

 , Bin Xu 3,*
, Bin Xu 3,*1 The Second Clinical Medical College of Zhejiang Chinese Medical University, 310000 Hangzhou, Zhejiang, China
2 Department of Neurology, Zhejiang Hospital Affiliated to Zhejiang University, 310000 Hangzhou, Zhejiang, China
3 Department of Neurology, The Second Affiliated Hospital of Zhejiang Chinese Medical University, 310000 Hangzhou, Zhejiang, China
Abstract
An individual's quality of life is greatly affected by Parkinson's disease (PD), a prevalent neurological degenerative condition. Rapid eye movement (REM) sleep behavior disorder (RBD) is a prominent non-motor symptom commonly associated with PD. Previous studies have shown a close relationship between PD and RBD. In addition to being a prodromal symptom of PD, RBD has a major negative impact on the prognosis of PD patients. This intrinsic connection indicates that there is a bidirectional relationship between PD and RBD. This paper provides a comprehensive review of the pathological mechanism related to PD and RBD, including the α-synuclein pathological deposition, abnormal iron metabolism, neuroinflammation, glymphatic system dysfunction and dysbiosis of the gut microbiota. Increasing evidence has shown that RBD patients have the same pathogenic mechanisms that underlie PD, but relatively little research has been done on how RBD contributes to PD progression. Therefore, a more thorough investigation is warranted to characterise how RBD affects the course of PD, in order to prepare for future therapeutic trials.
Keywords
- α-Synuclein
- glymphatic system
- intestinal flora
- iron metabolism
- neuroinflammation
- Parkinson's disease
- REM sleep behavior disorder
Parkinson’s disease (PD) is a common neurodegenerative illness. The main motor impairments of this disease are static tremor, muscle rigidity, bradykinesia, and postural instability. Non-motor symptoms include psychiatric disorders, sleep disorders, and olfactory dysfunction [1, 2, 3]. Among these symptoms, sleep issues were the second most frequent non-motor symptom after psychiatric illnesses, accounting for 64% of newly diagnosed PD patients in the largest survey of non-motor symptoms [3]. Sleep issues include excessive daytime sleepiness, rapid eye movement (REM) sleep behavior disorder (RBD), difficulty in initiating and maintaining sleep, parasomnias, restless leg syndrome, periodic limb movements of sleep, and obstructive sleep apnea [4]. Currently, RBD is considered to be a premotor marker for neurodegenerative disorders such as PD, multiple system atrophy (MSA), and dementia with Lewy bodies (DLB) [5].
REM sleep without atonia (RSWA) and the presence of aberrant behaviors that are
usually connected to dream content during REM sleep, are the hallmarks of
RBD [6]. Patients with RBD frequently have abnormal motor actions, such as
limb jerks, punching, kicking, or biting, as well as abnormal vocalisations, such
as shouting, swearing, laughing, or sobbing. Patients may potentially endanger
their bedmates, and even themselves by falling out of bed. RBD patients
frequently experience unpleasant or violent dreams, usually involving animal or
human attacks or pursuits. These dreams are intimately linked to the anomalous
actions that occur during REM sleep [7, 8, 9]. Despite the fact that we have
been aware of RBD symptoms for a long time, the precise pathogenic causes are
still unknown [10]. In rodents, there are two pathways that induce atonia
during REM sleep: (1) the sublaterodorsal nucleus (SLD) (equivalent to subcoeruleus nucleus in humans) in which
glutamatergic/
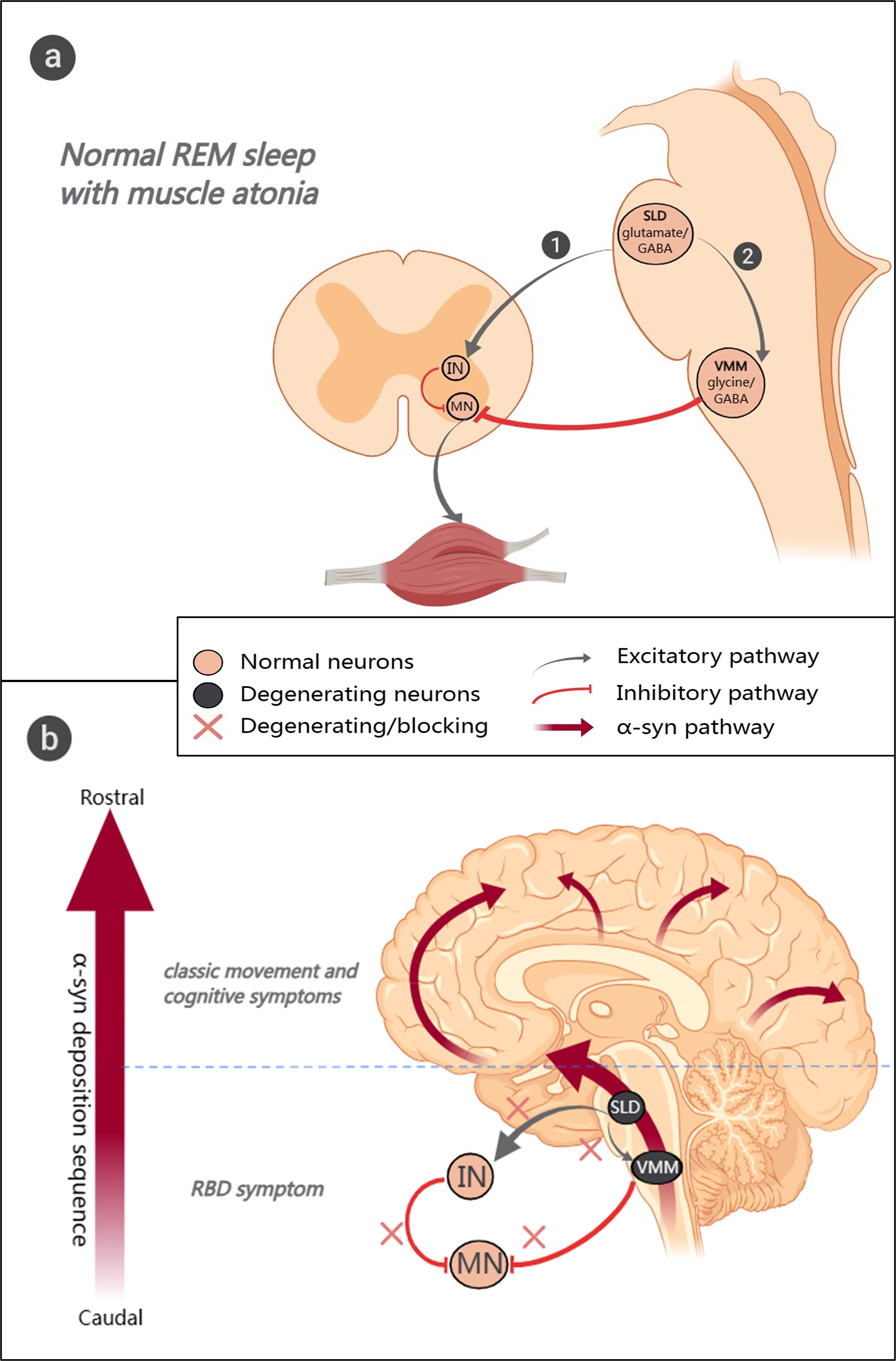 Fig. 1.
Fig. 1.
The normal circuits of REM sleep with atonia and the course of
In recent years, there has been increasing research interest in the possible association between PD and RBD, aiming to explore the mechanisms underlying their relationship and provide new insights for treatments that delay disease progression [15, 16, 17]. Therefore, this paper provides a comprehensive review of the relationship between the two, as well as the mechanisms involved.
On the one hand, RBD is considered as a prodromal symptom of PD and a clinical
marker for predicting PD development [18, 19]. The majority of patients with
idiopathic RBD (iRBD) eventually had developed synucleinopathies, including PD,
DLB, and MSA, at a 15-year follow-up [13]. A multicentre study involving 1280
iRBD patients showed an annual conversion rate of 6.3% from iRBD to
neurodegenerative diseases, with over half progressing to PD [20]. Braak
stages I through VI are used to categorise the pathological course of PD based on
the deposition sequence of
On the other hand, RBD symptoms are often associated with the severity and prognosis of individuals with PD. A recent study [25] compared the sequence of involvement of critical brain regions in PD patients with the onset of RBD occurring after motor symptoms (PD-postRBD), patients with RBD preceding motor symptoms (PD-preRBD), and patients without RBD symptom (PD-nonRBD). The researchers found that the PD-preRBD group and the PD-postRBD group shared a similar spatiotemporal sequence of neurodegeneration, but neither of them as PD-nonRBD is, which indicates that the occurrence of RBD symptoms in the course of PD reflects the different spatial and temporal progressions of the lesion in the brain. PD patients with accompanying RBD symptom (PD-RBD) exhibit a more aggressive progression of neurodegeneration [26, 27, 28]. Those patients often have more severe muscle rigidity and axial motor symptoms, and a higher risk of developing levodopa-induced dyskinesia. Moreover, they experience more severe autonomic dysfunction and a higher risk of cognitive impairment [29, 30, 31]. The results suggest that the pathological course of PD proceeds more quickly when paired with RBD, suggesting a particular pattern of neurodegeneration.
In conclusion, there is a bidirectional relationship between PD and RBD, and the underlying mechanisms behind this relationship require further investigation.
The primary pathogenic characteristic of PD is the aberrant aggregation of
amyloid-like protein (
Since most iRBD patients eventually develop PD and DLB, which are marked by
The loss of those dopaminergic neurons in the substantia nigra pars compacta
that contain neuromelanin, along with iron deposition, is another characteristic
neuropathological hallmark of PD [43]. Abnormal iron metabolism can induce
oxidative stress, hasten
Abnormal iron deposition in the locus coeruleus and substantia nigra of iRBD patients has been found as well [24, 51], although the increase in iron content in the substantia nigra is not as significant as in PD patients, and the iron content in the substantia nigra is positively correlated with disease duration and severity [24]. However, two other studies [52, 53] did not find statistically significant differences in iron content between iRBD patients and healthy controls. The reason for this variation could be the use of different imaging assessment methods to quantify brain iron content in the studies. Relevant investigations have consistently reported lower sensitivity of neuromelanin-sensitive signals in the substantia nigra and locus coeruleus-subcoeruleus complex of individuals with iRBD than in healthy controls [53, 54, 55, 56, 57]. Furthermore, temporal alterations in neuromelanin, dopaminergic neurons, and iron metabolism of iRBD patients follow a similar pattern to those of PD patients [51, 54]. These findings suggest that aberrant iron metabolism is present in both PD and iRBD, and more study is required to examine iron metabolism in PD-RBD patients.
Neuroinflammation typically refers to a chronic immune response in the central
nervous system. In the neurodegenerative process of PD,
Positron emission tomography (PET) study has found neuroimaging evidence of
central immune activation in iRBD patients [64], accompanied by lower
dopamine activity levels in the nigrostriatal pathway. Nonetheless, no
discernible variations in peripheral pro-inflammatory mediators (IL-1
After exchanging solutes with interstitial fluid and entering the brain parenchyma through the perivascular space of the arteries, cerebral spinal fluid from the subarachnoid space leaves the brain through the perivascular spaces of veins. Here, it functions as a “lymphatic fluid” in the central nervous system (CNS), eliminating waste products from the brain and supporting immunological surveillance [68, 69]. There has been evidence of lymphoid system dysfunction in PD patients, as evidenced by enlarged perivascular space (EPVS) [70], decreased diffusion tensor image analysis along the perivascular space (DTI-ALPS) index [71], reduced signal intensity in global blood-oxygen-level-dependent (gBOLD) [72], and degenerative changes in nigrostriatal dopaminergic neurons linked to EPVS in PD patients [70]. As of now, aquaporin 4 (AQP4) depolarisation or fluid flow obstruction-related lymphatic exchange problems are thought to be pathogenic and progressing components of PD [73].
The lymphoid-like system changes that are associated with iRBD have drawn significant attention as a potential precursor manifestation of synucleinopathies. Si et al. [74], by evaluating magnetic resonance image (MRI)-visible EPVS loads, found that EPVS loads were higher in patients with iRBD than in controls and in patients with PD, a phenomenon that may be related to the compensatory role of EPVS loads in the progression of iRBD to PD. Three other studies [75, 76, 77] assessed the lymphoid system in iRBD patients using DTI-ALPS, and consistently showed that the DTI-ALPS index of iRBD patients was lower than that of healthy controls, suggesting that iRBD patients had lymphoid system dysfunction. In two of the studies showing contradictory results, Bae et al. [77] did not detect a statistically significant difference between the ALPS indices of PD patients and iRBD patients, whereas Si et al. [76] observed that the ALPS index of PD patients was lower than that of iRBD patients. Even so, in Si’s study the mean ALPS index for PD patients was still lower than that of iRBD patients. During the follow-up of patients with iRBD and PD, researchers found a negative correlation between ALPS index and the risk of phenotypic transition from iRBD to PD [77], as well as a negative correlation between ALPS index and the severity of clinical symptoms in both groups [76]. Those studies suggested that lymphoid system dysfunction is closely related to the course of neurodegeneration in patients with iRBD. However, it is still necessary to explore whether there are alterations in AQP4 polarity and hemodynamics in the intracranial lymphoid system of patients with iRBD, with a goal of early identification of neuromodifying therapies that slow down the progression of iRBD to PD.
Studies examining how RBD lesions impair the lymphoid system and exacerbate PD
pathologic alterations are, nevertheless, comparatively rare. In the above EPVS
study, Si et al. [74] found no significant difference in EPVS load
between the PD-RBD and PD-nonRBD groups, a finding which, the authors believed,
was related to the consistent difference in severity of clinical symptoms of the
two groups. In addition, recently, Wang’s team [78] adopted the low-frequency
band (
Gut microbiota dysbiosis is believed to play an important role in the
pathogenesis of PD. Several studies [79, 80, 81] have reported the impact of the
microbiota-gut-brain axis on the pathophysiology of PD, emphasising its crucial
role in the occurrence and progression of Lewy body diseases. In PD patients,
alterations in the gut microbiota may result in a reduction of short-chain fatty
acids, T-cell activation, and an increase in intestinal permeability, all of
which can trigger an inflammatory response. Through these pathways,
According to a number of studies [80, 81, 87], patients with iRBD have changes in their intestinal flora similar to those of patients who have PD, including the enrichment of Desulfovibrio and Collinsella, and the depletion of Butyricicoccus, Faecalibacterium, and Lachnospira. The researchers found that the depletion of bacteria of Butyricococcus and Fecalobacteria was a specific marker to distinguish patients with iRBD and PD-RBD from patients with PD-nonRBD, and also found a negative correlation between the amount of these two bacteria and RBDQ-HK scores [87], so the authors believed that these two bacteria were specific markers of RBD symptoms. Furthermore, the characteristic features of PD-like gut microbiota dysbiosis (depletion of Lachnospira and Butyricicoccus) have been observed even in the prodromal stage of RBD and in first-degree relatives of RBD patients [81]. These studies suggest that changes in intestinal flora associated with the development of PD not only occur in the early stages of PD but even manifest in the early stages of RBD, thus necessitating early screening and intervention in populations at high risk for synucleinopathies. Although existing studies have found similar gut microbiota changes in RBD and PD, the underlying pathogenic mechanisms of these microbiota in the phenotypic transition of iRBD and the progression of PD remain unclear.
RBD symptoms can not only be regarded as a warning sign for the development of PD, but also as an adverse factor affecting its prognosis, indicating a bidirectional relationship between PD and RBD. Previous research has shown that iRBD patients exhibit various pathological changes similar to PD; future research should focus on improving and supplementing the inadequate understanding of the pathological processes by which RBD exacerbates the evolution of PD. The exploration of the relationship between PD and RBD, and the possible mechanisms behind it, has suggested the possibility that we can target neuromodification therapy and long-term management of the disease to delay, or even block, further disease progression.
PD, Parkinson’s disease; REM, Rapid eye movement; RBD, Rapid eye movement sleep behavior disorder; DLB, Dementia with Lewy bodies; MSA, Multiple system atrophy; RSWA, REM sleep without atonia; SLD, Sublaterodorsal nucleus; GABA,
YZ conceptualizad the main idea and writing framework, investigated the research progress in the field, wrote the original draft and drawn figure. XL participated in resources acquisition, validation and supervision. BX formulated the research goal, provided research materials, revised original manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.