achieved, with microscope (DM2500, Leica)
validation of myelin integrity (bright blue signal). The specific parameters are
as follows: Bright field, 4
On the 7th day after SCI, the mice in the SCI and SCI+POL (30 mg/kg) groups were
euthanized, and their spinal cords removed. RNA was extracted from the spinal
cords using TRIzol reagent (EF0131, YiFeiXue, Nanjing, Jiangsu, China). The cDNA
library construction and RNA sequencing were carried out by GeneChem (Shanghai,
China). Seven days post-SCI, euthanized SCI and SCI+POL (30 mg/kg) cohort mice
underwent spinal cord extraction. RNA isolation employed TRIzol reagent
(YiFeiXue). cDNA library preparation and RNA sequencing were executed by
GeneChem. Total RNA (1 µg/sample) underwent library construction using the
NEBNext® Ultra RNA Library Prep Kit (E7530, New England
Biolabs, Ipswich, MA, USA). Poly(A)+ mRNA enrichment utilized
oligo(dT)-immobilized magnetic beads, followed by fragmentation in
NEBNext® First Strand Buffer (94 °C, 15 min).
First-strand cDNA synthesis deployed random hexamers with M-MuLV Reverse
Transcriptase; second-strand synthesis employed DNA Polymerase I. After end
repair and 3′ adenylation, NEBNext® adaptors were ligated.
Size selection (250–300 bp) was performed using AMPure XP beads (A63880, Beckman
Coulter, Brea, CA, USA), followed by USER enzyme treatment (37 °C, 15
min) and PCR amplification with Phusion polymerase. Final libraries were
quality-controlled on an Agilent 2100 Bioanalyzer (G2939A, Agilent Technologies,
Santa Clara, CA, USA). Transcript quantification derived from fragments per
kilobase per million mapped reads. Differential gene expression screening
implemented the DESeq2 algorithm. Genes exhibiting adjusted p-values
Western blot (WB) employed a total protein extraction kit (KGP2100, KeyGEN BioTECH, Nanjing, Jiangsu, China) to isolate proteins from murine spinal cords and microglial cells separately. Protein concentration quantification utilized an enhanced BCA assay kit (P0012, Beyotime, Shanghai, China). Prior to electrophoresis, gel formulation deployed a polyacrylamide gel electrophoresis (PAGE) gel rapid preparation kit (PG211, Epizyme, Shanghai, China). Protein transfer onto polyvinylidene fluoride (PVDF) membranes (IPVH00010, Millipore, Billerica, MA, USA) occurred via rapid transfer solution (35 min duration). Post-transfer, PVDF membranes underwent ddH2O rinsing followed by immersion in freshly constituted 5% skimmed milk (1160GR500, BioFroxx, Guangzhou, Guangdong, China) for 1.5 hours at ambient temperature to effect blocking. Subsequently, membranes underwent primary antibody incubation overnight at 4 °C, succeeded by secondary antibody co-incubation at room temperature for 1 hour the ensuing day. Antibody specifications appear in Table 1. Protein signal acquisition employed an automated chemiluminescence imaging platform (Tanon 5200, Tanon Science & Technology, Shanghai, China), with quantification executed via ImageJ software (Version 1.54r, NIH, Bethesda, MD, USA).
| Antibodies name #Cat. No | Source | Species | Application | Dilution rate |
| iNOS Polyclonal antibody #22226-1-AP | Proteintech | Rb | WB, IF | 1:1000, 1:500 |
| COX2/Cyclooxygenase 2/PTGS2 Monoclonal antibod #66351-1-Ig | Proteintech | Ms | WB, IF | 1:1000, 1:200 |
| TNF-alpha Monoclonal antibody #60291-1-Ig | Proteintech | Ms | WB, IF | 1:1000, 1:200 |
| IL-1 beta Polyclonal antibody #26048-1-AP | Proteintech | Rb | WB, IF | 1:1000, 1:200 |
| IL-6 Rabbit pAb#500286 | Zenbio | Rb | WB, IF | 1:1000, 1:150 |
| NOX2 Polyclonal antibody #19013-1-AP | Proteintech | Rb | WB | 1:2000 |
| NOX4 Polyclonal antibody #14347-1-AP | Proteintech | Rb | WB, IF | 1:2000, 1:500 |
| AKT Polyclonal antibody #10176-2-AP | Proteintech | Rb | WB | 1:1000 |
| Phospho-AKT(Ser473) Monoclonal antibody #66444-1-Ig | Proteintech | Ms | WB | 1:1000 |
| MBP Polyclonal antibody #10458-1-AP | Proteintech | Ms | WB, IF | 1:1000, 1:200 |
| IBA1 Polyclonal antibody #10904-1-AP | Proteintech | Rb | IF | 1:300 |
| Phospho-PI3 Kinase p85/p55 (Tyr467/Tyr199) Rabbit pAb#341468 | Zenbio | Rb | WB | 1:1000 |
| PI3 Kinase p85 alpha (1C8) Mouse mAb #251221 | Zenbio | Ms | WB | 1:1000 |
| Phospho-mTOR (Ser2448) Antibody #2971 | CST | Rb | WB | 1:1000 |
| mTOR Monoclonal antibody #66888-1-Ig | Proteintech | Ms | WB | 1:1000 |
| GFAP(GA5) Mouse mAb #3670 | CST | Ms | IF | 1:600 |
| Neurofilament-H (E7Z7G) Rabbit mAb #30564 | CST | Rb | IF | 1:200 |
| ActivAbTMAnti-GAP43 Monoclonal Antibody #K200056M | Solarbio® LIFE SCIENCES | Ms | WB | 1:2000 |
| Beta Actin Monoclonal antibody #66009-1-Ig | Proteintech | Ms | WB | 1:10,000 |
| Goat Anti-Mouse IgG Secondary antibody (H + L), HRP # YFSA01 | YIFEIXUE BioTech | Goat | WB | 1:10,000 |
| Goat Anti-Rabbit IgG Secondary antibody (H + L), HRP # YFSA02 | YIFEIXUE BioTech | Goat | WB | 1:10,000 |
| Alexa Fluor® 594 AffiniPure Fab Fragment Goat Anti-Rabbit IgG (H + L) #111-587-003 | Jackson ImmunoResearch | Goat | IF | 1:500 |
| Alexa Fluor® 488 AffiniPure Fab Fragment Goat Anti-Rabbit IgG (H + L) #111-547-003 | Jackson ImmunoResearch | Goat | IF | 1:500 |
| Alexa Fluor® 594 AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgG (H + L) #115-586-003 | Jackson ImmunoResearch | Goat | IF | 1:500 |
| Alexa Fluor® 488 AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgG (H + L) #115-546-003 | Jackson ImmunoResearch | Goat | IF | 1:500 |
®: the registered trademark; #: the antibody number.
Abbreviations: iNOS, inducible Nitric Oxide Synthase; COX-2, Cyclooxygenase-2;
TNF-
For the spinal cord paraffin sections, the first step is to deparaffinize and
hydrate, then use the sodium citrate fixative solution (P0081, Beyotime) for
thermal antigen retrieval, followed by adding the endogenous peroxidase strong
blocking solution (P0100B, Beyotime) and incubating at room temperature for 30
min. For the cell slide sections, the spinal cord paraffin sections and cell
slide sections start to be consistent from the following experimental steps. For
immunofluorescence, specimens received immunostaining blocking solution (P0102,
Beyotime) incubated at ambient temperature for 1 hour. Primary antibody
application preceded overnight incubation at 4 °C. The subsequent day witnessed
incubation with corresponding fluorescent secondary antibodies at ambient
temperature for 1 hour (under light-restricted conditions). Antibody particulars
are cataloged in Table 1. 4′,6-Diamidino-2-phenylindole (DAPI, G1012, Servicebio)
solution application ensued until complete specimen coverage, followed by
10-minute ambient incubation. Conclusively, anti-fluorescence quenching mounting
medium (G1401, Servicebio) was applied before coverslip-mediated sample sealing.
The sections were observed with a microscope (DM2500, Leica). The specific
parameters are as follows: Fluorescence imaging, 20
BV-2 microglial cells (CL-0493, Wuhan Pricella Life Science & Technology, Wuhan, Hubei, China) were sustained in Dulbecco’s Modified Eagle Medium (DMEM; KGM12800, KeyGEN, Nanjing, Jiangsu, China) enriched with 10% fetal bovine serum (FBS; 10099141, Gibco, Waltham, MA, USA) and 1% Penicillin-Streptomycin (15140122, Gibco, Grand Island, NY, USA) under 37 °C/5% CO2 conditions. The microglial BV-2 cell line was authenticated by short tandem repeat (STR) profiling. Mycoplasmic contamination was absent in the authenticated BV2 line by using both PCR-based and colorimetric assay methods. To establish a neuroinflammatory paradigm, cultures received 20 µM POL (HY-N0033, MedChemExpress, Woodbridge, NJ, USA) pretreatment for 24 hours preceding 1 µg/mL lipopolysaccharide (LPS; L2880, Sigma-Aldrich) stimulation for 6 hours.
Cellular viability underwent quantification via Cell Counting Kit-8 (CCK-8; C6005, NCM Biotech, Suzhou, Jiangsu, China). BV-2 progeny were subjected to 5 µM, 10 µM, 20 µM, 40 µM, and 80 µM POL exposure for 6 hours, succeeded by CCK-8 incubation (10 µL, 2 hours). Absorbance measurements (450 nm) utilized a Biotek microplate reader (Synergy H1, Agilent BioTek, Winooski, VT, USA), with untreated specimens representing 100% viability controls.
The 3D structure of Poliumoside was obtained from PubChem in structured data
file (SDF) format and converted to mol2 format using OpenBabel (Version 2.4.1,
Open Babel development community). Protein targets (PI3K, AKT, and mTOR) were
retrieved from the UniProt database, and their protein data bank (PDB) files were
processed in PyMOL (Version 2.5.x, Schrödinger, LLC, New York, NY, USA) to remove
water molecules and ligands. Molecular docking was performed using AutoDock Vina
(v1.5.6, Olson Lab, La Jolla, CA, USA), where protein structures were
hydrogenated, and the ligand (POL) was optimized by assigning charges and
rotatable bonds. Molecular docking positioned the search box centrally within the
active site. Optimal binding conformations were selected according to docking
scores (kcal/mol) and visualized via PyMOL. We considered the target proteins
(PI3K, AKT, and mTOR) to exhibit strong binding affinity with Poliumoside when
the binding energy was
Statistical analyses deployed GraphPad Prism (Version 10.4.1, GraphPad Software,
San Diego, CA, USA). Data represent mean
Histopathological interrogation of principal organs (brain, spinal cord, heart, liver, spleen, lung, kidney) via H&E staining. Murine subjects received POL (15 or 30 mg/kg) across seven days. Treated cohorts manifested no significant pathological deviations relative to controls, indicating absent POL-induced organic toxicity at examined dosages (Fig. 1C).
To evaluate the therapeutic effects of POL treatment on motor function recovery
in SCI mice, we used BMS scoring, hindlimb reflex assessment, and footprint
analysis. POL-treated SCI mice demonstrated significantly higher BMS scores than
did untreated SCI controls, with particularly notable improvements at 21 and 28
dpi (p
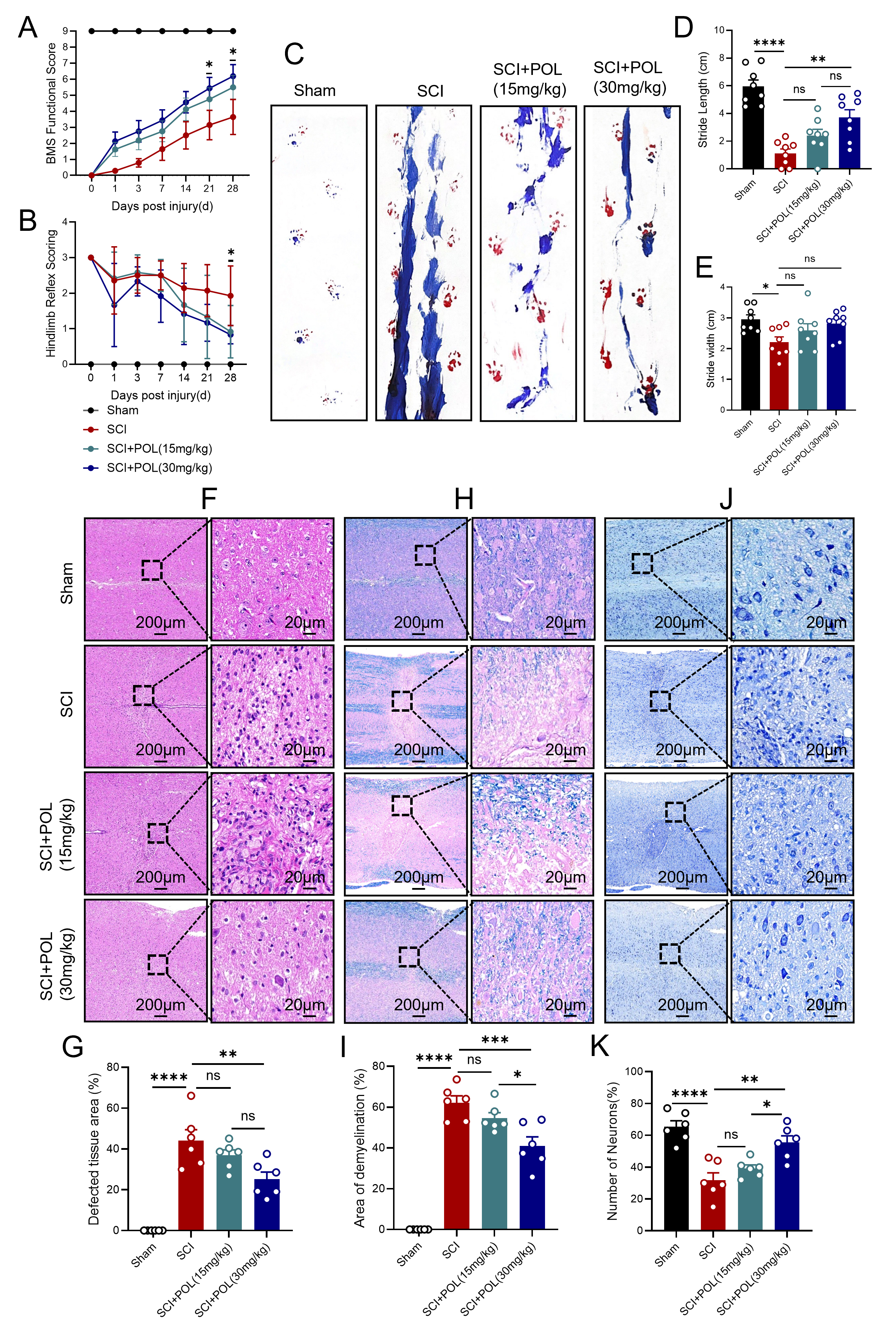 Fig. 2.
Fig. 2.
POL treatment alleviated pathological changes and improved motor
function after SCI. (A) BMS metrics within 28 dpi across sham, SCI, POL-treated
SCI cohorts. (B) Hindlimb reflex metrics at 28 dpi. (C) Exemplar plantar imprints
at 28 dpi. Quantitative stride length (D) and base width (E) analyses. (F)
H&E-stained transverse sections (28 dpi). Scale bars = 200 µm, 20
µm. (G) Lesion area quantification. (H) LFB-stained longitudinal sections
depicting myelination (28 dpi). Scale bars = 200 µm, 20 µm. (I)
Demyelinated area quantification. (J) Nissl-stained longitudinal sections (28
dpi). Scale bars = 200 µm, 20 µm. (K) Surviving neuron
quantification. Data: mean
To further elucidate POL’s mechanistic action in SCI intervention,
transcriptomic interrogation was conducted. Sequencing outcomes delineated 28,940
differentially expressed genes distinguishing SCI cohorts from SCI+POL
counterparts, comprising 1194 substantially upregulated and 1966 markedly
downregulated genes (p
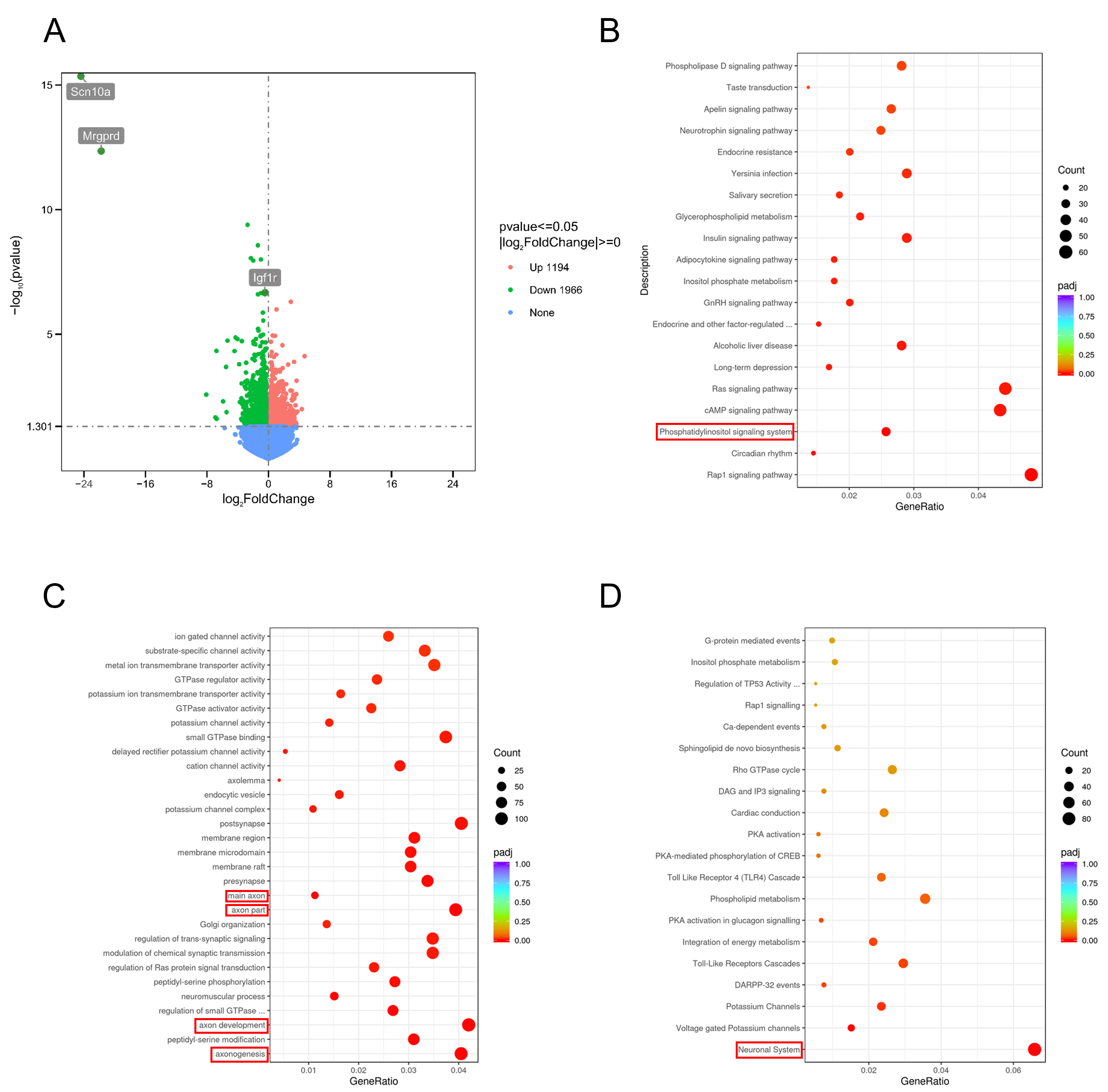 Fig. 3.
Fig. 3.
POL treatment modulated gene expression and regulated axon regeneration-associated pathways after SCI. (A) Differential expression volcano map between SCI and SCI+POL cohorts. (B) KEGG pathway enrichment profiling. (C) Gene Ontology enrichment assessment. (D) Reactome pathway enrichment analysis. KEGG, Kyoto encyclopedia of genes and genomes. In panels B, C, and D, the red boxes highlight the KEGG pathway, GO term, and Reactome pathway, respectively, that were highly enriched and most directly related to the axon and neuroinflammation under investigation. In the three parentheses in the part of the figure references of the results, we have also added “red box” accordingly.
Following spinal cord injury, axonal reconstitution and myelin sheath remodeling
constitute pivotal processes for neural function restoration. Our investigation
documented significantly elevated neurofilament 200 (NF200) axonal density within
lesion areas of POL-treated specimens versus SCI controls through
immunofluorescence, indicating enhanced axonal regeneration (p
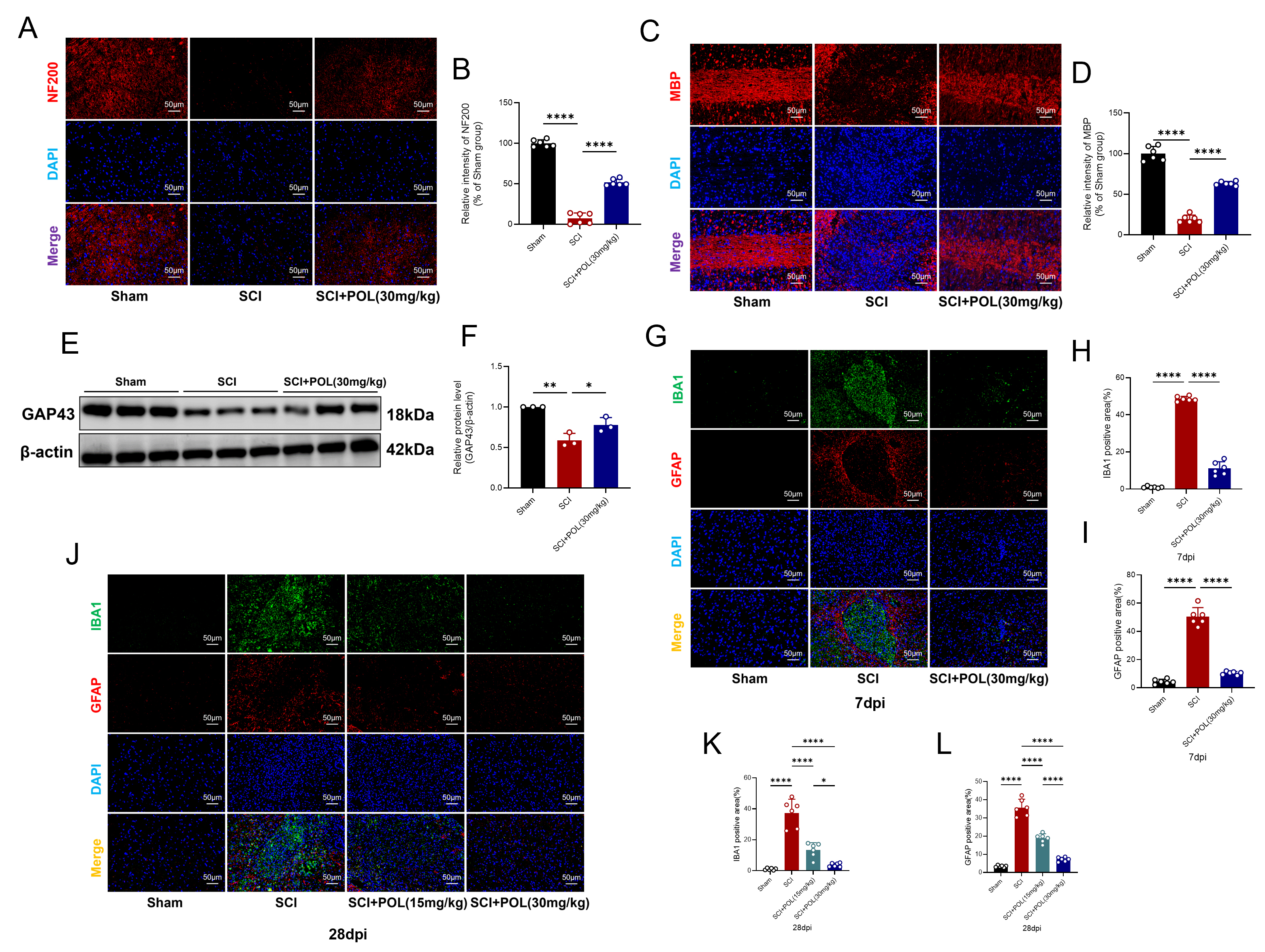 Fig. 4.
Fig. 4.
POL treatment enhanced axonal regeneration and myelin repair
while reducing glial scar after SCI. (A) NF200 axonal distribution (red) in
longitudinal sections (7 dpi). Scale bar = 50 µm. (B) NF200 fluorescence
quantification. (C) MBP myelination (red; 7 dpi). Scale bar = 50 µm. (D)
MBP fluorescence quantification. (E) Representative GAP43 WB (3 dpi).
(F) GAP43 expression quantification. (G,J) Dual IBA1 microglia (green)/GFAP
astrocytes (red) immunofluorescence at 28 dpi (H) and 7 dpi (K). Scale bar = 50
µm. (H,K) IBA1 fluorescence quantification. (I,L) GFAP fluorescence
quantification. Blot data: mean
Following spinal cord injury, activated microglia propagate inflammatory
mediators iNOS and COX-2 while secreting cytokines TNF-
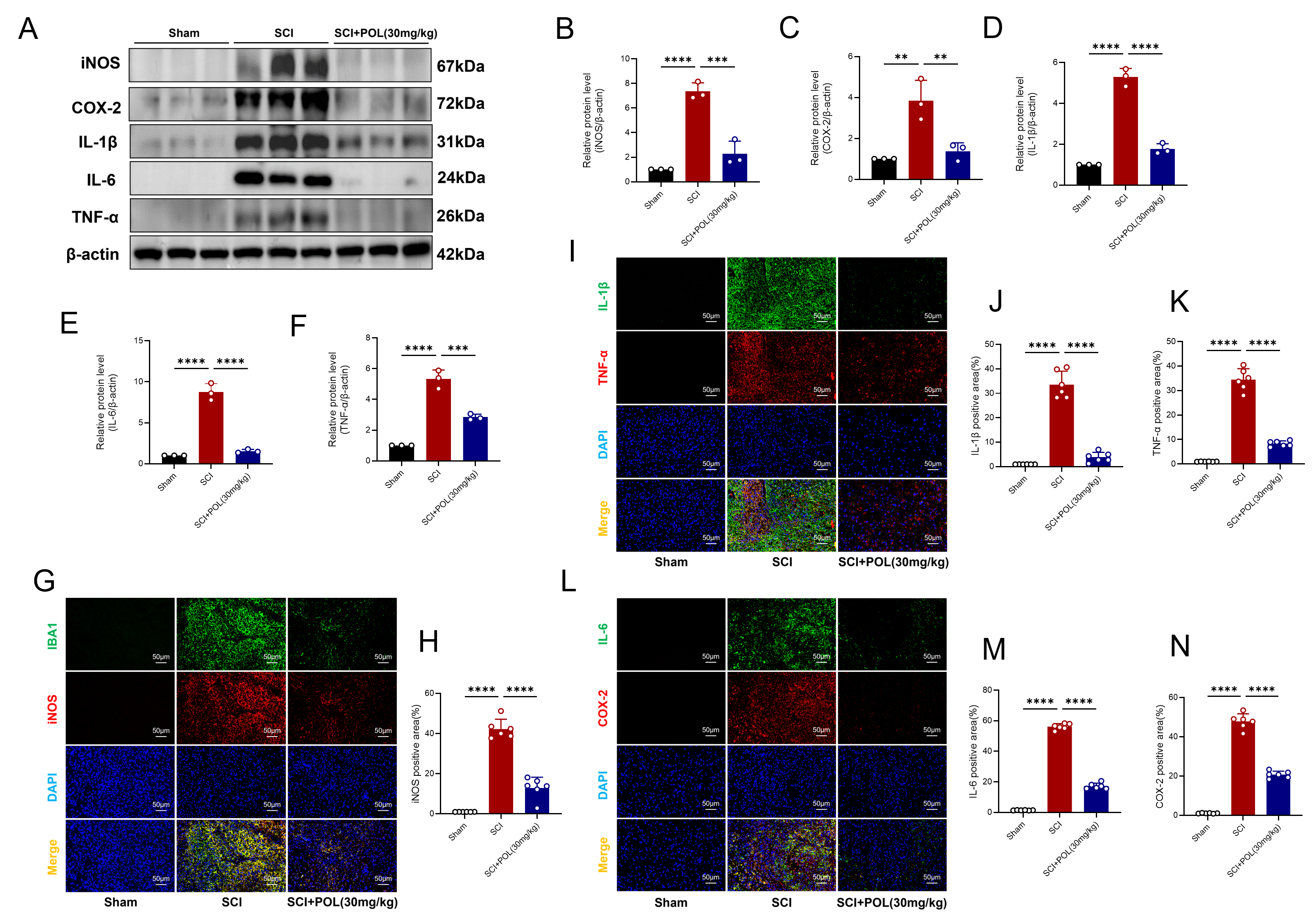 Fig. 5.
Fig. 5.
POL treatment attenuated microglial-mediated neuroinflammation
after SCI. (A) Representative iNOS, COX-2, IL-1
Compared to the injury cohort, POL administration post-SCI significantly
mitigated levels of the oxidative stress-associated proteins NOX2 and NOX4 (NOX2:
p = 0.0406; NOX4: p = 0.0128) (Fig. 6A–C). For all original
WB images in Fig. 6A see the Supplementary material. The
microglial biomarker IBA1 underwent concomitant staining with NOX4.
Immunofluorescence analysis revealed POL substantially diminished NOX4
fluorescence intensity co-localized with IBA1 relative to injured controls (NOX4:
p
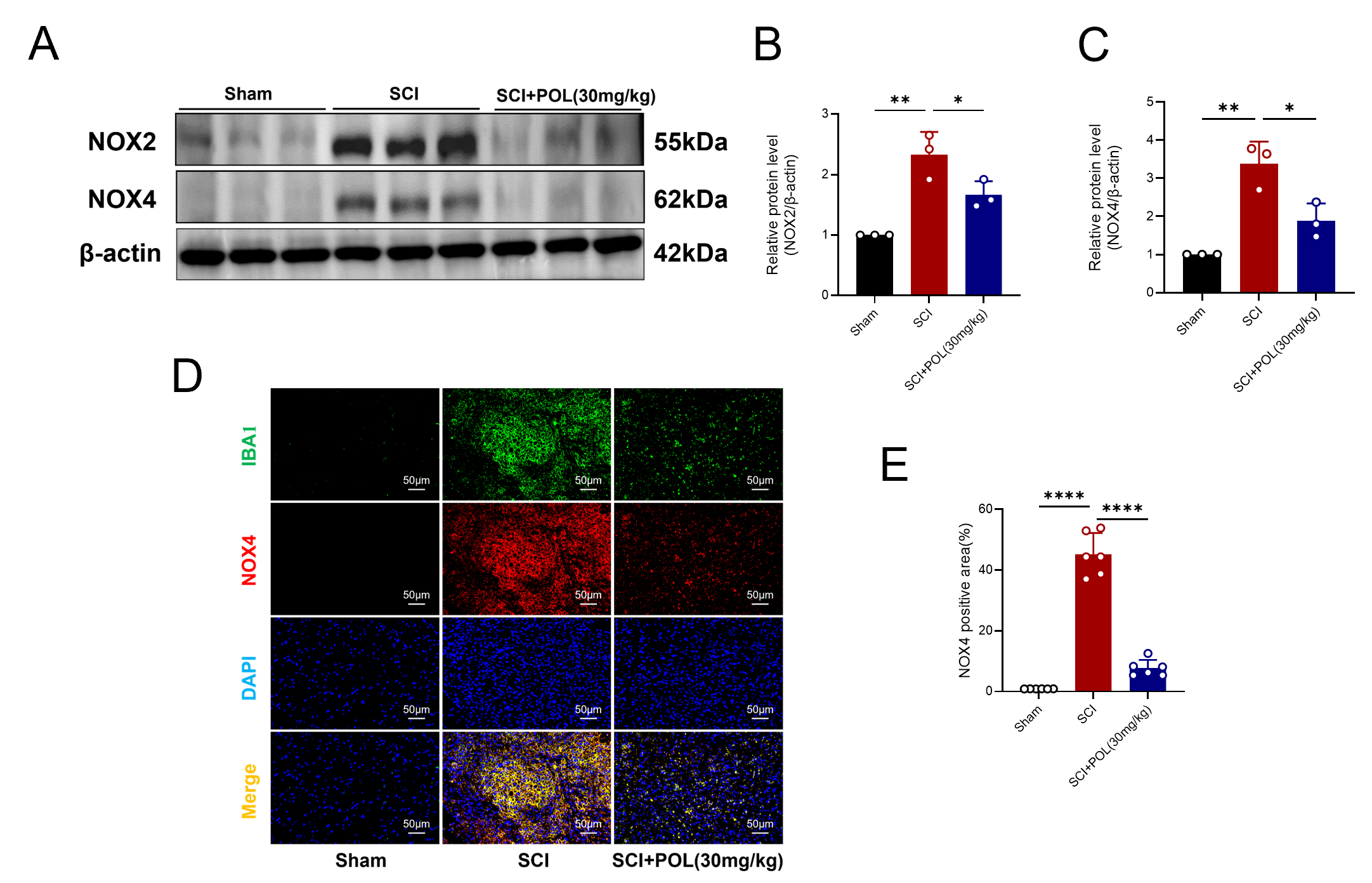 Fig. 6.
Fig. 6.
POL treatment attenuated microglial-mediated oxidative stress
after SCI. (A) Representative WB depict NOX2 and NOX4 levels within
the spinal cord at 3 dpi. (B) Quantitative assessment of NOX2 expression. (C)
Quantitative assessment of NOX4 expression. (D) Dual immunofluorescence labeling
visualized microglia via IBA1 (green) and NOX4 (red) at 7 dpi. Scale bar = 50
µm. (E) Quantification of NOX4 fluorescence intensity. WB
quantification reflects mean
BV-2 microglial cellular viability under varying POL dosages underwent assessment via CCK-8 methodology. Incubation with POL exclusively (5, 10, 20, 40, 80 µM) for 24 hours manifested no viability alterations (Fig. 7A). This investigation consequently elected 20 µM POL for ensuing cellular assays. LPS activation of BV-2 microglia in vitro replicated post-spinal cord injury neuroinflammatory cascades. WB analyses revealed LPS markedly amplified iNOS and COX-2 protein quantities, which POL administration substantially attenuated (iNOS: p = 0.0388; COX-2: p = 0.0207; Fig. 7B–D). For all original Western blot images in Fig. 7B see the Supplementary material. Immunofluorescence imaging demonstrated inflammatory markers iNOS and COX-2 intensification 6 hours post-LPS provocation. Following 20 µM POL intervention, LPS-induced iNOS levels declined (Fig. 7E), with COX-2 exhibiting analogous diminishment (Fig. 7F). These outcomes validate POL’s neuroinflammation-inhibiting efficacy within microglia.
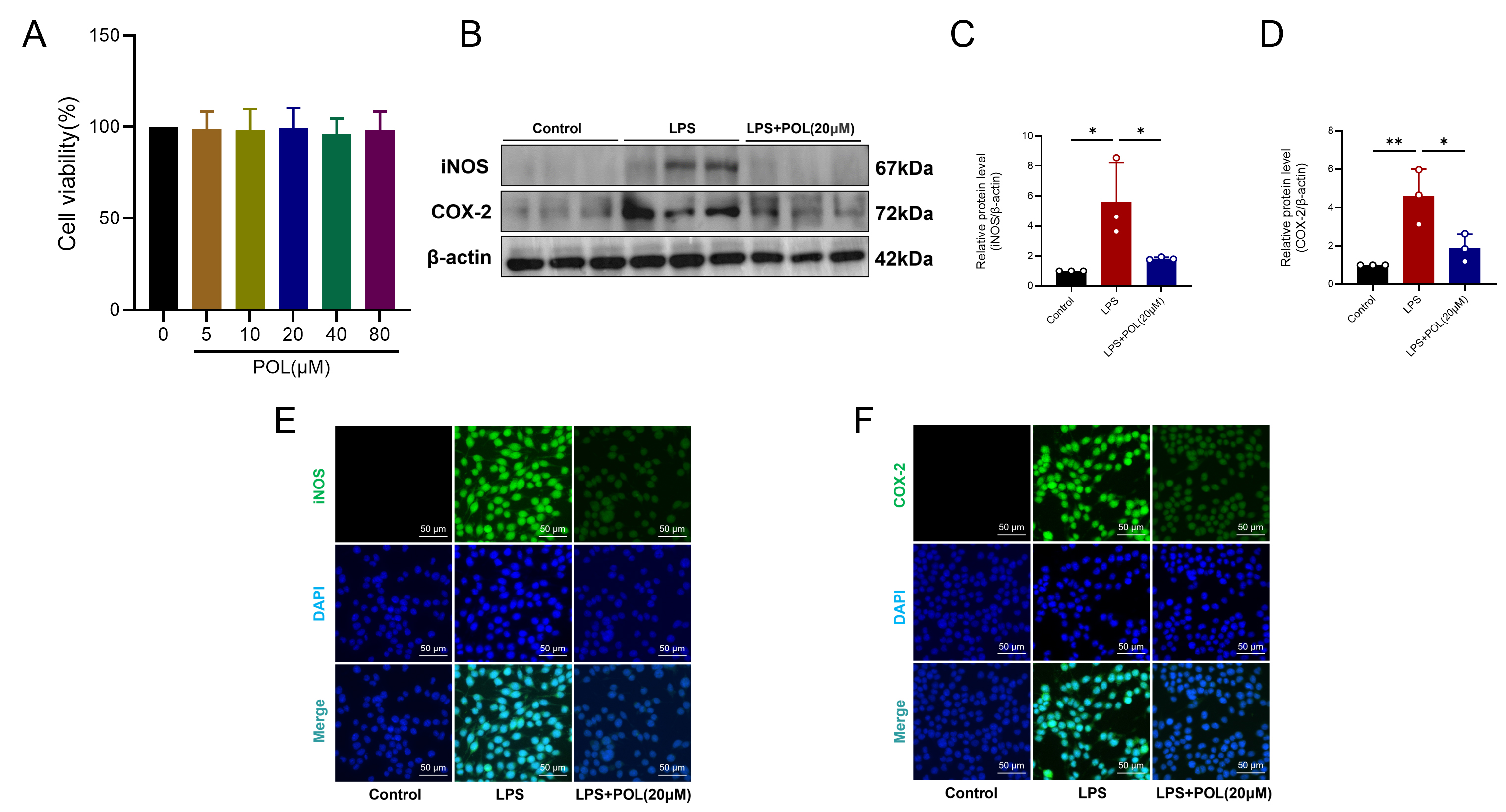 Fig. 7.
Fig. 7.
POL treatment suppressed LPS-induced neuroinflammation in BV-2
microglial cells. (A) CCK-8 assay quantifying cellular viability. (B)
Representative WB depicting iNOS/COX-2 levels in BV-2 microglia after
24-hour POL exposure and 6-hour LPS stimulation. (C) iNOS expression quantitative
evaluation. (D) COX-2 expression quantitative evaluation. (E) Representative
immunofluorescence visualizing iNOS (green) in BV-2 microglia post-treatment.
Scale bar = 50 µm. (F) Representative immunofluorescence visualizing COX-2
(green) under identical conditions. Scale bar = 50 µm. CCK-8 data: mean
To delineate the precise molecular actions of POL against SCI, guided by RNA-Seq
indications, molecular docking assessed POL’s binding capacity towards three
pivotal targets: PI3K, AKT, and mTOR, followed by evaluation of the PI3K/AKT/mTOR
cascade both in vivo and in vitro. Docking simulations revealed
substantial binding affinities between POL and core PI3K/AKT/mTOR constituents,
exhibiting energies of –7.5 kcal/mol (PI3K), –8.8 kcal/mol (AKT), and –9.1
kcal/mol (mTOR), all surpassing the established threshold for potent molecular
binding (
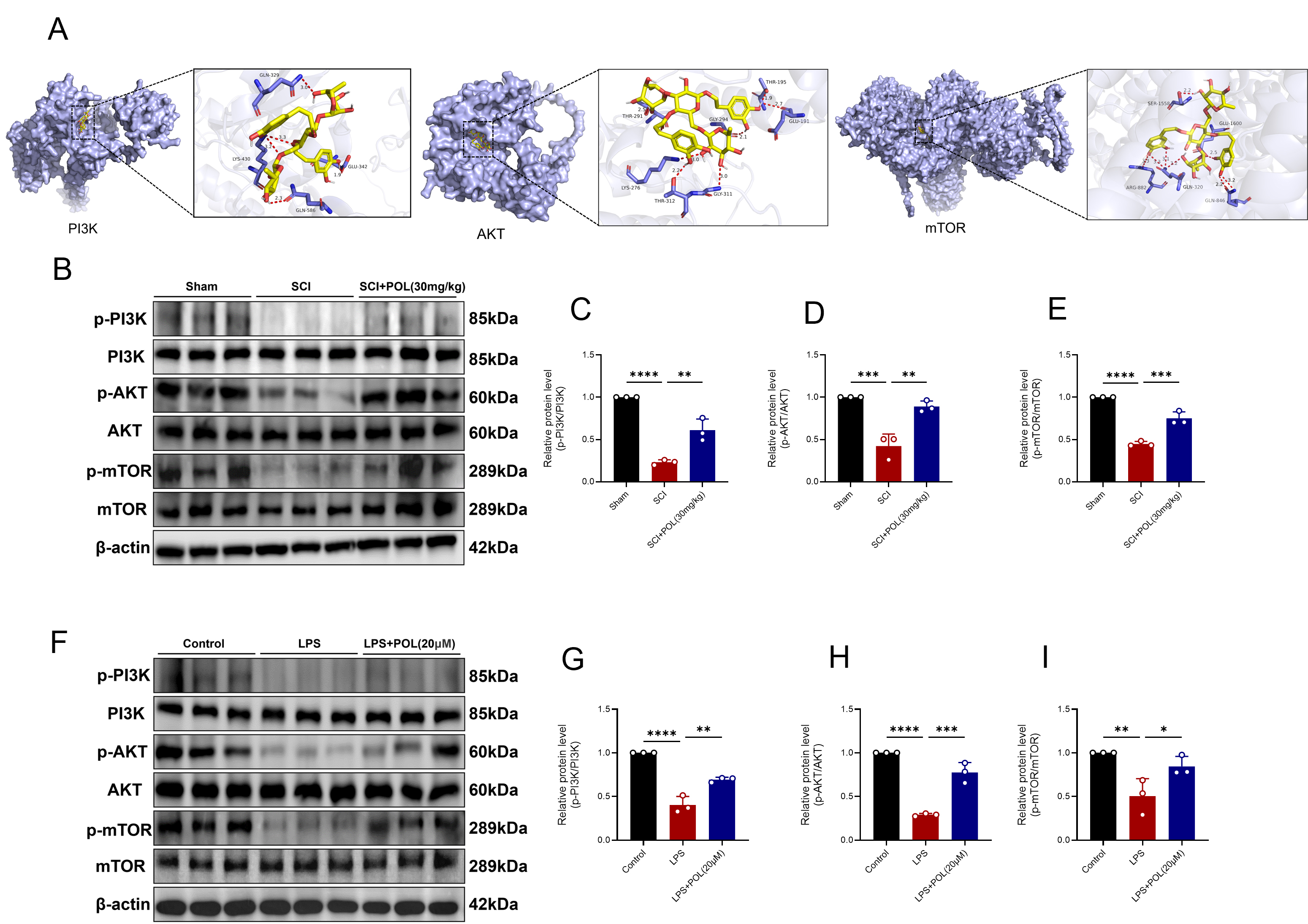 Fig. 8.
Fig. 8.
POL treatment activated the PI3K/AKT/mTOR signaling pathway
after SCI. (A) Molecular docking models depicting POL’s binding interactions
with PI3K, AKT, and mTOR. (B) Representative WB of phosphorylated and
total PI3K/AKT/mTOR proteins in spinal cord tissue at 3 dpi. (C) Quantification
of p-PI3K/PI3K expression levels. (D) Quantification of p-AKT/AKT expression
levels. (E) Quantification of p-mTOR/mTOR expression levels. (F) Representative
immunoblots depicting phosphorylation states and corresponding total PI3K, AKT,
and mTOR proteins in BV-2 microglial cells following 24-hour POL administration
and 6-hour LPS exposure. (G) Quantification of p-PI3K/PI3K expression levels. (H)
Quantification of p-AKT/AKT expression levels. (I) Quantification of p-mTOR/mTOR
expression levels; Findings are presented as mean
SCI constitutes a devastating, persistent, irreversible disability marked by profound sensory and motor deficits [26]. SCI pathophysiology encompasses two discrete stages: primary mechanical damage and secondary injury—the latter denoting progressive pathological cascades initiated post-trauma, involving neuroinflammation, oxidative stress, motor neuron apoptosis, tissue edema, and blood-spinal cord barrier (BSCB) compromise [27]. Consequently, formulating effective early-stage therapeutic interventions to alleviate secondary injury remains imperative. This investigation initially establishes POL administration mediates neuroprotection and enhances functional restoration post-SCI through mitigation of inflammatory processes and oxidative damage. Specifically, in vivo POL treatment substantially diminished neuronal apoptosis, microglia-driven neuroinflammation, oxidative injury, demyelination, and glial scarring, while concurrently promoting axonal regrowth. In vitro, POL treatment attenuated LPS-induced microglial inflammatory reactions, aligning with prior observations [17]. Mechanistically, our evidence suggests POL likely orchestrates these safeguarding outcomes via PI3K/mTOR/AKT signaling pathway activation. Taken together, these discoveries furnish novel mechanistic understanding of POL’s neuroprotective function during SCI recuperation, underscoring its early-intervention therapeutic promise.
POL, a phenylethanoid glycoside, possesses anti-inflammatory, antioxidant, anti-apoptotic, and neuroprotective properties [28]. Prior research examining Forsythoside B (a structural analog among phenylethanoid glycosides) for SCI management utilized animal doses of 10 mg/kg and 40 mg/kg [29]. Consequently, this investigation employed two analogous POL concentrations (15 mg/kg and 30 mg/kg) administered intraperitoneally. These doses also were consistent with those used in recent research that demonstrated POL’s efficacy in mitigating microglia-mediated neuroinflammation after ischemic stroke at 5, 10, and 15 mg/kg doses [19]. Given the absence of prior toxicity data for POL, we first evaluated its systemic effects in healthy mice, particularly in CNS tissues. H&E staining confirmed the absence of toxicological effects at both 15 mg/kg and 30 mg/kg doses. Behavioral tests directly demonstrated the ability of POL treatment to enhance motor function recovery post-SCI, and histological analyses revealed reduced demyelination, increased neuronal survival, and diminished lesion areas. Notably, the 30 mg/kg dose exhibited significantly better therapeutic outcomes than did 15 mg/kg, which warranted its selection for subsequent experiments. Beyond established anti-inflammatory and antioxidant effects, RNA-Seq analysis was conducted to elucidate POL’s mechanistic underpinnings in SCI. Results indicated POL’s robust promotion of axonal regeneration and active involvement in nervous system biological processes, potentially mediated by downstream PI3K/AKT/mTOR pathway modulation.
Axons, constituting fundamental structural and operational units within neural circuitry, serve an indispensable function in neuronal signal transmission. Thus, injuries affecting either the central or peripheral nervous systems mandate axonal regrowth to reinstate neurological capabilities [30]. Axonal remodeling and regeneration represent pivotal determinants for neural network reconstitution following spinal cord trauma [31]. Substantial evidence indicates microglia-driven neuroinflammation and oxidative stress provoke liberation of neurotoxic agents post-SCI, subsequently yielding excessive glial proliferation, cicatrix formation, suppression of axonal regeneration, and exacerbation of neural tissue destruction [32, 33], thereby impeding motor function recovery. These observations align with our experimental outcomes demonstrating POL treatment attenuated microglia-mediated neuroinflammation and oxidative stress after SCI, consequently inhibiting post-injury glial scar development while facilitating axonal regrowth and myelin restoration.
The PI3K/AKT/mTOR signaling cascade—comprising PI3K, AKT, and mTOR—serves as a master regulator of fundamental cellular processes including growth, proliferation, survival, and metabolic homeostasis [34]. Accumulating evidence underscores its neuroprotective potential following SCI [22, 35]. For example, EX-netrin1 regulated inflammation and axon growth after SCI via the Unc5b/PI3K/AKT/mTOR pathway [36]. Similarly, a neural regeneration-enhanced triple-network hydrogel promotes neuronal differentiation, inhibits astrocyte differentiation, and supports axonal regeneration via activating the PI3K/AKT/mTOR signaling pathway [37]. Our in vivo and in vitro findings corroborate these neurorestorative properties. Intriguingly, KEGG pathway enrichment analysis suggests that POL’s therapeutic mechanism in SCI may also involve the Ras signaling pathway. Ras, a small GTP-binding protein, functions as a molecular switch at the cell membrane to initiate signaling cascades promoting cell growth and proliferation, with Raf kinase serving as its primary effector [38]. This is supported by recent work of serum response factor (SRF)-mediated axonal regeneration via Ras/Raf/cofilin signaling demonstrating [39]. Moreover, LNT-UsSeNPs directly regulated the PI3K/AKT/mTOR and Ras/Raf/MEK/ERK signaling pathways by regulating selenoproteins to achieve non-immunosuppressive anti-inflammatorytherapy [40]. Therefore, the potential involvement of POL in modulating the Ras signaling pathway opens a promising new avenue for comprehensively elucidating its therapeutic mechanisms in SCI.
Based on RNA-Seq results, we observed significant downregulation of Igf1r, an axonal regeneration-related target gene, following POL treatment. Igf1r encodes a transmembrane tyrosine kinase receptor orchestrating multifunctional participation within SCI pathophysiology, governing cell proliferation, survival, metabolic processes, and axonal regeneration [41]. Prior research established that combining twelve weeks of treadmill training with OPN overexpression enhanced axonal regeneration and motor capabilities through activation of the IGF-1R/Akt/mTOR pathway. Analogously, we postulate that POL potentially activates the PI3K/AKT/mTOR signaling cascade by targeting Igf1r, thereby augmenting axonal regeneration; this offers crucial perspectives for discerning upstream therapeutic targets of POL in SCI management. Furthermore, Scn10a and Mrgprd constituted the two most substantially downregulated genes. Specifically, Scn10a encodes Nav1.8, a voltage-gated sodium channel exhibiting predominant expression within sensory neurons; within these neurons, Nav1.8 proves indispensable for generating and conducting action potentials. Nav1.8 plays a critical role in nociception due to its high expression in nociceptive neurons [42]. Mrgprd is primarily expressed in sensory neurons, particularly those mediating pain transmission, and functions to recognize and respond to specific neuropeptides and chemical substances that act as pain-signaling mediators [43]. In the chronic phase of SCI, approximately 90% of patients develop neuropathic pain below the injury level, significantly hindering rehabilitation [44]. Given that persistent inflammation across spinal segments contributes to post-SCI neuropathic pain, it is worth further investigating whether POL alleviates neuropathic pain by targeting Scn10a and Mrgprd and whether this effect is linked to its anti-inflammatory properties.
However, this study also has some limitations. First, in the cell experiments, we only verified the therapeutic effect of POL on BV2-mediated neuroinflammation, but did not explore the oxidative stress phenotype at the cellular level. Second, the optimal drug concentration of POL in cell experiments was not studied in detail. And last, the activation of the PI3K/AKT/mTOR signaling pathway was only simply verified; its association with neuroinflammation, oxidative stress, and axonal regeneration was not verified with a rescue experiment. Therefore, the directions for future research are whether POL affects other biological processes and related targets related to SCI, as well as other specific regulatory mechanisms related to POL’s regulation of axonal regeneration. Subsequent investigations will examine combinations of POL with hydrogels, emerging nanomaterials, stem cells, and additional therapeutic agents to assess drug targeting efficacy and optimize delivery systems; relevant clinical trials will be integrated to accelerate clinical translation.
POL administration mitigated neuroinflammation and oxidative stress consequent to murine SCI through activation of the PI3K/AKT/mTOR signaling cascade. Furthermore, POL intervention diminished glial scar extent within injury epicenters while stimulating axonal regeneration and myelin restoration. Consequently, POL therapy enhances post-SCI motor function and facilitates neural functional recovery.
The data during the research are available from the corresponding author on reasonable request.
QL conceived and designed the study, performed the experiments, and drafted the manuscript. HH and GX contributed to the study design. MX and TW were responsible for data collection. ZZ and JH carried out data analysis. QF contributed to the experimental design, provided suggestions and assistance in establishing the mouse model of SCI, and participated in the preparation and organization of the figures. ZC made substantial contributions to the conception and design, provided critical revision of the manuscript for important intellectual content, and approved the final version. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal study was reviewed and approved by the Animal Experiment Ethics Committee of Nantong University (No. S20250318-004) and use Committee and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
We would like to express our gratitude to all the reviewers for their constructive suggestions for this research.
This research was funded by Project of Jiangsu Administration of Traditional Chinese Medicine (No. MS2024092, MS2022090) and Jiangsu Provincial Health Commission Project (No. ZQ2024026).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN43900.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.